

Abstract
Background
High-throughput short read sequencing is revolutionizing genomics and systems biology research by enabling cost-effective deep coverage sequencing of genomes and transcriptomes. Error detection and correction are crucial to many short read sequencing applications including de novo genome sequencing, genome resequencing, and digital gene expression analysis. Short read error detection is typically carried out by counting the observed frequencies of kmers in reads and validating those with frequencies exceeding a threshold. In case of genomes with high repeat content, an erroneous kmer may be frequently observed if it has few nucleotide differences with valid kmers with multiple occurrences in the genome. Error detection and correction were mostly applied to genomes with low repeat content and this remains a challenging problem for genomes with high repeat content.
Results
We develop a statistical model and a computational method for error detection and correction in the presence of genomic repeats. We propose a method to infer genomic frequencies of kmers from their observed frequencies by analyzing the misread relationships among observed kmers. We also propose a method to estimate the threshold useful for validating kmers whose estimated genomic frequency exceeds the threshold. We demonstrate that superior error detection is achieved using these methods. Furthermore, we break away from the common assumption of uniformly distributed errors within a read, and provide a framework to model position-dependent error occurrence frequencies common to many short read platforms. Lastly, we achieve better error correction in genomes with high repeat content. Availability: The software is implemented in C++ and is freely available under GNU GPL3 license and Boost Software V1.0 license at “http://aluru-sun.ece.iastate.edu/doku.php?id=redeem”.
Conclusions
We introduce a statistical framework to model sequencing errors in next-generation reads, which led to promising results in detecting and correcting errors for genomes with high repeat content.
Background
High throughput next generation sequencing has revolutionized genomics, making it possible to sequence new genomes or resequence individual genomes at a manifold cheaper cost and in an order of magnitude less time than earlier Sanger sequencing. With this technology, ambitious genome sequencing projects target many organisms rather than a few, and large scale studies of sequence variation become feasible [1]. Many next-geneneration sequencing technologies have been developed, including systems currently in wide use, such as the Illumina Genome Analyzer (earlier known as Solexa) and Applied Biosystems SOLiD, as well as more recent and new offerings from companies such as Complete Genomics and Pacific Biosciences [2]. Many next-generation sequencing systems produce short reads, e.g., the widely used Illumina Genome Analyzer systems typically produce 35 - 150bp reads. Short read technologies have been widely adopted for both genome sequencing and resequencing applications; hence, development of high quality short read assemblers (e.g., [3-7]) and short read mapping tools that map reads to a reference genome [8,9] are important.
Short reads of novel genomes are typically assembled using de Bruijn graphs that represent observed kmers as nodes and length (k – 1) overlaps as edges. In the absence of errors, the size of such a graph is bounded by the length of the genome, but can be as high as 4k in the presence of errors. In the mapping process, a read with sequencing errors may map to multiple locations, or sometimes nowhere at all. Thus, error removal or correction is necessary to keep the size of the graph manageable [7,10] and simplify non-repetitive read mapping [11].
Many approaches have been proposed to identify and sometimes correct sequencing errors in next-generation sequencing data. More recent ones include SAP (Spectral Alignment Problem)-based methods [4,10], SHREC [12] and Reptile [13]. SAP-based methods identify any kmer occurring less than a constant, user-specified frequency threshold to be erroneous. Chin et al. [14] have shown that an optimum threshold can be derived analytically, assuming the genome to be a random sequence and that errors are independently and uniformly distributed in the reads. SHREC, a suffix trie based method, classifies any substring that occurs less often than an analytically calculated threshold to contain errors based on the same assumptions as in [14]. An erroneous base, identified as an infrequent branch of the suffix trie, is corrected to one of its siblings when applicable. Reptile explores read decompositions and makes corrections to any substring whenever an unambiguous choice can be made. In contrast to the previous two approaches, erroneous substrings are inferred based on assessing their frequencies relative to the frequencies of the alternative (error free) substrings. This accommodates under-sampled genomic regions. All of these methods are mainly suitable for genomes with a low degree of repetitive sequences.
Repeats in genomes can lead to mishandling of errors in many ways. Nearly identical repeats can easily be mistaken to be sequencing errors. Even when errors are rare, an erroneous kmer may appear at a moderate frequency if it has few nucleotide differences from one or more valid kmers that have a high frequency of occurrence in the genome. The problem of detecting and correcting sequencing errors among reads in the presence of repeats has so far not been adequately addressed. Nevertheless, repeats are widely prevalent, even in some viral genomes such as N. meningitidis. Other genomes, like those of plants, are known for their high repeat content; for instance, an estimated 65-80% of the maize genome is spanned by repeats, which makes the assembly, mapping and error detection and correction tasks difficult. Although packages like FreClu [11] and Recount [15] could be potentially adapted to consider repeats, they are specifically designed for transcriptome data and correct read counts rather than identify and correct erroneous bases within reads. Moreover, insufficient replication of full length reads in genomic data prevents these methods from accurately estimating model parameters.
In this paper, we address the problem of identifying and correcting sequencing errors in short reads from genomes with different levels of repetition, particularly for reads produced by the widely used Illumina Genome Analyzer platform. Similar to existing approaches, we decompose the input reads into kmer substrings and count the number of times Yl each kmer xl occurs in the reads. However, instead of inferring erroneous kmers based on these observed occurrence frequencies [10,12], we developed a maximum likelihood estimate of the expected number Tl of attempts to read xl, including both attempts that resulted in error-free reads and erroneous reads. In addition, we propose a new method to choose the threshold, which can be used to identify erroneous kmers as those xl’s for which Tl’s are lower than the threshold. We demonstrate that using estimates of read attempts enables more accurate detection of sequencing errors than using observed frequencies for a wide choice of thresholds. We further develop an error correction method to transform erroneous bases in each read to the correct ones and compare the results with SHREC [12] and Reptile [13], two of the most recent error correction methods. The results demonstrate significant improvement in error correction capabilities for genomes with high repeat content. The proposed method is made available through the software package REDEEM (Read Error DEtection and Correction via Expectation Maximization) at “http://aluru-sun.ece.iastate.edu/doku.php?id=redeem”.
Methods
Let G denote the reference genome to be sequenced, and let R = {r1, r2,…, rN} be the collection of resulting short reads. For simplicity, we assume each read has a fixed length L. The sequence coverage is 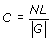 , where |G| is the genome length. Define the k-spectrum of a read r to be the set rk = {r[i : i + k – 1] | 0 ≤ i <L – k + 1}, where r[i : j] is the substring from position i to j in r. The k-spectrum produced by all the reads is
, where |G| is the genome length. Define the k-spectrum of a read r to be the set rk = {r[i : i + k – 1] | 0 ≤ i <L – k + 1}, where r[i : j] is the substring from position i to j in r. The k-spectrum produced by all the reads is 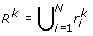 .
.
Each kmer xl ∈ Rk has αl occurrences in G and Yl instances observed in read set R. Define 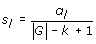 , the probability that a random k-length fragment in the genome is kmer xl. Occurrence frequency αl, or equivalently sl, is unobserved, but of paramount interest. Indeed, if we knew sl = 0, but observed Yl > 0, then we would know each observed instance of xl contains at least one misread base. Under the assumption that errors are rare, it makes sense to label kmers xl with Yl <M as errors, where M is chosen such that P(Yl <M | sl > 0) is reasonably small. Since sl is unknown, the threshold M is set ad hoc, based on training or simulated data [10], or analytical calculations [12,14] assuming the genome to be a random sequence and errors to be distributed uniformly in the reads. In practice, these assumptions do not hold true. Moreover, the problem of misread kmers contributing to the observed frequencies Yl (see Fig. 1) is exacerbated in repetitious genomes where kmers with high genomic occurrence may result in generation of the same misread multiple times [10].
, the probability that a random k-length fragment in the genome is kmer xl. Occurrence frequency αl, or equivalently sl, is unobserved, but of paramount interest. Indeed, if we knew sl = 0, but observed Yl > 0, then we would know each observed instance of xl contains at least one misread base. Under the assumption that errors are rare, it makes sense to label kmers xl with Yl <M as errors, where M is chosen such that P(Yl <M | sl > 0) is reasonably small. Since sl is unknown, the threshold M is set ad hoc, based on training or simulated data [10], or analytical calculations [12,14] assuming the genome to be a random sequence and errors to be distributed uniformly in the reads. In practice, these assumptions do not hold true. Moreover, the problem of misread kmers contributing to the observed frequencies Yl (see Fig. 1) is exacerbated in repetitious genomes where kmers with high genomic occurrence may result in generation of the same misread multiple times [10].
Figure 1.

A kmer neighborhood. The neighborhood of trimer AAA is the collection of trimers in R3 that have a nonvanishing chance of being misread as AAA, in this case trimers with at most one substitution.
We will develop a model that estimates the expected number of attempts Tl to read each kmer xl. The threshold is then applied to each estimated Tl instead of the corresponding observed Yl. The model we propose is similar to that of RECOUNT [15] used to correct next generation short read counts. Both models derive from a method originally meant to detect sequencing errors in SAGE libraries [16]. Our model differs from the previous models in that it works with kmers rather than full reads, since there is insufficient replication of full length reads in genomic data (as compared to transcriptome data). In addition, instead of assuming the misread bases to be drawn from {A, C, G, T} with equal probability, we propose a parametric error model that can be trained from the reads produced by the control lane (e.g.using the Illumina Genome Analyzer) in the same experiment. This strategy has already proven to be useful in several pioneering works [11,17]. In addition, we will show that the model is somewhat robust to incorrect assumptions in the underlying error model. A further step is to modify erroneous bases to their true forms in each read. This task has rarely been attempted previously for repetitive regions. We propose a method that utilizes transition probabilities and the contexual information of individual reads to achieve this goal. Like others, we ignore insertion and deletion errors assuming they are rarely produced by next-generation sequencing technology, which is true for reads from the Illumina Genome Analyzer [18].
Error model
The simplest error model posits that sequencing errors occur independently at all sites with constant probability pe. Let pe(xm,xl) be the probability that xm is misread as xl. This model produces symmetric misread probabilities:
| (1) |
where d(·, ·) denotes the Hamming distance between two kmers. It is known, however, that short read technology produces errors with distinct patterns [18]. As a first approximation, we assume that errors strike sites in the kmer independently, but with varying probabilities. For example, we observe in dataset preparation section that errors cluster in the 3′ portion of reads and, consequently, kmers. Let qi(α,β) be the probability that nucleotide α at position i of a kmer is (mis)read as nucleotide β, with ∑β qi(α, β) = 1. Then, the misread probability is
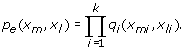 |
These misread probabilities are no longer symmetric, and can be arranged into a 4k × 4k matrix Pe, where non-zero entries in the lth row identify all possible ways to (mis) read kmer xl.
We now discuss some ways to reduce and simplify the calculations. We observe substitution errors are relatively rare, so misread kmers generally contain far fewer than k errors. Thus, when considering possible origins of a misread kmer, we can safely restrict our attention to kmers within some Hamming distance dmax from the current kmer. Capping the maximum distance between kmers at dmax induces a sparse Pe, whose entries are normalized by dividing each row by the corresponding row sum. Finally, we ignore kmers that are not observed in the data (i.e. Ym = 0 or, equivalently, xm ∉ Rk), so the (incomplete) neighborhood of kmer xl, denoted by 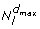 , is given as {xm ∈ Rk : d(xl,xm) ≤ dmax}. Failure to include unobserved kmers could bias estimation of αl by ignoring kmers actually present in G and capable of contributing to Yl. However, the bias cannot be large since αm must be small, otherwise Ym would not be zero.
, is given as {xm ∈ Rk : d(xl,xm) ≤ dmax}. Failure to include unobserved kmers could bias estimation of αl by ignoring kmers actually present in G and capable of contributing to Yl. However, the bias cannot be large since αm must be small, otherwise Ym would not be zero.
After considering errors and applying the simplifications, the counts Yl follow a Multinomial distribution
Y = (Y1,…,Y|Rk|) ~ Multinomial(N(L – k + 1),p),
but unobserved kmers are ignored and the probability vector p = (p1,p2 …,pl …,p|Rk|) depends on the kmer neighborhood. Specifically,
where s = (s1,…,s|Rk|) is restricted to the set of observed kmers Rk. It becomes clear that when xl is surrounded by highly repetitious xm with large sm, then Yl may exceed threshold M because of high misread occurrence with probability smpe(xm,xl). Thus, when errors combine with repeats, it is more appropriate to apply a threshold to estimates of the parameters sl than observed Yl.
The observed log likelihood, l(s |Y), involves a mixture over the neighborhood (Fig. 1) of kmers that could be (mis) read as kmer xl,
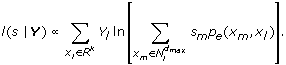 |
This setup lends itself to maximum likelihood estimation via the EM algorithm [19]. The update equations are adapted from [16] using a different error model and are given as follows:
The expectations of hidden data Ylm obtained in the E step are
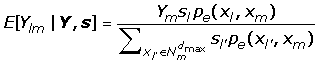 |
The M step yields maximum likelihood estimates
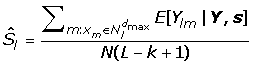 |
Notice the estimated expected number of attempts to read kmer xl is Tl = ŝlN(L – k + 1), directly proportional to ŝl and sitting on the same scale as Yl. In fact, by observing the E step is unchanged and the log likelihoood l(s |Y) is computed up to an additive constant when sl is replaced with Tl, we use the EM algorithm to compute Tl directly. For inference, we apply the threshold to estimates T = (T1,…,T|Rk|) rather than ŝ, to more easily compare our method with thresholding on Y. The algorithm is initialized by setting Tl =Yl and iterating until the log likelihood converges.
Error detection and correction
Error detection, in practice, requires a method to choose a threshold M that minimizes the number of wrong decisions when classifying kmers as erroneous or not. We discuss a model-free method for estimating the threshold M in the Appendix, but no results presented in this paper use estimated thresholds.
To correct errors, consider each of the nucleotides in a read r. Each nucleotide appears in at least one and up to k kmers. Suppose the nucleotide at position 1 ≤ i ≤ L of the read appears at position 1 ≤ t ≤ k of kmer xl. The probability that the true nucleotide at position t was b prior to possible misread is
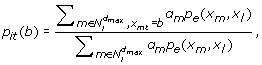 |
where estimates Tm are substituted for the unknown αm. Since multiple overlapping kmers provide non-independent information about the base at position i, we average across available t to obtain distribution pi(b). If argmaxbpi(b) ≠ r[i : i], then we declare nucleotide r[i : i] misread and correct it to argmaxbpi(b). To limit computations, we apply this method to reads likely to contain at least one erroneous kmer, as identified with a liberal threshold M.
Results and discussion
Dataset preparation
In order to test our model, we compiled various simulated and real datasets (Table 1). The datasets are classified into the following types (Table 1, column 2). Type 1 are simulated Illumina reads from (a) synthetically constructed genomes embedded with various types of non-overlapping repeats, and (b) previously sequenced genomes known to be rich in repeats. Type 2 are actual Illumina reads from a previously sequenced genome with a low degree of repetition.
Table 1.
Experimental datasets
| Dataset | Type | Reference genome | Genome length | Repeats | Repeat Types (length, multiplicity) | C | Number of reads |
|---|---|---|---|---|---|---|---|
| D1 | 1(a) | - | 1M | 20% | (1000, 200) | 80x | 2.2M |
| D2 | 1(a) | - | 1M | 50% | (500, 400), (1500, 200) | 80x | 2.2M |
| D3 | 1(a) | - | 1M | 80% | (500, 400), (1500, 200) | 80x | 2.2M |
| (3000, 100) | |||||||
| D4 | 1(b) | N. meningitidis | 2.1M | - | - | 80x | 4.8M |
| D5 | 1(b) | Maize | 418K | - | - | 80x | 0.92M |
| D6 | 2 | E. coli | 4.6M | - | - | 160x | 20.7M |
‘-’ denotes the information that is not quantified; K: thousand; M: million.
Reference genome preparation
The reference genomes of type 1(a) were initially generated using the nucleotide distribution of a piece of B73 maize genome (A: 28% C:23% G: 22% T: 27%). Then, repeat regions of different lengths and multiplicities (Table 1, column 6) derived from the same nucleotide distribution were embedded at random locations in these reference genomes. The reference genome N. meningitidis (NC_013016) of D4 is known to be a small, repeat rich, viral genome. The maize genome is known to contain up to 80% repeats and only the relatively unique regions have been fully assembled. Hence, we concatenated the first 20 contigs from Chromosome 1 of the B73 assembly, and removed all non-ACGT characters to form the reference genome of D5.
Short read preparation
The simulated Illumina reads (type 1) were produced by first estimating an error distribution from a real Illumina short read dataset, then simulating uniformly distributed reads of the reference genomes with these error rates. We used the RMAP software [9] to map Illumina data (Sequence Read Archive ID: SRX000429) to the reference genome E. coli str. K-12 allowing up to three mismatches. We were able to map 98.5% of reads; this percent is increased to 99.1% by allowing up to ten mismatches. However, allowing more mismatches increases the chance of a mismapped read since reads are only 36bp, and typically, mapping software can work at full sensitivity for up to two mismatches. Unmapped reads were discarded, and all remaining reads were assumed correctly mapped. By comparing the mapped reads to the reference genome, we estimated L 4 × 4 misread probability matrices M = (M1,M2,…,ML), where L is the read length and each entry (α, β) (α, β ∈ {A, C, G, T}) in misread probability matrix Mi (1 ≤ i ≤ L) is the probability a nucleotide α on the reference genome is (mis)read as β at position i in the read. This is calculated as the total number of times α is read to be β at position i among all mapped reads, divided by the number of times the corresponding position of the reference genome is α. Finally, we simulated Illumina sequencing to generate N reads by applying M to N uniformly distributed L-substrings in the reference genome.
Rationale
Simulated data are essential because highly repetitive genomic regions, for which our error model is designed, are often masked prior to assembly. Even when assembly can be done, accurate mapping of sequenced reads back to the assembly is difficult when genomes are repetitive [20]. Under these conditions, only simulation can provide unambiguous error information. Type 1(a) datasets were prepared such that they emulate repeat content ranging from a microbial genome with low repeats to a highly repetitive plant genome. However, to inject reality wherever possible, the reference genomes of Type 1(b) were selected from the previous assemblies. Lastly, the type 2 dataset demonstrated the applicability of our model to real, although non-repetitive, real read data.
Error detection and correction results
Our model accommodates sequencing errors via the misread probabilities pe(xm, xl) between any two kmers xm and xl. To calculate pe(xm,xl), we need to specify the position specific misread probabilities, qi(α, β), 1 ≤ i ≤ k, α, β ∈ {A, C, G, T}, for each position of a kmer. Ideally, we would set qi(·, ·) to match the errors in the current dataset inferred from reads in the control lane [11,17]. When such information is not available, we can rely on information derived from other read data generated on the same platform. In the worst case, we can use the simple error model of Eq. (1), which only requires specification of the average error rate pe.
Based on these choices, we tested our datasets using four types of sequencing error (misread) distributions: tIED, wIED, tUED, and wUED (defined below). Our simulation procedure introduced errors according to the misread probability matrices M estimated from dataset SRX000429, so the true error distribution, tIED, was obtained by estimating qi(·, ·) from the same dataset SRX000429. The estimation procedure is similar to the one used for estimating M (defined in the previous section), except each read is decomposed into L – k + 1 kmers and the count of each type of misread nucleotide at each kmer position is determined. (Note, the same nucleotide contributes counts in up to k distinct kmers.) Since, the estimated qi(·, ·) represent fewer parameters than M, qi(·, ·) only approximates the true misread probability matrices M, which themselves only approximate true read errors. The wrong Illumina error distribution, wIED, is the situation encountered when Illumina data are only available from a different experiment (and often different lab). To emulate this case, we derived a second set of error probabilities qi(·, ·) from Illumina reads of Acinetobacter sp. ADP1 (Short Read Archive acc. SRX001814, 17.7M reads of 36bp length). The error rates differ at kmer position i = 11 (Table 2) and others (not shown) in the E. coli and A. sp. ADP1 short read datasets, demonstrating that wIED is indeed the wrong error distribution. Finally, in the absence of detailed error information, we can use the uniform error distribution with constant error probability pe. When the average error rate pe = 0.006 is estimated from dataset SRX000429, the error distribution is the true uniform error distribution (tUED). When the error rate is overestimated at pe = 0.02, above the published rate of 0.01–0.015 [21], it is the wrong uniform error distribution, wUED.
Table 2.
Estimated error probabilities qi(·, ·), position i = 11
| E. coli str. K-12 substr. | Acinetobacter sp. ADP1 | |||||||||
| ×10–2 | A | C | G | T | ×10–2 | A | C | G | T | |
| A | 98.96 | 0.63 | 0.18 | 0.23 | A | 96.18 | 2.53 | 0.19 | 1.10 | |
| C | 0.15 | 99.60 | 0.10 | 0.15 | C | 0.20 | 99.32 | 0.08 | 0.40 | |
| G | 0.05 | 0.17 | 99.25 | 0.53 | G | 0.12 | 0.30 | 97.60 | 1.98 | |
| T | 0.05 | 0.19 | 0.18 | 99.58 | T | 0.09 | 0.18 | 0.13 | 99.60 | |
The same measures as in [14] are used for evaluation, where a false positive (FP) denotes an error free kmer has been considered as erroneous and a false negative (FN) denotes an unidentified erroneous kmer. Table 3 reports the minimum number of wrong predictions (WPs), FP+FN, achieved by applying optimum thresholds on observed Y , used by existing methods, or by applying thresholds on the estimated number of attempts to read T, used in our method. The results of our method are shown for the four types of error distributions in columns tIED, wIED, tUED, and wUED. Bolded entries indicate where lower minima were achieved with our method compared to the standard method. Given the true error distribution, our method committed over 95% fewer WPs for all datasets except D6, where our method still managed 7% fewer WPs. Interestingly, using the wrong Illumina error distribution (column wIED) achieved at least 33% fewer WPs in all repetitive genomes except D4, where our wIED method performed about on par with applying the threshold on Y . The minimum WPs achieved by the true uniform error model tUED are two- to three-fold smaller than the corresponding values in column Y . However, using elevated error rate pe = 0.02 led to higher minimum WPs, except in dataset D3, the most highly repetitive simulated genome.
Table 3.
A comparison of minimum error rates.
| Data | Minimum (FP + FN) Value | ||||
|---|---|---|---|---|---|
| Y | tIED | wIED | tUED | wUED | |
| D1 | 2212 | 18 | 984 | 1020 | 4648 |
| D2 | 6392 | 23 | 1300 | 3150 | 6729 |
| D3 | 6809 | 19 | 1300 | 2696 | 3124 |
| D4 | 216 | 10 | 236 | 80 | 719 |
| D5 | 552 | 14 | 373 | 297 | 1346 |
| D6 | 14236 | 13275 | 13441 | 13671 | 18793 |
A comparison of the minimum number of wrong predictions achieved by applying optimum thresholds to observed occurrences Y , and our model with each of the error distributions tested. Bold numbers indicate that our model outperforms.
Even though we presented a method to choose the threshold value (see Appendix), it is not possible for any method to guarantee the optimal threshold. Ideally, the error detection methods should be relatively insensitive to choice of threshold. To compare methods across many thresholds, we plot log(FP + FN), with respect to a wide range of thresholds (Fig. 2). The plot in every case resembles a U-shape since many error kmers are missed (FN) when the threshold is too low and many correct kmers are declared errors (FP) when the threshold is too high. Our method achieved fewer WPs for datasets D1,D2,D3 and Maize with error distributions tIED, wIED, and tUED at all thresholds. The improvement in error detection increased with the degree of repetition, seen in simulations D1 to D3 and also in comparing N. meningitidinis and Maize. Our method tended to flatten the bottom of the U-shape such that a wider range of thresholds often beat even the minimum error obtained under Y thresholding. In all datasets, our method often shifted the U-shape leftward, such that for very small thresholds, our WPs were far less than the Y -based methods, regardless of the error distribution used. As the threshold increases, WPs for all methods eventually converge to the same constant, where all kmers are considered erroneous. For moderately large thresholds, our method sometimes resulted in higher WPs, especially for the wrong error distribution wUED, and sometimes wIED, and genomes with a low degree of repeats.
Figure 2.

Plots of log(FP + FN) vs. threshold for all datasets. In each plot, we compare the results by applying thresholds to Y and to T estimated by our model using the tIED, wIED, tUED and wUED error distributions.
REDEEM misclassified the fewest kmers when using the “true” error model, but even in our simulations, there was a mismatch between the simulated errors and the estimated “true” error model. The position-specific error probabilities used to compute kmer misread probabilities are not the true error probabilities that vary by position in the full length read. The difference is exacerbated as reads get longer relative to the kmer length. Since it would be possible to compute misread probabilities pe(xm, xl) using read-derived position probabilities, this mismatch between kmer and read errors can be eliminated with more sophisticated error models that account for the position of each kmer in the read. Since data to estimate the read error properties can be collected in parallel on a known, control genome, we contend that estimating the true error model is not an undue burden in practical applications [11,17]. Quality scores may also inform on errors [15] and could be incorporated in the REDEEM error model.
As discussed previously, only simulated data with different degrees of repeats can be utilized to measure error correction results for repeat-rich genomic regions due to the fact that mapping short reads from such regions uniquely to the reference genome, and the assembly of genomes with high repeat content, remain open problems. We compare our correction results with SHREC [12] and Reptile [13] using datasets D1, D2 and D3 with increasing degrees of repeat content. The results are shown in Table 4. To be self-contained, we reproduce the evaluation measures from [13]: A True Positive (TP) is any erroneous base that is changed to the true base, a False Positive (FP) is any true base changed wrongly, a True Negative (TN) is any true base left unchanged, and a False Negative (FN) is any erroneous base left unchanged. Sensitivity = TP / (TP + FN) and Specificity = TN / (TN + FP). Gain = (TP - FP) / (TP + FN) denotes the total percentage of erroneous bases removed from the dataset post-correction.
Table 4.
Error correction results
| Data | Method (d) | Sensitivity | Specificity | Gain | CPU Time(min) | Memory(GB) |
|---|---|---|---|---|---|---|
| D1 | SHREC | 81.2% | 99.9% | 80.3% | 23.9 | 5.9 |
| Reptile | 78.9% | 99.9% | 78.9% | 0.6 | 0.19 | |
| REDEEM | 71.3% | 99.9% | 51.5% | 114.1 | 2.5 | |
| D2 | SHREC | 54.0% | 99.9% | 52.7% | 22.7 | 5.8 |
| Reptile | 57.8% | 99.9% | 57.8% | 0.5 | 0.16 | |
| REDEEM | 78.6% | 99.9% | 64.6% | 72.7 | 1.6 | |
| D3 | SHREC | 29.3% | 99.9% | 26.7% | 21.7 | 5.8 |
| Reptile | 46.8% | 99.9% | 46.8% | 0.5 | 0.13 | |
| REDEEM | 86.4% | 99.9% | 79.4% | 31.2 | 0.63 | |
REDEEM is designed to specifically target error correction for repeat-rich genomes. While both SHREC and Reptile do not explicitly model the effect of repeats, the variable length suffixes captured by SHREC and different read decompositions explored by Reptile can provide richer and more precise information about errors. Currently, REDEEM does not utilize all such information. Therefore, in genomes with low repeat content, both SHREC and Reptile outperform. However, as the repetition within the genome increases, REDEEM significantly outperforms both methods by accounting for the repetition in the kmer neighborhood. Further experiments show that error correction results are affected mainly by the percentage of the length of the genome spanned by repeats, rather than repeat types and lengths. This can serve as a yardstick in deciding when to use REDEEM over conventional error correction. It is also possible to combine the features of a conventional error correction method such as Reptile with the explicit modeling of repeats as done in REDEEM to produce an error-correction method that is superior both when sampling low repeat and highly-repetitive genomes.
All experiments were carried out on 3.16GHz Intel Xeon Processors; run time and memory usage of all three programs are shown in the last two columns in Table 4. As expected, the run time of REDEEM is longer due to the complexity of modeling repeats explicitly. The largest simulation, D6, took 120 minutes and 9 GB. No error detection/correction method except naïve thresholding on observed counts yet scales to practical next-generation applications, but REDEEM is at least comparable to existing, non-repeat-aware methods.
Conclusions
There have been some attempts to formally characterize repeats in genomes [22], but generally, the term “repeat” is used loosely in the literature, with meaning varying by context. In this paper, we consider kmer xl a repeat when its genomic occurrence αl is higher than what is expected in a random genomic sequence. Because genomes are not random, all genomes display some degree of repetition. Perhaps such cryptic repetition explains why we can achieve lower false prediction rates at optimal thresholds even on genomes like E. coli, which according to the Ir measure of [22] is only somewhat repetitive.
In summary, we have presented a new method that improved error detection and correction when sampling repeat-rich genomic regions using next-generation sequencers. Important future work includes better models and algorithms to simultaneously estimate error parameters from the data, to consider variation in coverage along the genome, to speed up computations, and to handle larger datasets through better memory management.
Competing interets
The authors declare that they have no competing interests.
Authors contributions
XY and KD developed and implemented the model and algorithmic solutions. SA helped conceive of and coordinated the study. All authors contributed to the development and revision of the manuscript.
Appendix
The true expected number of attempts to read kmer xl, 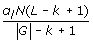 , are constant multiples of the discrete genome occurrences αl ∈ {0,1,…,}, where the coverage-related constant
, are constant multiples of the discrete genome occurrences αl ∈ {0,1,…,}, where the coverage-related constant 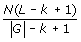 is unknown. The estimated Tl vary from these true values because of sampling and estimation error. A histogram of estimated Tl (see Fig. 3 for the E. coli dataset) thus reveals peaks corresponding to αl = 0, αl = 1, and αl = 2. The constant multiple is about 57, which can also be verified from Table 1. The kmers with Tl near 0 have no occurrences in the genome. One approach to model multi-modal distributions, such as that of Fig. 3, is to use a mixture model. Then, erroneous kmers would be those derived from the mixture component corresponding to the first mode. We propose mixture distribution
is unknown. The estimated Tl vary from these true values because of sampling and estimation error. A histogram of estimated Tl (see Fig. 3 for the E. coli dataset) thus reveals peaks corresponding to αl = 0, αl = 1, and αl = 2. The constant multiple is about 57, which can also be verified from Table 1. The kmers with Tl near 0 have no occurrences in the genome. One approach to model multi-modal distributions, such as that of Fig. 3, is to use a mixture model. Then, erroneous kmers would be those derived from the mixture component corresponding to the first mode. We propose mixture distribution
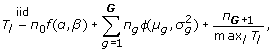 |
(2) |
Figure 3.

Histogram of estimated Tl for E. coli dataset.
where the mixing probabilities π0,…,πG+1 sum to 1. The first component f(α, β) of the mixture is a Gamma distribution, corresponding to the erroneous kmers. The second through (G + 1)th components are a series of normal distributions fitting the subsequent peaks for αl = 1,2,…,G. The last component is a uniform distribution over the observed range of Tl. We use the uniform distribution to account for the few kmers with large αl > G. One particular parameterization of the normal components is justified as follows. Under the assumption of reads distributed uniformly throughout the genome, the true Tl are Poisson random variables, where the mean depends on the identity of kmer xl and the error model. Rather than model the errors again, we hypothesize these means are Gamma random deviates. Then, Tl follows a Negative Binomial distribution [24], with means µg = gµp/(1 – p) and variances 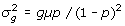 when αl = g ∈ {1,…,G}. Finally, by the Central Limit Theorem, the Negative Binomial is well-approximated by the Normal distribution with matching means and variances when the coverage-related parameter
when αl = g ∈ {1,…,G}. Finally, by the Central Limit Theorem, the Negative Binomial is well-approximated by the Normal distribution with matching means and variances when the coverage-related parameter 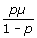 is large.
is large.
We can obtain the maximum likelihood estimate of the parameter vector θ = (π0,…,πG+1, α, β, µ,p) using another EM algorithm. Let Zlg, l ∈ {1,…,|Rk|}, g ∈ {0,…,G + 1} indicate if kmer xl is in group g. Then, the complete log likelihood for the proposed mixture model (Eq. 2) is
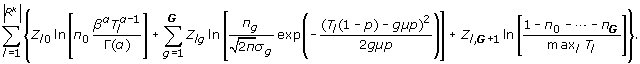 |
Let 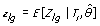 be the probability xl belongs in group g given the current estimate
be the probability xl belongs in group g given the current estimate 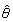 of all model parameters. For computational efficiency, at each iteration, we first compute
of all model parameters. For computational efficiency, at each iteration, we first compute
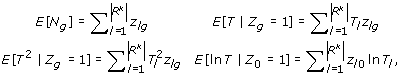 |
the first three for all g = 0,…,G + 1. Here, Ng is the number of kmers in group g, T is the number of attempts to read a random kmer, and Zg indicates if this kmer is in group g. Then, the update equations for the parameters are
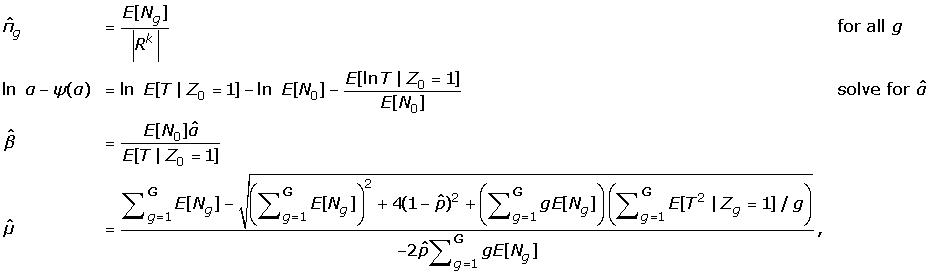 |
where 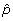 is found as the root of
is found as the root of
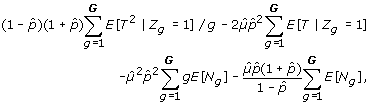 |
given 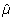 above. This and the second equation are implicit functions for
above. This and the second equation are implicit functions for 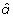 and
and 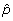 that can be solved using any one-dimensional root finder. To choose the best number of groups
that can be solved using any one-dimensional root finder. To choose the best number of groups 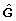 , we compute and minimize the BIC [23] over a range of plausible G. Members of the gamma component that represent kmers not present in the genome are identified as those with
, we compute and minimize the BIC [23] over a range of plausible G. Members of the gamma component that represent kmers not present in the genome are identified as those with 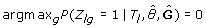 .
.
Contributor Information
Xiao Yang, Email: xyang@iastate.edu.
Srinivas Aluru, Email: aluru@iastate.edu.
Karin S Dorman, Email: kdorman@iastate.edu.
Acknowledgements
This research is supported in part by NSF CCF-0811804.
This article has been published as part of BMC Bioinformatics Volume 12 Supplement 1, 2011: Selected articles from the Ninth Asia Pacific Bioinformatics Conference (APBC 2011). The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/12?issue=S1.
References
- Stratton M. Genome resequencing and genetic variation. Nature Biotechnology. 2008;26(1):65–66. doi: 10.1038/nbt0108-65. [DOI] [PubMed] [Google Scholar]
- Perkel JM. Sanger Who? Sequencing the Next Generation. Science. 2009;10:275–279. [Google Scholar]
- Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Research. 2008;18(5):810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M, Pevzner P. Short read fragment assembly of bacterial genomes. Genome Research. 2008;18(2):324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson B, Regennitter M, Yang X, Schnable P, Aluru S. Parallel de novo Assembly of Large Genomes from High-Throughput Short Reads. 24th IEEE International Parallel & Distributed Processing Symposium. 2010. pp. 1–10.
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Research. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Xuan Z, Zhang MQ. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics. 2008;9:128–135. doi: 10.1186/1471-2105-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M, Pevzner P, Tang H. Fragment assembly with short reads. Bioinformatics. 2004;20(13):2067–2074. doi: 10.1093/bioinformatics/bth205. [DOI] [PubMed] [Google Scholar]
- Qu W, Hashimoto S, Morishita S. Efficient frequency-based de novo short-read clustering for error trimming in next-generation sequencing. Genome Research. 2009;19(7):1309–15. doi: 10.1101/gr.089151.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder J, Schröder H, Puglisi SJ, Sinha R, Schmidt B. SHREC: a short-read error correction method. Bioinformatics. 2009;25(17):2157–2163. doi: 10.1093/bioinformatics/btp379. [DOI] [PubMed] [Google Scholar]
- Yang X, Dorman KS, Aluru S. Reptile: Representative tiling for short read error correction. Bioinformatics. 2010;26(20):2526–2533. doi: 10.1093/bioinformatics/btq468. [DOI] [PubMed] [Google Scholar]
- Chin FYL, Leung HCM, Li WL, Yiu SM. Finding optimal threshold for correction error reads in DNA assembling. BMC Bioinformatics. 2009;10(Suppl 1):S15. doi: 10.1186/1471-2105-10-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijaya E, Frith MC, Suzuki Y, Horton P. Recount: expectation maximization based error correction tool for next generation sequencing data. Genome Informatics. 2009;23(1):189–201. [PubMed] [Google Scholar]
- Beissbarth T, Hyde L, Smyth GK, Job C, Boon WM, Tan SS, Scott HS, Speed TP. Statistical modeling of sequencing errors in SAGE libraries. Bioinformatics. 2004;20(Suppl 1):i31–i39. doi: 10.1093/bioinformatics/bth924. [DOI] [PubMed] [Google Scholar]
- Weese D, Emde AK, Rausch T, Doring A, Reinert K. et al. RazerS–fast read mapping with sensitivity control. Genome Research. 2009;19(9):1646–1654. doi: 10.1101/gr.088823.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Research. 2008;36(16):e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. Journal of the Royal Statistical Society, Series B. 1977;39:1–38. [Google Scholar]
- Zhi D, Keich U, Pevzner P, Heber S, Tang H. Correcting base-assignment errors in repeat regions of shotgun assembly. IEEE/ACM Trans Comput Biol Bioinform. 2007;4(1):54–64. doi: 10.1109/TCBB.2007.1005. [DOI] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Haubold B, Wiehe T. How repetitive are genomes? BMC Bioinformatics. 2006;7:541. doi: 10.1186/1471-2105-7-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the Dimension of a Model. The Annals of Statistics. 1978;6(2):461–464. [Google Scholar]
- McCullagh P, Nelder JA. Generalized Linear Models. 2. New York: Chapman & Hall; 1989. [Google Scholar]