

Abstract
Background
In circulating influenza viruses, gradually accumulated mutations on the glycoprotein hemagglutinin (HA), which interacts with infectivity-neutralizing antibodies, lead to the escape of immune system (called antigenic drift). The antibody recognition is highly correlated to the conformation change on the antigenic sites (epitopes), which locate on HA surface. To quantify a changed epitope for escaping from neutralizing antibodies is the basis for the antigenic drift and vaccine development.
Results
We have developed an epitope-based method to identify the antigenic drift of influenza A utilizing the conformation changes on epitopes. A changed epitope, an antigenic site on HA with an accumulated conformation change to escape from neutralizing antibody, can be considered as a "key feature" for representing the antigenic drift. According to hemagglutination inhibition (HI) assays and HA/antibody complex structures, we statistically measured the conformation change of an epitope by considering the number of critical position mutations with high genetic diversity and antigenic scores. Experimental results show that two critical position mutations can induce the conformation change of an epitope to escape from the antibody recognition. Among five epitopes of HA, epitopes A and B, which are near to the receptor binding site, play a key role for neutralizing antibodies. In addition, two changed epitopes often drive the antigenic drift and can explain the selections of 24 WHO vaccine strains.
Conclusions
Our method is able to quantify the changed epitopes on HA for predicting the antigenic variants and providing biological insights to the vaccine updates. We believe that our method is robust and useful for studying influenza virus evolution and vaccine development.
Background
Influenza viruses occur all over the world and cause significant morbidity and mortality [1]. The surface proteins hemagglutinin (HA) and neuraminidase (NA) are the primary targets of the protective immune system. In circulating influenza viruses, gradually accumulated mutations on the HA, which interacts with infectivity-neutralizing antibodies, lead to the escape of immune system (called antigenic drift). The antibody recognition of HA is highly correlated to the conformation changes on the antigenic sites (epitopes). To quantify a changed epitope escaping from neutralizing antibodies is the basis to study the antigenic drift for the vaccine development [2-5].
Most of methods measuring the antigenic variances on HA focused on amino acid position mutations, such as hamming distance [6] or phylogenic distance [2]. An antibody often utilized complementarily-determining regions (CDRs) to bind two specific sites (called epitopes) on the antigen (HA) [7]. The HA consists of five epitopes and each epitope has ~20 structural neighbour amino acids locating on the protein surface [8]. Recently, few studies discussed the relationships between the epitopes and vaccine efficiency [9].
Here, we have proposed a method to identify the antigenic drift of influenza A by quantifying the conformation change of an epitope. Our method is able to predict antigenic variants of a given pair HA sequences which are often a vaccine strain and a circulating strain. Our model was evaluated to measure the antigenic drifts and vaccine updates on 3,331 circulating strains (from year 1982 to 2009) and to predict the antigenic variants on two data sets (i.e. 343 and 31,878 HI assays). These results demonstrate that our model is able to reflect the biological meanings and can explain the selections of WHO vaccine strains.
Materials and methods
Figure 1 presents the overview of our method for the antigenic drift of human influenza A (H3N2) viruses by quantifying changed epitopes. We first identified the critical amino acid positions based on both the antigenic variant and genetic diversity. We then measured a changed epitopes by calculating the accumulated conformation change based on amino acid mutations on an epitope. Finally, we evaluated our model for predicting antigenic variants and selecting the WHO vaccines.
Figure 1.

Overview of our method for the antigenic drift. (A) The overview of our method. (B) The structural locations of selected 64 critical amino acid positions on five epitopes (Epitope A in red; B in purple; C in orange; D in cyan; E in green). The sialic acid is in green. All structures are presented by using PyMOL.
Changed epitopes
The changed epitope is the core of our method. Here, we defined a changed epitope as follows: an antigenic site (epitope) on HA with accumulated amino acid mutations induces the conformation change to escape from the neutralizing antibody. The conformation change of a mutation depends on its position on HA structure and the mutation rate during 40 years. A changed epitope can be considered as a "key feature" for measuring antigenic variants of a pair HA sequences. Here, a changed epitope can be used to predict antigenic variants and antigenic drifts for the selections of vaccine strains. The definition of five epitopes including 131 positions was proposed by Wilson et al.[8] and Bush et al.[2].
Data sets
To describe and evaluate the ability of the changed epitopes for predicting antigenic variants, we collected hemagglutination inhibition (HI) assays, describing the antigenic variants and similar viruses of the current global influenza surveillance system. The HI assay describes whether one (e.g. circulating) strain will be recognized by an antibody against the vaccine strain. We collected 343 H3N2 virus HI assays (Data available at http://gemdock.life.nctu.edu.tw/influenza/File1.xls) with 125 HA sequences from Weekly Epidemiological Record (WER) [10], World Health Organization (WHO) collaborating center [11] and related publications [12-14] (Table S1 in additional file 1). Each pair includes the HI assay value (i.e. antigenic distance) and a pair of HA sequences (329 amino acids). In general, an influenza vaccine should be updated if an antigenic distance is more than 4.0 between the current vaccine strain and the circulating strain in next season [3][15]. Among 343 pairs of HA sequences, 225 pairs with antigenic distance ≥ 4 are considered as "antigenic variants" and 118 pairs are considered as "similar viruses". For example, the antigenic distance of the pair HA sequences, A/England/42/72 and A/PortChalmers/1/73, is 12 and this pair is considered as "antigenic variants". Conversely, the antigenic distance of the pair HA sequences, A/Wuhan/359/95 and A/Nanchang/933/95, is 1 and this pair is considered as "similar viruses". In addition, we prepared another HI assay data set to independently evaluate our model for predicting antigenic variants proposed by Smith et al.[3]. We assume that a virus-pair in the same antigenic group is considered as a "similar viruses" pair and a virus-pair in different groups is considered as "antigenic variants" pair. Finally, we yielded 31,878 HI measurements from the supporting materials [3].
To study the antigenic drifts and WHO vaccine updates, we collected 3,331 HA sequences (Data available at http://gemdock.life.nctu.edu.tw/influenza/File2.xls) from influenza virus resource [16] and influenza sequence database [17]. These sequences were assigned into 38 influenza seasons according to their collection dates.
Identify antigenic critical positions on HA
Recently, we proposed a method to identify antigenic critical positions [5] by utilizing both antigenic variants and genetic diversity. The Shannon entropy and information gain (IG) were used to measure genetic diversity and antigenic discriminating score for amino acid positions on HA, respectively. Here, we based on these rules to select 64 amino acid positions as the critical positions (Table S2 in additional file 1).
Models for antigenic variants based on changed epitopes
To address the issue of measuring accumulated mutations on an epitope to escape from neutralizing antibody, we proposed 4 models considering the number of amino acid mutations on 329 amino acids and 64 selected critical positions of HA (Table 1). Models one and two regarded an epitope as "changed" if there are more than 1 and 2 mutations within an epitope, respectively, based on 329 amino acids. A changed epitope of Model three is defined as two amino acid mutations on 64 critical positions. Models one, two, and three regarded a pair HA sequences as "antigenic variants" if there are more than two changed epitopes. Conversely, one changed epitope is viewed as "similar viruses".
Table 1.
Summary of four models
| Model | Regarding HA positions | Changed epitope | Antigenic variants |
|---|---|---|---|
| Model one | 329 positions | ≥1 mutation | ≥2 changed epitopes |
| Model two | 329 positions | ≥2 mutations | ≥2 changed epitopes |
| Model three | 64 selected positions | ≥2 mutations | ≥2 changed epitopes |
| Model four | 64 selected positions | ≥3 mutations (epitope B) ≥2 mutations (others) | ≥1 (epitopes A or B) ≥2 (others) |
Model four treated one changed epitope (A or B) as "antigenic variants". Epitopes A and B, which are near to the receptor binding site, often play the key role for escaping from neutralizing antibody. Here, the epitopes A and B (denoted as "B+") were regarded as "changed" if there are more than 2 and 3 mutations, respectively. For the pair A/Mississippi/1/85 and A/Leningrad/360/86 (Table 2), the numbers of mutations were 1, 3, 0, 1, and 1 on epitopes A, B, C, D and E, respectively. The numbers of changed epitopes for Models one and two are 4 (epitopes A, B, D, and E) and 1 (epitope B), respectively. Models three and four regarded the epitope B as a changed epitope because these three mutations (i.e. positions 156, 159 and 188) were the selected critical positions.
Table 2.
The changed epitopes and mutations of 11 virus-pairs under 4 models
| Virus A | Virus B | Type1 | Changed epitopes | HD2 | Mutation positions | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model one | Model two | Model three | Model four | Epitope A | Epitope B | Epitope C | Epitope D | Epitope E | ||||
| A/PortChalmers/1/73 | A/Singapore/4/75 | S | ABCDE | B | B | B | 9 | 1263 | 160, 189 | 278 | 242 | 83 |
| A/Nanchang/933/95 | A/NewYork/43/96 | S | ABCE | E | none | none | 6 | 122 | 190 | 275 | 57, 92, 262 | |
| A/Alaska/10/95 | A/France/75/97 | S | ABCDE | BC | none | none | 12 | 135 | 128, 165 | 275, 312 | 226 | 262 |
| A/Sydney/5/97 | A/Ireland/10586/99 | S | ABDE | ABD | none | none | 7 | 137, 142 | 192, 194 | 172,226 | 57 | |
| A/Mississippi/1/85 | A/Leningrad/360/86 | V | ABDE | B | B | B+ | 6 | 138 | 156, 159, 188 | 226 | 88 | |
| A/Guizhou/54/89 | A/Beijing/353/89 | V | ABC | A | A | A | 5 | 135, 144, 145 | 159 | 44, | ||
| A/Wellington/1/2004 | A/Victoria/505/2004 | S | ABDE | AD | none | none | 10 | 138, 145 | 189 | 219, 226, 227 | 94 | |
| A/Shangdong/9/93 | A/Pennsylvania/9/93 | S | ABCD | CD | C | C | 12 | 135 | 164 | 53, 276 | 214, 219, 226, 229, 238 | |
| A/England/42/72 | A/PortChalmers/1/73 | V | BDE | B | B | B+ | 6 | 160, 188, 193 | 208 | 63 | ||
| A/NewYork/55/2004 | A/Anhui/1239/2005 | V | ABD | B | B | B+ | 7 | 138, | 156, 160, 193 | 219, | 138, | |
| A/Shanghai/16/89 | A/Beijing/353/89 | V | AB | A | A | A | 3 | 135, 145 | 159 | |||
1V represents antigenic variant and S represents similar virus.
2 denote hamming distance of a pair sequences
3 Bold is the antigenic critical position.
Finally, we compared our models with two related methods [4,8] for predicting antigenic variants. Wilson & Cox [8] suggested that a viral variant usually contains more than 4 residue mutations located on at least two of the five epitopes. Lee & Chen [4] proposed a model based on the hamming distance (HD) of 131 positions on five epitopes to predict antigenic variants. Their models predicted a pair of HA sequences as "antigenic variants" if the number of mutation is more than 6.
Variant ratio for measuring the antigenic drift
We used the variant ratio (VR) to measure the vaccine efficiency on year y. The VR is defined as 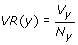 , where Ny is total number of circulating strains in the year y and Vy is the number of circulating strains which are "antigenic variants" against the vaccine strain in the year. Here, we considered an influenza vaccine should be updated and the antigenic variants are emerging if the VR value is ≥ 0.5.
, where Ny is total number of circulating strains in the year y and Vy is the number of circulating strains which are "antigenic variants" against the vaccine strain in the year. Here, we considered an influenza vaccine should be updated and the antigenic variants are emerging if the VR value is ≥ 0.5.
Results
Antigenic critical positions
In this study, we followed our previous work to select the critical positions [5] having high IGs, statistically derived from 343 HI assays, and high entropies, which were calculated using 125 HA sequences. 64 positions on HA were selected as critical positions (Table S2 in additional file 1). Among these 64 critical positions, 54 positions locate on the epitopes (54/64) and 53 positions locate on the HA surface (Fig. 1B). Additionally, 13 and 42 of these 64 critical positions were the positive selections [2] and cluster substitutions [3], respectively.
Changed epitopes for antigenic variants
Currently, several methods measured a changed epitope to escape from neutralizing antibody [8]. Here, we utilized the degree of accumulated mutations within an epitope to evaluate a changed epitope according to 329 positions and 64 selected positions. Figures 2 and 3 show the relationships between changed epitopes and antigenic variants on 4 models.
Figure 2.

The relationships between number of changed epitopes and antigenic variants on 4 models. (A) The first model considered an epitope as changed if there is at least one mutation within it. (B) The second model considered an epitope as changed if there are at least two mutations within it. (C) The third model considered an epitope as changed if there are at least two critical mutations within it. (D) The fourth model was derived from model three and further defined "1+" type if there are at least 2 and 3 critical mutations in epitope A and B. respectively.
Figure 3.

The changed-epitope composition and antigenic variants on 4 models. (A) Model one. (B) Model two (C) Model three. (D) Model four.
Models one and two: Changed epitopes on 329 positions
Figures 2A (Model one) and 2B (Model two) show the relationships between number of changed epitopes and "antigenic variants" on 343 pair HA sequences with HI assays. Among these 343 pairs for Model one, the changed epitopes of 225 "antigenic variants" pairs range from 1 to 5 and the changed epitopes of 118 "similar viruses" pairs range from 0 to 5. Among 34 similar viruses with more than 4 changed epitopes for Model one, we observed the following results: (1) the average number of changed epitopes was 4.2; (2) the average number of changed epitopes with only one mutation was 2.02 and 33 pairs have more than one changed epitope with only one mutation. For example, the virus pair, A/PortChalmers/1/73 and A/Singapore/4/75, has four changed epitopes with one mutation (i.e. Epitopes A, C, D, and E) (Table 2). In general, these 34 similar viruses should be regarded as "antigenic variants" because there are more than four changed epitopes. This result shows that the Model one is not reasonable.
For Model two, the average number of changed epitopes was 2.2 for these 34 similar viruses. According to the distribution (Figure 2B), Model two achieved the highest accuract if more that two changed epitopes was considered as "antigenic variants". The accuracies were 74.9% (257/343) and 92.2% (29410/31878) for predicting antigenic variants on the training set and independent set, respectively. This result was similar to the previous work [8].
Model three: Changed epitopes on 64 selected positions
Model three considered a changed epitope when the number of mutations on the 64 selected critical positions is more than 2. In Model two, the numbers of "antigenic variants" and "similar viruses" with ≥ 3 changed epitopes were 119 and 16, respectively (Fig. 2B). The averages of changed epitopes with ≥ 2 mutations on 329 positions for "antigenic variants" and "similar viruses" were 3.8 and 3.2, respectively. The averages of changed epitopes with ≥ 2 mutations on 64 selected critical positions for "antigenic variants" and "similar viruses" were 3.2 and 1.5, respectively (Fig. 2C). These results show that Model three using mutations on 64 critical positions is better than Model two to discriminate "antigenic variants" from "similar viruses". For the "similar viruses", A/Alaska/10/95 and A/France/75/97, there are 12 mutations to drive zero changed epitope because no epitope with ≥ 2 mutations on selected 64 positions (Table 2).
Three HA/antibody complex structures can be used to provide structural evidences for the changed epitopes [18] (Fig. S1 in additional file 1). Among these complexes, two antibodies bind to epitopes A and B (PDB code 1KEN [19] and 2VIR [20]), while the third binds to epitopes C and E (PDB code 1QFU [21]). The antibodies consistently bind to two epitopes and this result agrees to Models two and three. HA/antibody structures and Models two and three show that two position mutations often induce the conformational change of an epitope to escape from the antibody recognition. However, the numbers of changed epitopes of 48 "similar viruses" pairs are 2 (35 pairs) and 3 (13 pair) for Model two (Fig. 2B). Conversely, 14 "similar viruses" pairs have more than 2 changed epitopes for Model three (Fig. 2C).
Model four
Among 72 "antigenic variants" pairs with one changed epitope based on Model three, 70 pairs change on epitopes A or B. The single changed epitope on A or B, which can cause "antigenic variants", agreed to HA/antibody complex structures and the experiments. The receptor binding site, surrounded by epitopes A and B, is a basis for HA protein for the neutralizing mechanism [19,22] (Fig. 1B).
Based on this observation, the epitopes A and B play a key role for neutralizing antibodies. Model four based on Model three considered a pair HA sequences as "antigenic variants" when ≥ 2 changed epitopes or ≥ 1 changed epitope on A or B. In Model four, a pair HA sequences with ≥ 3 mutations on 64 critical positions for the epitope B is regarded as "antigenic variants". Thus, we annotated a virus-pairs with single changed epitope on A or B as "1+" type (Fig. 3D). For example, the pair, A/Guizhou/54/89 and A/Beijing/353/89, occurs the changed epitope on A (i.e. mutation positions 135, 144 and 145) (Table 2). The accuracies of Model four were 81.6% and 94.0% on the training set and independent set, respectively. This model outperformed two compared methods, i.e. Wilson & Cox (89.7%) [8] and Lee & Chen (92.4%) [4], on the independent data set (Fig. S2 in additional file 1).
In the HA/antibody structure complex (PDB code 1KEN [19]), the antibody binds on epitopes A and B using two CDRs (i.e. CDR1 and CDR3) on the heavy chain and one CDR (i.e. CDR2) on the light chain (Fig. 4). The interface of antibody and HA consists of 13 and 5 contacted residues locating on epitopes B and A, respectively. Among these 13 positions, 7 positions were selected as critical positions. Based on Model four, 46 "antigenic variants" pairs have one changed epitope B with 3 mutations on epitope B, denoted as "B+". This result suggested a single changed epitope B can cause antigenic variants. For example, the pair virus strains, A/NewYork/55/2004 and A/Anhui/1239/2005, have three critical mutations on epitope B (i.e. positions 156, 160 and 193) (Table 2). According to the HA/antibody structure (Fig. 4), the residue 156 interacts to CDR2 (position 55 on the antibody) and the residue 193 interacts with three residues on CDR2 (positions 50, 55 and 57) and one residue on CDR3 (position 105). This structure suggested that mutations on residues 156, 160 and 193 can induce the conformation change on epitope B to escape from CDR2 and CDR3 of the neutralizing antibody.
Figure 4.

The HA/antibody structure and interface. (A) The antibody (pale green) and HA trimer (PDB code 1KEN). (B) The interface of the antibody and HA. The selected critical positions on epitope B and the CDRs in the heavy (red) and light (pink) chains of the antibody are labelled.
Antigenic drift and epitope evolution
We utilized the changed epitopes to study the antigenic drift on 3,331 circulating strains ranging from 1982 to 2009 (38 influenza seasons). One of WHO surveillance network's purposes is to detect the emergence and spread of antigenic variants that may signal a need to update the composition of influenza vaccine [1,3]. Here, we considered an emerging antigenic variant according to WER strain, which was the dominant strain in each influenza season [6] (Table S1 in additional file 1). For a selected season, we applied Model four, measuring changed epitopes for the pairs between the vaccine and circulating strains for "antigenic variants", and the variant ratio (VR) to detect the emerging antigenic variants.
Among 38 seasons (1982~2009), our model detected 12 seasons with emerging antigenic variants (VR ≥ 0.5) and 10 of them followed by the update of WER strain in the next season (Fig. 5A). For example, the 85-86 season, 80% of the circulating strains with changed epitope "B+" (Fig. 5B), is the first emerging antigenic variants and the WER strain updated in the next season (i.e. from A/Mississippi/1/85 to A/Leningrad/360/86). In addition, among seven "emerging antigenic variants" seasons (matching WHO vaccine updates), four seasons (i.e. 89-90, 91-92, 95-96 and 02-03) matched the antigenic cluster transitions proposed by Smith et al. [3]. The other three seasons, which were detected by one changed epitope on A or B, are consistent to the WER strain updates (i.e. 87-88, 85-86 and 99). These results suggested that "emerging antigenic variants" with ≥ 2 changed epitopes may cause the major antigenic drift while "emerging antigenic variants" with one changed epitope on A or B may cause the minor antigenic drift.
Figure 5.

The epitope evolution and antigenic drift. (A) The distributions of variant ratios of WER and Smith vaccine strains from 1982-1983 to 2009 seasons. 10 seasons with emerging variants and followed by the update of WER strain in the next season are labelled (red arrow) (B) The average hamming distances (HD) of 5 epitopes from 1982-1983 to 2009 seasons.
To observe the epitope evolution, Figure 5B illustrates the hamming distance (HD) on 64 critical positions of five epitopes. For example, the VR of the season 85-86 was 0.8 (Fig. 5A) and the epitope with the largest HD was epitope B (HD is 3.4). For 16 seasons with WER strain updates, the average HDs of epitopes A, B, C, D and E were 1.2, 2.1, 0.4, 0.4 and 0.5 respectively. These results showed that epitopes A and B change more frequently in vaccine update seasons and they play a key role for antigenic drift.
Discussion
According to the distribution of antigenic variants of Model four (Fig. 3D), it is interesting that the major pairs (209/225 pairs) of the antigenic variants have the changed epitope on epitopes A or B which are closed to the receptor binding site. Furthermore, many experiments suggested that the occlusion of the receptor binding site by antibodies bound to the HA molecule forms the dominant neutralizing mechanism [19,22]. These results implied that a pair of viruses often is "similar virus" if the epitopes A and B are not changed.
Among 225 "antigenic variants" pairs, 13 pairs have no changed epitopes (Table S3 in additional file 1). 11 pairs of these 13 pairs have contradict antigenic types by two antiseras, which suggested a more powerful experimental assay is required to verify the antigenic types. For example, the antibody against the A/Alaska/10/95 strain can't inhibit the A/Idaho/4/95 strain; while the antibody against the A/Idaho/4/95 strain inhibits the A/Alaska/10/95 strain.
Conclusions
This study demonstrates our model is robust and feasible for quantifying the changed epitopes. According to the distribution of antigenic variants in HI assays and HA/antibody complex structures, we found that two critical position mutations with high genetic diversity and antigenic scores can induce the conformation change of an epitope. Epitopes A and B, closing the receptor binding site of HA, play a key role for neutralizing antibodies. In addition, two changed epitopes often drive the antigenic drift and can be used to explain the selections of 24 WHO vaccine strains. We believe that our method is useful for the vaccine development and studying the evolution of human influenza A virus.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Conceived and designed the experiments: JWH and JMY. Performed the experiments and analyzed the data: JWH and JMY. Wrote the paper: JWH and JMY
Supplementary Material
The supplementary information
Contributor Information
Jhang-Wei Huang, Email: popblue.bi93g@nctu.edu.tw.
Jinn-Moon Yang, Email: moon@faculty.nctu.edu.tw.
Acknowledgements
J.-M. Yang was supported by National Science Council and partial support of the ATU plan by MOE. Authors are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
This article has been published as part of BMC Bioinformatics Volume 12 Supplement 1, 2011: Selected articles from the Ninth Asia Pacific Bioinformatics Conference (APBC 2011). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/12?issue=S1.
References
- Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC. et al. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine. 2008;26(Suppl 4):D31–D34. doi: 10.1016/j.vaccine.2008.07.078. [DOI] [PubMed] [Google Scholar]
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286(5446):1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Lee MS, Chen JS. Predicting antigenic variants of influenza A/H3N2 viruses. Emerging infectious diseases. 2004;10(8):1385–1390. doi: 10.3201/eid1008.040107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JW, King CC, Yang JM. Co-evolution positions and rules for antigenic variants of human influenza A/H3N2 viruses. BMC Bioinformatics. 2009;10(Suppl 1):S41. doi: 10.1186/1471-2105-10-S1-S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Dushoff J, Levin SA. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(9):6263–6268. doi: 10.1073/pnas.082110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HP, Dimmock NJ. Competitive binding of neutralizing monoclonal and polyclonal IgG to the HA of influenza A virions in solution: only one IgG molecule is bound per HA trimer regardless of the specificity of the competitor. Virology. 1994;205(1):360–363. doi: 10.1006/viro.1994.1653. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annual Review of Immunology. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- Gupta V, Earl DJ, Deem MW. Quantifying influenza vaccine efficacy and antigenic distance. Vaccine. 2006;24(18):3881–3888. doi: 10.1016/j.vaccine.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Weekly Epidemiological Record. 2007. 45, 46, 48, 49, 50, 51, 52, 55, 57, 58, 60, 61, 62, 63, 65, 66, 67, 68, 69, 70, 71, 73, 74, 78, 79, 80, 71, 82.
- Centers for Disease Control and Prevention. Information for the Vaccines and Related Biological Products Advisory Committee, CBER, FDA. 2007. p. 28, p.18, p.19, p.17.
- Ellis JS, Chakraverty P, Clewley JP. Genetic and antigenic variation in the haemagglutinin of recently circulating human influenza A (H3N2) viruses in the United Kingdom. Archives of virology. 1995;140(11):1889–1904. doi: 10.1007/BF01322680. [DOI] [PubMed] [Google Scholar]
- Both GW, Sleigh MJ, Cox NJ, Kendal AP. Antigenic Drift in Influenza Virus-H3 Hemagglutinin from 1968 to 1980 - Multiple Evolutionary Pathways and Sequential Amino-Acid Changes at Key Antigenic Sites. Journal of Virology. 1983;48(1):52–60. doi: 10.1128/jvi.48.1.52-60.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiras MT, Aguilar JC, Galiano M, Carlos S, Gregory V, Lin YP, Hay A, Perez-Brena P. Rapid molecular analysis of the haemagglutinin gene of human influenza A H3N2 viruses isolated in Spain from 1996 to 2000. Archives of virology. 2001;146(11):2133–2147. doi: 10.1007/s007050170025. [DOI] [PubMed] [Google Scholar]
- Webster R, Cox NJ, Stohr K. WHO Manual on Animal Influenza Diagnosis and Surveillance. WHO/CDS/CSR/NCS/20025. 2002. Rev.1.
- Bao YM, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the national center for biotechnology information. Journal of Virology. 2008;82(2):596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macken C, Lu H, Goodman J, Boykin L. The value of a database in surveillance and vaccine selection. Options for the Control of Influenza Iv. 2001;1219:103–106. [Google Scholar]
- Knossow M, Gaudier M, Douglas A, Barrere B, Bizebard T, Barbey C, Gigant B, Skehel JJ. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302(2):294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- Barbey-Martin C, Gigant B, Bizebard T, Calder LJ, Wharton SA, Skehel JJ, Knossow M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294(1):70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376(6535):92–94. doi: 10.1038/376092a0. [DOI] [PubMed] [Google Scholar]
- Fleury D, Barrere B, Bizebard T, Daniels RS, Skehel JJ, Knossow M. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nature Structural Biology. 1999;6(6):530–534. doi: 10.1038/9299. [DOI] [PubMed] [Google Scholar]
- Ndifon W, Wingreen NS, Levin SA. Differential neutralization efficiency of hemagglutinin epitopes, antibody interference, and the design of influenza vaccines. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8701–8706. doi: 10.1073/pnas.0903427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The supplementary information