

Abstract
Anywhere water is in the liquid state, bacteria will exist as biofilms, which are complex communities of cells cemented together. Although frequently associated with disease and biofouling, biofilms are also important for engineering applications, such as bioremediation, biocatalysis and microbial fuel cells. Here we review approaches to alter genetic circuits and cell signaling toward controlling biofilm formation, and emphasize utilizing these tools for engineering applications. Based on a better understanding of the genetic basis of biofilm formation, we find that biofilms may be controlled by manipulating extracellular signals and that they may be dispersed using conserved intracellular signals and regulators. Biofilms could also be formed at specific locations where they might be engineered to make chemicals or treat human disease.
Genetic basis of biofilm formation and dispersal
Bacteria alternate between planktonic (free-swimming) and sessile states, with dense, multi-cellular communities called ‘biofilms’ being the more important state [1]. Nearly all cells make biofilms [1, 2], which are formed in aquatic environments by the attachment of bacteria to submerged surfaces, to the air/liquid interface, and to each other. Biofilms attach via appendages, such as fimbriae and flagella [3], and microcolonies are formed by the production of microbial products including polysaccharides, proteins, lipids, and DNA [4]. Hundreds of genes are differentially controlled during biofilm development including stress-associated genes [5-8]; hence, these systems present an interesting challenge in terms of their control.
Biofilms are dynamic in that cells might detach from the biofilm matrix and disperse [9]. Active dispersal is initiated by the bacteria via a highly regulated process, whereas passive dispersal is mediated by external forces such as fluid shear and abrasion [9]. Changes in environmental conditions (e.g., nutrition level and oxygen depletion), whether favorable or unfavorable, can lead to biofilm dispersal [9] since it is beneficial to increase the biofilm at nearby locations when nutrients are plentiful and beneficial to colonize further locations when nutrients are scarce [3]. For example, Pseudomonas aeruginosa biofilms undergo dispersal in response to a sudden decrease [10] or a sudden increase [11] in nutrients. Furthermore, reproducible, periodic dispersal occurs in Actinobacillus actinomycetemcomitans [9], P. aeruginosa [12] and Serratia marcescens [13]; hence, biofilm dispersal is important for the survival of the species as it allows the bacterial population to expand. For many pathogenic bacteria, biofilm dispersal also plays a critical role in the transmission of bacteria from environmental reservoirs to human hosts, in the transmission of bacteria between hosts, and in the exacerbation and spread of infection within a single host [9].
Although complex and not fully understood (for example, many bacteria possess redundant means to form biofilms), biofilm formation is an ordered process that is dependent on the response of the cell to environmental cues, which in turn regulates specific genes. Stages of biofilm formation include motility to the surface, attachment, formation of clusters, development of differentiated structures, and dispersal. In P. aeruginosa, these stages of biofilm formation depend in part on an ordered response of three two-component systems: BfiSR, BfmSR and MifSR [14]. Two-component regulators usually consist of a sensing an environmental signal via a membrane-bound histidine protein-kinase receptor, followed by communicating that change via phosphorylation of a response regulator. The three two-component regulators are sequentially phosphorylated. Inactivation of BfiSR causes biofilm formation to cease at the stage of irreversible attachment, whereas inactivation of BfmSR and MifSR stop biofilm maturation at later stages (∼ 24 h and ∼72 h, respectively).
Biofilm formation and dispersal are ultimately genetic processes; therefore, they can be manipulated like other genetic systems using synthetic biology tools [15]. As an example, stress increases biofilm formation [16-18]; In Gram-negative bacteria, this occurs via the impact of stress on the concentration of the intracellular second messenger, c-di-GMP (3,5-cyclic diguanylic acid) (Box 1). High concentrations of c-di-GMP lead to low motility and biofilm formation, whereas low concentrations of c-di-GMP lead to high motility and dispersal [9, 19]. Several lines of evidence indicate that this control of c-di-GMP must be elegant and temporal: for most cells to form biofilms, they must interact with the surface so motility should be high [20]. Indeed, inactivation of diguanylate cyclases YeaI, YedQ and YfiN of Escherichia coli, which should reduce c-di-GMP levels, increases initial biofilm formation dramatically (T. Wood, unpublished). In addition, c-di-GMP levels are inversely related to extracellular DNA levels and are reduced by a tyrosine phosphatase [21]. Extracellular DNA is an important matrix component required for initial attachment [22] (when c-di-GMP should be low) and for biofilm maturation in order to develop a mushroom-shaped structure [23]. Hence, c-di-GMP levels should be low for enhanced motility and surface attachment, high for biofilm formation and maturation, and then low again for biofilm maturation and dispersal.
Box 1. c-di-GMP: ubiquitous signal for controlling biofilm formation.
c-di-GMP is an internal messenger that exists in almost all bacteria. It is the central regulator of biofilm formation because it mediates the switch between motile and sessile forms in Gram-negative bacteria [3]. High levels of c-di-GMP increase biofilm formation, whereas low levels reduce biofilm formation. c-di-GMP helps biofilms mature by increasing exopolysaccharide production, cell size, and cell aggregation, while decreasing swimming motility and extracellular DNA production [63]. c-di-GMP also binds to the PilZ domains of proteins to regulate exopolysaccharide synthesis, twitching motility, and flagella activity [19]; for example, binding to the PilZ domain of YcgR of E. coli leads to decreased swimming motility via reduced flagella activity, in turn increasing biofilm formation. This molecule plays a significant role in biofilm dispersal events as well; decreases in c-di-GMP concentrations induce biofilm dispersal by decreasing exopolysaccharide production and increasing motility [9].
c-di-GMP is synthesized from two guanosine-5′-triphosphate molecules by diguanylate cyclase (DGC), and is degraded by phosphodiesterase (PDE) into 5′-phosphoguanylyl-(3′-5′)-guanosine and guanosine monophosphate [19]. There are 29 putative DGCs and PDEs in E. coli [63], although many of these proteins have not been studied. The duplication of enzymatic activity (synthesizing or degrading c-di-GMP) is unusual with respect to internal signals (e.g. intracellular cyclic AMP is synthesized by a single adenylate cyclase and degraded by a specific PDE [19]), but is a consistent feature for c-di-GMP in many genomes. One explanation is that DGCs and PDEs are individually activated and inactivated under different conditions, although the propagation of these environmental signals is not understood thoroughly. For example, although BdcA is not a PDE, it binds to c-di-GMP and primarily works toward biofilm dispersal. Consistently, its gene expression is the highest at stationary phase (8 h) [63]. Similarly, we hypothesize that for initial biofilm formation, c-di-GMP levels are low for surface attachment, and some specific DGCs may be inactivated (or its gene transcription is low) or some specific PDEs (or c-di-GMP-binding proteins) may be activated (or its gene transcription is high). During biofilm maturation, this gene regulation/protein modification controlling c-di-GMP levels is reversed. Hence, if we can understand the regulation of each DGC/PDE pair, we might be able to manipulate each stage of biofilm development under various growth conditions.
Biofilms and disease
Biofilms are important contributors to the state of human disease, considering 80% of bacterial chronic inflammatory and infectious diseases involve biofilms [24]. As the most common infection, biofilms of uropathogenic E. coli cause urinary tract infections that result in 8 million trips annually to physicians in the United States [25]. Enterohemorrhagic E. coli (EHEC) infections in the gastrointestinal (GI) tract are also important, as there are over 76 million food-related infections annually in the U.S. that lead to 325,000 hospitalizations [26], 5,000 deaths [26], and an economic cost up to $152 billion USD [27]. Furthermore, P. aeruginosa is an opportunistic pathogen that is responsible for many biofilm infections, including those associated with ventilator-associated pneumonia, urinary and peritoneal dialysis catheters, bacterial keratitis, otitis externa, burns, and lungs [28]. Another opportunistic pathogen, Staphylococcus epidermidis, frequently infects catheter materials; nearly 80% of the cells involved in biomaterial-associated infections are S. epidermidis [29].
Biofilms and engineering
The robust nature of biofilms (i.e. their ability to withstand chemical and physical stresses more than their planktonic counterparts [30]) makes them superior for many beneficial biotechnology applications. Their resiliency stems primarily from the physical barrier provided by the matrix to some chemicals [31] and predators as well as from the physiological heterogeneity [32] caused by concentration gradients, mutation (e.g. enhanced genetic exchange [33]), and stochastic/environment-induced gene expression that leads to dormancy [34]; this sleeping state allows cells to withstand otherwise lethal environments because they are not metabolizing.
Biofilms have been important for over a century for wastewater treatment [35] (Table 1) and for a recent extension of this process known as bioremediation. In bioremediation applications, biofilms are used for the removal of various environmental contaminants (e.g. substituted aromatics, heavy metals) [30] (Table 1) as well as myriad processes, including rhizoremediation, where biofilms on the roots of plants degrade chlorinated ethenes [36]. Beneficial biofilms have also been proposed to decrease biocorrosion [37], as well as hold promise for other applications, such as biocatalysis [38], which includes the production of biofuels, specialty/bulk chemicals, biologics, and food additives (Table 1). For biofuels such as ethanol from Zymomonas mobilis, greater productivity has been achieved in biofilms than by planktonic cells [38] (Table 1). Furthermore, biofilms are also important for bioelectrochemical systems such as microbial fuel cells where communities form on the surface of electrodes to produce power [39].
Table 1. Representative applications of engineered biofilms.
| Application | Description | Refs. |
|---|---|---|
| Bioremediation | Removing hydrocarbons and heavy metals from the environment. | [30] |
| Wastewater treatment | Removing organic pollutants and ammonia from wastewater. | [35] |
| Biocorrosion control | Inhibiting the biocorrosion of steel, copper, and aluminum. | [37, 40-44] |
| Biofuels | Producing ethanol with Zymomonas mobilis. | [38] |
| Disease treatment | Reducing EHEC and Pseudomonas infections with indole and 7-hydroxyindole. | [52, 74] |
| BioMEMS | Spatially controlling the placement of bacterial “nanofactories” in a device; synthesizing QS signals or QQ compounds to control cellular behavior. | [71] |
| Chemicals | Producing specialty and bulk chemicals in a biorefinery. | This review |
| Pharmaceutical testing | Testing drugs and pro-drugs. | This review |
Early engineered biofilm systems: population control
To take advantage of the potential of biofilms, their formation must be controlled. The first engineered biofilm for any application was a consortium to reduce the biocorrosion of 1018 mild steel and 304 stainless steel [40]. Because it has only one membrane, Bacillus subtilis was engineered to secrete the human peptide antimicrobial bactenecin to inhibit the growth of the deleterious sulfate-reducing bacterium Desulfovibrio vulgaris. Growth of the corrosion-causing D. vulgaris was reduced by 36-fold in the biofilm, and corrosion was reduced by 12-fold [37]. This work was corroborated by using natural bacterial strains, such as Bacillus brevis 18-3 and Bacillus licheniformis, that produce the antimicrobial gramicidin and γ-polyglutamate, respectively, to inhibit the corrosion of stainless steel and mild steel by sulfate-reducing bacteria [41] and by iron-oxidizing bacteria [42]; to inhibit the corrosion of copper [43]; and to inhibit the corrosion of aluminum [44] (Table 1). These studies are important in that they employed a consortium rather than a single species, demonstrated that bacterial populations could be controlled in biofilms, and showed that cells could be engineered to achieve a biotechnology aim.
Using a synthetic circuit, an external stressor (UV light) has been applied to control the biofilm formation of a single E. coli strain (Figure 1a) [45]. UV light effectively damages DNA, which in turn activates the protease activity of RecA. RecA then cleaves the lambda cI repressor, which de-represses traA – the conjugation plasmid pilin gene under control of the pL promoter. Biofilm formation increases as a result of conjugation. This was one of the first reports showing that biofilm formation could be manipulated by an external stimulus, and that “plug and play” genetic circuits can be developed (Figure 1a).
Fig. 1.
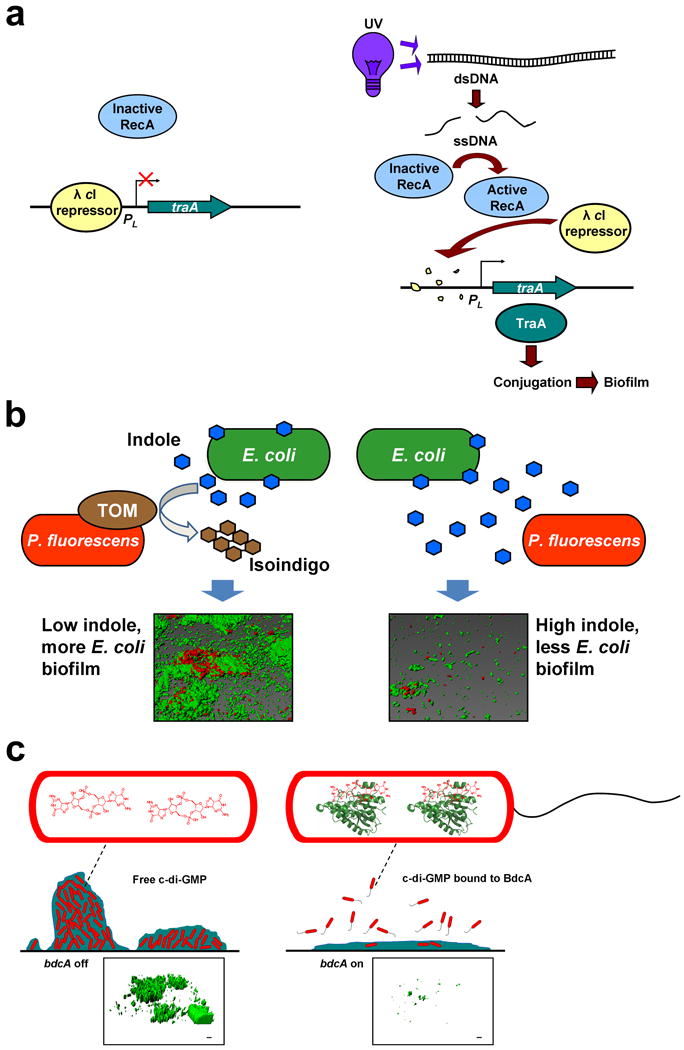
Engineered genetic circuits to control biofilm formation. (a) Genetic circuit to control E. coli biofilm formation using an environmental signal (e.g. UV light). Phage λ cI repressor silences traA transcription by binding its promoter. Application of UV light converts double-stranded DNA (dsDNA) into single-stranded DNA (ssDNA), which activates the RecA protease. Active RecA degrades λ cI, which induces traA. traA expression increases biofilm formation by enhancing conjugation. Reproduced with permission from Ref. [45]. (b) Genetic circuit to control dual-species biofilm formation using QS. Toluene o-monooxygenase (TOM) is produced by cloning tomA0A1A2A3A4A5 of Burkholderia cepacia G4 into Pseudomonas fluorescens using a constitutive promoter, which converts indole into insoluble isoindigo. Once indole is removed by the pseudomonad, indole no longer inhibits E. coli biofilm formation. Representative confocal consortial biofilm images are shown in which P. fluorescens is tagged with a red fluorescent protein and E. coli is tagged with a green fluorescent protein. Reproduced with permission from Ref. [49]. (c) Genetic circuit to control E. coli biofilm dispersal using a c-di-GMP-binding protein. Engineered BdcA variant binds c-di-GMP, which decreases the concentration of free c-di-GMP. Lower c-di-GMP concentrations lead to increased motility and reduced adhesin production, resulting in biofilm dispersal. Reproduced with permission from Ref. [63]. Representative confocal biofilm images are shown in which E. coli is tagged with a green fluorescent protein.
Engineering quorum sensing circuits to control biofilm formation
Bacterial cells communicate via quorum sensing (QS) (Box 2): signals are secreted, their concentrations build extracellularly as a function of cell density, then they are internalized or detected to coordinate gene expression via response regulators [46]. Building on this understanding, coordinated cell communication has been used to control fluorescence in biofilms between two E. coli populations via QS circuits [47]. The circuitry was flexible and should allow for future work with genes – other than those encoding fluorophores – to be manipulated in an analogous fashion [48].
Box 2. Quorum sensing and quenching for controlling biofilms.
Quorum sensing (QS) is cell communication using extracellular signals that are secreted during specific growth stages. The external signal is then internalized or detected by surface proteins, which results in changes in the regulation of usually several genes. In effect, the cell is able to respond to changes in population density or changes in hydrodynamics that dictate signal concentrations. Typical signaling molecules include N-acylhomoserine lactones (AHL) in Gram-negative bacteria, and autoinducer-2 (AI-2) in both Gram-negative and -positive bacteria [46]. AHL signaling is usually species-specific, whereas AI-2 signaling is common to many bacteria, but is not universal. AI-2 increases biofilm formation by enhancing flagella motility [72].
Since QS is integral to biofilm formation, it is possible to interfere or quench QS (QQ) to control biofilm formation, which would be beneficial for biofouling applications. Unlike antimicrobials, QQ compounds (also known as anti-virulence compounds) are utilized to disrupt cell communication without affecting growth; these compounds hold much promise because there should be less Darwinian selection pressure for resistance to them [77]. By disrupting communication, pathogens become more susceptible to the host immune system and build less-effective biofilms. Biofilms of pathogens are notoriously difficult to eradicate with antibiotics. To this end, brominated furanones have been utilized to reduce biofilm formation in E. coli via QQ of both AHL- and AI-2-mediated signaling [78]. Also, nitric oxide disperses biofilm formation by disrupting c-di-GMP internal signaling [79], and D-amino acids trigger biofilm dispersal by causing the release of amyloid fibers that link cells together in the biofilm [80].
The first synthetic circuit to control biofilm formation via QS utilized indole [49], which inhibits E. coli biofilm formation [49-53] in a QS fashion [52]. Based on these fundamental studies, a simple circuit was built in which the extracellular concentration of indole in a consortium containing E. coli and Pseudomonas fluorescens was altered by cloning toluene o-monooxygenase into the chromosome of the pseudomonad. Constitutive expression of the monooxygenase in the pseudomonad served to decrease the concentration of extracellular indole available to E. coli by converting it into insoluble isoindigo. The 22-fold reduction in the extracellular concentration of the biofilm inhibitor indole led to an 12-fold increase in the biofilm formation of E. coli (Figure 1b) [49], demonstrating that the level of biofilm formation of a specific member of a consortium can be controlled by varying the concentration of a QS compound.
Protein engineering to control biofilm formation
SdiA (suppress cell division inhibitors) detects N-acylhomoserine lactone (AHL) signals [54] – although E. coli makes no AHL of its own – and is required for sensing the biofilm signal indole [49, 53]. Hence, SdiA helps E. coli respond to the QS signals of other bacteria as well as to its own QS signal indole. Cementing the role of indole in E. coli biofilm formation and its relation to SdiA, protein engineering of SdiA was performed via error-prone PCR [55]. Random mutagenesis of genes is a powerful approach to create proteins with novel functions; with this approach, knowing the structure of the protein is not necessary, because beneficial mutations are found everywhere – both near and far from active sites – and they are hard to find by rational approaches [56]. The key is to develop screens, or even better, selection methods to identify the beneficial mutations that lead to enhanced protein activity [57]. Using this approach, 4457 colonies were screened for altered biofilm formation to identify a SdiA variant with amino acid replacements F7L, F59L, Y70C, M94K and K153X (Figure 2a); this variant had reduced biofilm formation by 20-fold relative to wild-type SdiA after 24 h owing to an increase in indole concentrations [55]. Another SdiA variant has been identified with four amino acid replacements (E31G, Y42F, R116H and L165Q) that has led to 7-fold higher biofilm formation in the presence of N-octanoyl-D,L-homoserine lactone and N-(3-oxododecatanoyl)-L-homoserine lactone produced by pseudomonads; this activity is opposite that of wild-type SdiA, which reduces biofilm formation in the presence of AHL signals [55]. Hence, protein engineering may be used create tools to control biofilm formation. The regulator SdiA was not only converted into a more sensitive inhibitor of biofilm formation via its own signal indole, but also its activity was reversed via its conversion into a stimulator of biofilm formation in the presence of signals produced by other bacteria.
Fig. 2.
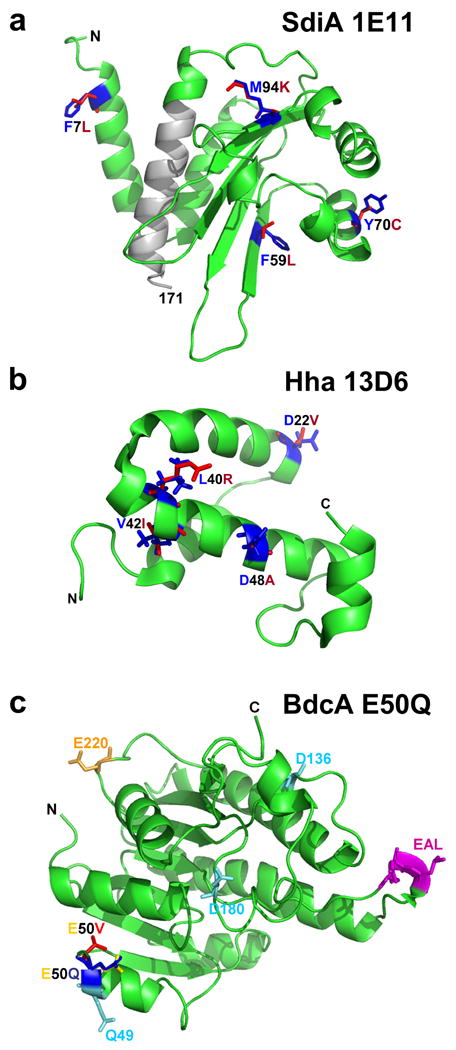
(a) Protein structure of evolved SdiA to decrease biofilm formation [55]. Truncated region (residues 153-171) is shown as a gray ribbon, and substituted residues of SdiA 1E11 (F7L, F59L, Y70C and M94K) are shown in red, while the original residues are shown in blue. (b) Protein structure of evolved Hha to disperse biofilms [62]. Substituted residues of Hha 13D6 (D22V, L40R, V42I, and D48A) are shown in red, while the original residues are shown in blue. (c) Protein structure of evolved BdcA to disperse biofilms [63]. The E50 residue is indicated in yellow. The replaced amino acid V in the BdcA E50V variant is indicated in red. The replaced amino acid Q in the BdcA E50Q variant is indicated in blue. The typical c-di-GMP phosphodiesterase contains EALXR for coordinating Mg2+, Q/R/D/D for c-di-GMP binding, and T/E for catalysis. Here the possible EAL-domain residues are indicated: EAL in pink as part of the EALXR motif, Q49/D136/D180 in turquoise as part of the Q/R/D/D motif, and E220 in orange as part of the T/E motif. The amino and carboxy termini are marked as N and C, respectively. Amino acid abbreviations: A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; I, isoleucine; K, lysine; L, leucine; M, methionine; Q, glutamine; R, arginine; T, threonine; V, valine; Y, tyrosine; X, any amino acid residue.
Engineering a global regulator to control biofilm formation
The first engineered regulator to control biofilm formation was the histone-like nucleoid structuring protein (H-NS) of E. coli [58]. H-NS is a global regulator that represses transcription by recognizing curved DNA sequences [59, 60] and is widely conserved in Gram-negative bacteria [61]. H-NS is very abundant, with more than 20,000 copies per cell [60], and consists of three domains: N-terminal oligomerization, C-terminal DNA-binding, and a flexible linker [59]. Using error-prone PCR, 2104 mutants were screened for altered biofilm formation, and H-NS variant K57N was identified as a mutant that reduces biofilm formation 10-fold compared to wild-type H-NS [58]. Hence, the activity was reversed: wild-type H-NS increases biofilm formation, whereas H-NS K57N reduces its formation (but does not alter dispersal). H-NS K57N reduces biofilm formation through its interaction with the nucleoid-associated proteins Cnu and StpA, which leads to enhanced excision of one out of nine resident cryptic prophages in E. coli – the defective prophage Rac (wild-type H-NS represses excision). Excision of the prophage results in cell lysis through the expression of a host killing toxin HokD [58] of the cryptic prophage Qin. A global regulatory system may therefore be evolved through a single amino acid change in the N-terminal oligomerization domain of a protein to control biofilm formation, prophage excision, and programmed cell death. Such tools may be important for helping to remove biofilms.
Engineering regulators to control biofilm dispersal
Arguably one of the loftiest goals related to biofilm formation is to be able to remove existing biofilms, either to treat disease or to control biofilm formation for engineering applications. Recently, two approaches have been attempted, using internal circuits to effect biofilms dispersal: one involved the global regulator Hha (high hemolysin activity) [62] and the other relied on a novel c-di-GMP-binding protein, BdcA (biofilm dispersal via c-di-GMP; formerly YjgI), that titrates c-di-GMP, thereby causing dispersal [63].
Hha regulates 162 loci via its partner, H-NS [64] and has been linked to biofilms. hha is induced 30-fold in E. coli biofilms [7], and Hha decreases initial biofilm formation by repressing the transcription of both rare codon tRNAs and fimbrial genes [65]. Hha is also toxic and leads to cell lysis and biofilm dispersal as a result of the activation of prophage lytic genes [66] and the induction of the protease ClpXP [65]. Hence, Hha is a global transcriptional regulator in biofilm formation so it was reasoned that it could be engineered to control biofilm dispersal [62]. Using random mutagenesis of hha and a direct screen for biofilm dispersal, Hha variant 13D6 (D22V, L40R, V42I and D48A) was obtained (Figure 2b). Hha 13D6 causes nearly complete (96%) biofilm dispersal in flow cells by increasing programmed cell death [62]. Importantly, it induces dispersal without affecting initial biofilm formation.
Unlike Hha 13D6, BdcA has been engineered to cause biofilm dispersal by decreasing the concentration of second messenger c-di-GMP [63] (Figure 1c). Once c-di-GMP levels are reduced, genetic cascades are influenced, thus leading to phenotypic changes such as increased motility and decreased adhesin formation (e.g. curli, cellulose) [19]. As a c-di-GMP-binding protein, BdcA exhibits pleiotropic effects, such as reduction in exopolysaccharide, cell length and aggregation, while increasing motility and extracellular DNA [63]. BdcA was discovered during a biofilm screen of uncharacterized genes related to the protein TqsA that exports the bacterial signal AI-2 [67]; the bdcA mutant was found to be deficient in biofilm dispersal [63]. Hence, it was reasoned that BdcA positively regulates biofilm dispersal. 31P NMR spectroscopy has been used to show that BdcA binds c-di-GMP, but does not act as a phosphodiesterase [63]. To increase biofilm dispersal, protein engineering was used to evolve BdcA for greater c-di-GMP binding [63]. A novel screen of 6000 mutants was developed that relies on the increase in swimming motility that occurs as c-di-GMP levels are decreased [63]. The best BdcA variant had the single amino acid change E50V (Figure 2c), which caused fourfold greater biomass dispersal in flow cells than the wild-type BdcA. Saturation mutagenesis was then employed to increase biofilm dispersal further by testing all 20 possible amino acid substitutions at this position [68] because a single mutation randomly placed in codons generates, on average, only 5.6 out of 19 possible substitutions, and multiple mutations in a single codon are rare [69]. Using this approach, BdcA E50Q was identified (Figure 2c), which causes nearly complete removal of biofilms in flow cells (18-fold better than wild-type BdcA), without affecting initial biofilm formation. Hence a second genetic switch was created based on a novel c-di-GMP-binding protein that may be used to remove biofilms after they are formed. Since c-di-GMP is used by nearly all bacteria, this approach may be useful in many strains.
Engineering bacteriophage to control biofilm dispersal
Bacteriophages have also been engineered to control bacterial biofilm dispersal. Dispersin B from A. actinomycetemcomitans, an enzyme which degrades the biofilm adhesion β-1,6-N-acetyl-d-glucosamine, has been cloned into a T7 phage, resulting in nearly 100% dispersal of an E. coli biofilm [70]. dspB was placed under control of the T7φ10 promoter so it was transcribed during infection. This demonstrated that combining a polysaccharide-degrading enzyme with normal phage lytic mechanisms is more effective than using an unmodified phage. Because different bacteria utilize different biofilm extracellular polymeric building blocks (some cells utilize more than one building block), and phages are usually very strain-specific, several enzymes and several types of phage would likely be required to disperse multi-species biofilms. However, the approach holds promise for removing one strain specifically in a multi-species biofilm.
Targeting gene expression and patterning in biofilms
Targeting gene expression within biofilms is desirable for certain biotechnology applications such as tumor-specific anti-cancer therapies and quorum quenching (QQ) measures (see QS/QQ text box). One successful approach in targeting gene expression has been to anchor cells at specific locations where chitosan had been previously deposited in a biological microelectromechanical systems (bioMEMS) device to create bacterial “nanofactories” that are targeted to a specific location, assemble, synthesize the QS signal AI-2, and detect AI-2 [71]. Pfs (S-adenosylhomocysteine nucleosidase) and LuxS (S-ribosylhomocysteinase) convert S-adenosylhomocysteine to AI-2, so a (His)6-protein G-LuxS–Pfs-(Tyr)5 fusion protein was used to target cells specifically to areas where chitosan was deposited via the Tyr tag. Protein G helped to assemble E. coli cells via an antibody against E. coli that binds to the fusion protein, and GFP was used to detect the AI-2 signal. Hence, spatially selective synthesis, capture and manipulation of a QS signal that is known to increase biofilm formation [72] was achieved within a bioMEMS device (Table 1). This opens doors to future studies involving the impact of AI-2 on biofilm formation as well as for the synthesis of QQ compounds at specific locations; i.e., within biofilms, to thwart pathogens using truly novel therapies.
Another approach to direct biofilm development to specific regions includes creating regions that resist biofilm formation. Using self-assembled monolayers on gold film that were terminated with D-mannitol, biofilm formation has been controlled for E. coli, P. aeruginosa, and Candida albicans for up to 26 days [73]. This achievement allows gaps at the micrometer scale to be formed between biofilms without a physical barrier so that gene expression may be studied as a function position relative to the gap. Also, bacterium-surface and bacterium-host interactions may be explored.
Future prospects
To engineer is to control. Therefore, an overarching goal is to discern the complete genetic basis of biofilm formation and dispersal so that this information may be used to control these communities of associated cells for biotechnological applications (Table 1). The studies highlighted in this review demonstrate that once individual biofilm regulatory circuits are understood, biofilm formation and dispersal may be manipulated.
Biofilms can be formed in a spatially selective manner; cells can be engineered to secrete compounds and QS signals to affect the behavior of neighboring cells in a consortium; and biofilms can be dispersed. Further refinements in this nascent field include the need to demonstrate that biofilms may be generated and removed as desired (i.e. a reversible process), which will facilitate the re-use of platforms. In addition, it should be demonstrated that beneficial cells can be engineered to displace or integrate into an existing biofilm. This might allow treatment of disorders in the complex GI tract, and allow one to prevent colonization by pathogens, such as EHEC and P. aeruginosa, by secreting compounds known to inhibit these strains [52, 74] (Table 1). The introduction of beneficial biofilms will also facilitate engineering applications, such as the reduction of biocorrosion.
Along with spatiotemporal control of biofilm formation and dispersal, gene expression might be controlled as a function of depth at a single position. This could perhaps be achieved by using oxygen-responsive promoters, because oxygen levels will drop to zero at the base of the biofilm in an aerobic environment. This depth-controlled expression can be monitored with new techniques, such as direct RNA sequencing [75] of cells taken from specific positions using laser capture microdissection [76]; as a result, biofilm synthetic capabilities might be optimized as a function of depth at each position. Synthetic biology and protein engineering approaches may be utilized to make these depth-controlled circuits. The ability to harness biofilms in a spatiotemporal manner via engineered cellular communication could allow the creation of sophisticated reactor systems. Such reactor systems could lead to formation of bio-refineries on large and small scales in which plant-derived feedstocks are converted into a wide array of products, thereby making simultaneous use of the growth compounds and metabolites produced from complex feedstocks.
Acknowledgments
This work was supported by the National Institutes of Health (R01 GM089999). T.W. is the T. Michael O'Connor Endowed Professor at Texas A & M University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolter R, Losick R. One for all and all for one. Science. 1998;280:226–227. doi: 10.1126/science.280.5361.226. [DOI] [PubMed] [Google Scholar]
- 3.Karatan E, Watnick P. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol Mol Biol Rev. 2009;73:310–347. doi: 10.1128/MMBR.00041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. 2010;8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 5.Beloin C, et al. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol. 2004;51:659–674. doi: 10.1046/j.1365-2958.2003.03865.x. [DOI] [PubMed] [Google Scholar]
- 6.Domka J, et al. Temporal gene-expression in Escherichia coli K-12 biofilms. Environ Microbiol. 2007;9:332–346. doi: 10.1111/j.1462-2920.2006.01143.x. [DOI] [PubMed] [Google Scholar]
- 7.Ren D, et al. Gene expression in Escherichia coli biofilms. Appl Microbiol Biotechnol. 2004;64:515–524. doi: 10.1007/s00253-003-1517-y. [DOI] [PubMed] [Google Scholar]
- 8.Schembri MA, et al. Global gene expression in Escherichia coli biofilms. Mol Microbiol. 2003;48:253–267. doi: 10.1046/j.1365-2958.2003.03432.x. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan JB. Biofilm dispersal: mechanisms, clinical implications, and potential therapeutic uses. J Dent Res. 2010;89:205–218. doi: 10.1177/0022034509359403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunt SM, et al. Hypothesis for the role of nutrient starvation in biofilm detachment. Appl Environ Microbiol. 2004;70:7418–7425. doi: 10.1128/AEM.70.12.7418-7425.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sauer K, et al. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J Bacteriol. 2004;186:7312–7326. doi: 10.1128/JB.186.21.7312-7326.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies DG, Marques CNH. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J Bacteriol. 2009;191:1393–1403. doi: 10.1128/JB.01214-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice SA, et al. Biofilm formation and sloughing in Serratia marcescens are controlled by quorum sensing and nutrient cues. J Bacteriol. 2005;187:3477–3485. doi: 10.1128/JB.187.10.3477-3485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrova OE, Sauer K. A novel signaling network essential for regulating Pseudomonas aeruginosa biofilm development. PLoS Pathog. 2009;5:e1000668. doi: 10.1371/journal.ppat.1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pai A, et al. Engineering multicellular systems by cell-cell communication. Curr Opin Biotechnol. 2009;20:461–470. doi: 10.1016/j.copbio.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landini P. Cross-talk mechanisms in biofilm formation and responses to environmental and physiological stress in Escherichia coli. Res Microbiol. 2009;160:259–266. doi: 10.1016/j.resmic.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 17.LaPaglia C, Hartzell P. Stress-induced production of biofilm in the hyperthermophile Archaeoglobus fulgidus. Appl Environ Microbiol. 1997;63:3158–3163. doi: 10.1128/aem.63.8.3158-3163.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XS, et al. YcfR (BhsA) influences Escherichia coli biofilm formation through stress response and surface hydrophobicity. J Bacteriol. 2007;189:3051–3062. doi: 10.1128/JB.01832-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 20.Wood TK, et al. Motility influences biofilm architecture in Escherichia coli. Appl Microbiol Biotechnol. 2006;72:361–367. doi: 10.1007/s00253-005-0263-8. [DOI] [PubMed] [Google Scholar]
- 21.Ueda A, Wood TK. Tyrosine phosphatase TpbA of Pseudomonas aeruginosa controls extracellular DNA via cyclic diguanylic acid concentrations. Environ Microbiol Rep. 2010;2:449–455. doi: 10.1111/j.1758-2229.2010.00171.x. [DOI] [PubMed] [Google Scholar]
- 22.Rice KC, et al. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc Natl Acad Sci USA. 2007;104:8113–8118. doi: 10.1073/pnas.0610226104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allesen-Holm M, et al. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol. 2006;59:1114–1128. doi: 10.1111/j.1365-2958.2005.05008.x. [DOI] [PubMed] [Google Scholar]
- 24.Sauer K, et al. Biofilms and biocomplexity. Microbe. 2007;2:347–353. [Google Scholar]
- 25.Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113:5–13. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- 26.Mead PS, et al. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scharff RL. Health-related costs from foodbourne illness in the United States. Produce Safety Project 2010 [Google Scholar]
- 28.Macé C, et al. Identification of biofilm-associated cluster (bac) in Pseudomonas aeruginosa involved in biofilm formation and virulence. PLoS ONE. 2008;3:e3897. doi: 10.1371/journal.pone.0003897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Götz F. Staphylococcus and biofilms. Mol Microbiol. 2002;43:1367–1378. doi: 10.1046/j.1365-2958.2002.02827.x. [DOI] [PubMed] [Google Scholar]
- 30.Singh R, et al. Biofilms: implications in bioremediation. Trends Microbiol. 2006;14:389–397. doi: 10.1016/j.tim.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Fux CA, et al. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 33.Ehrlich GD, et al. The distributed genome hypothesis as a rubric for understanding evolution in situ during chronic bacterial biofilm infectious processes. FEMS Immunol Med Microbiol. 2010;59:269–279. doi: 10.1111/j.1574-695X.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis K. Persister cells. Annu Rev Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 35.Wuertz S, et al., editors. Biofilms in wastewater treatment - an interdisciplinary approach. IWA Publishing; 2003. [Google Scholar]
- 36.Yee DC, et al. Rhizoremediation of trichloroethylene by a recombinant, root-colonizing Pseudomonas fluorescens strain expressing toluene ortho-monooxygenase constitutively. Appl Environ Microbiol. 1998;64:112–118. doi: 10.1128/aem.64.1.112-118.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayaraman A, et al. Corrosion inhibition by aerobic biofilms on SAE1018 steel. Appl Microbiol Biotechnol. 1997;47:62–68. [Google Scholar]
- 38.Rosche B, et al. Microbial biofilms: a concept for industrial catalysis? Trends Biotechnol. 2009;27:636–643. doi: 10.1016/j.tibtech.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Kiely P, et al. Anodic biofilms in microbial fuel cells harbor low numbers of higher-power-producing bacteria than abundant genera. Appl Microbiol Biotechnol. 2010;88:371–380. doi: 10.1007/s00253-010-2757-2. [DOI] [PubMed] [Google Scholar]
- 40.Jayaraman A, et al. Inhibiting sulfate-reducing bacteria in biofilms by expressing the antimicrobial peptides indolicidin and bactenecin. J Indust Microbiol Biotechnol. 1999;22:167–175. doi: 10.1007/s002530051520. [DOI] [PubMed] [Google Scholar]
- 41.Jayaraman A, et al. Inhibiting sulfate-reducing bacteria in biofilms on steel with antimicrobial peptides generated in situ. Appl Microbiol Biotechnol. 1999;52:267–275. doi: 10.1007/s002530051520. [DOI] [PubMed] [Google Scholar]
- 42.Zuo R, Wood TK. Inhibiting mild steel corrosion from sulfate-reducing and iron-oxidizing bacteria using gramicidin-S-producing biofilms. Appl Microbiol Biotechnol. 2004;65:747–753. doi: 10.1007/s00253-004-1651-1. [DOI] [PubMed] [Google Scholar]
- 43.Jayaraman A, et al. Axenic aerobic biofilms inhibit corrosion of copper and aluminum. Appl Microbiol Biotechnol. 1999;52:787–790. doi: 10.1007/s002530051592. [DOI] [PubMed] [Google Scholar]
- 44.Örnek D, et al. Pitting corrosion inhibition of aluminum 2024 by Bacillus biofilms secreting polyaspartate or γ-polyglutamate. Appl Microbiol Biotechnol. 2002;58:651–657. doi: 10.1007/s00253-002-0942-7. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi H, et al. Programmable cells: interfacing natural and engineered gene networks. Proc Natl Acad Sci USA. 2004;101:8414–8419. doi: 10.1073/pnas.0402940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jayaraman A, Wood TK. Bacterial quorum sensing: signals, circuits, and implications for biofilms and disease. Annu Rev Biomed Eng. 2008;10:145–167. doi: 10.1146/annurev.bioeng.10.061807.160536. [DOI] [PubMed] [Google Scholar]
- 47.Brenner K, et al. Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium. Proc Natl Acad Sci USA. 2007;104:17300–17304. doi: 10.1073/pnas.0704256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brenner K, et al. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 2008;26:483–489. doi: 10.1016/j.tibtech.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Lee J, et al. Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiol. 2007;7:42. doi: 10.1186/1471-2180-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bansal T, et al. Differential effects of epinephrine, norepinephrine, and indole on Escherichia coli O157:H7 chemotaxis, colonization, and gene expression. Infect Immun. 2007;75:4597–4607. doi: 10.1128/IAI.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Domka J, et al. YliH (BssR) and YceP (BssS) regulate Escherichia coli K-12 biofilm formation by influencing cell signaling. Appl Environ Microbiol. 2006;72:2449–2459. doi: 10.1128/AEM.72.4.2449-2459.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J, et al. Enterohemorrhagic Escherichia coli biofilms are inhibited by 7-hydroxyindole and stimulated by isatin. Appl Environ Microbiol. 2007;73:4100–4109. doi: 10.1128/AEM.00360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee J, et al. Indole cell signaling occurs primarily at low temperatures in Escherichia coli. ISME J. 2008;2:1007–1023. doi: 10.1038/ismej.2008.54. [DOI] [PubMed] [Google Scholar]
- 54.Lindsay A, Ahmer BMM. Effect of sdiA on biosensors of N-acylhomoserine lactones. J Bacteriol. 2005;187:5054–5058. doi: 10.1128/JB.187.14.5054-5058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J, et al. Reconfiguring the quorum-sensing regulator SdiA of Escherichia coli to control biofilm formation. Appl Environ Microbiol. 2009;75:1703–1716. doi: 10.1128/AEM.02081-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van den Heuvel RHH, et al. Laboratory-evolved vanillyl-alcohol oxidase produces natural vanillin. J Biol Chem. 2004;279:33492–33500. doi: 10.1074/jbc.M312968200. [DOI] [PubMed] [Google Scholar]
- 57.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hong SH, et al. Controlling biofilm formation, prophage excision, and cell death by rewiring global regulator H-NS of Escherichia coli. Microb Biotechnol. 2010;3:344–356. doi: 10.1111/j.1751-7915.2010.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorman CJ. H-NS: a universal regulator for a dynamic genome. Nat Rev Microbiol. 2004;2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- 60.Rimsky S. Structure of the histone-like protein H-NS and its role in regulation and genome superstructure. Curr Opin Microbiol. 2004;7:109–114. doi: 10.1016/j.mib.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Tendeng C, Bertin PN. H-NS in Gram-negative bacteria: a family of multifaceted proteins. Trends Microbiol. 2003;11:511–518. doi: 10.1016/j.tim.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Hong SH, et al. Engineering global regulator Hha of Escherichia coli to control biofilm dispersal. Microb Biotechnol. 2010;3:717–728. doi: 10.1111/j.1751-7915.2010.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma Q, et al. Engineering a novel c-di-GMP-binding protein for biofilm dispersal. Environ Microbiol. 2010 doi: 10.1111/j.1462-2920.2010.02368.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baños RC, et al. Differential regulation of horizontally acquired and core genome genes by the bacterial modulator H-NS. PLoS Genet. 2009;5:e1000513. doi: 10.1371/journal.pgen.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.García-Contreras R, et al. Protein translation and cell death: the role of rare tRNAs in biofilm formation and in activating dormant phage killer genes. PLoS ONE. 2008;3:e2394. doi: 10.1371/journal.pone.0002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, et al. Control and benefits of CP4-57 prophage excision in Escherichia coli biofilms. ISME J. 2009;3:1164–1179. doi: 10.1038/ismej.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herzberg M, et al. YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J Bacteriol. 2006;188:587–598. doi: 10.1128/JB.188.2.587-598.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rui L, et al. Saturation mutagenesis of toluene ortho-monooxygenase of Burkholderia cepacia G4 for enhanced 1-naphthol synthesis and chloroform degradation. Appl Environ Microbiol. 2004;70:3246–3252. doi: 10.1128/AEM.70.6.3246-3252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bylina EJ, et al. Solid-phase enzyme screening. ASM News. 2000;66:211–217. [Google Scholar]
- 70.Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fernandes R, et al. Biological nanofactories facilitate spatially selective capture and manipulation of quorum sensing bacteria in a bioMEMS device. Lab on a Chip. 2010;10:1128–1134. doi: 10.1039/b926846d. [DOI] [PubMed] [Google Scholar]
- 72.González Barrios AF, et al. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022) J Bacteriol. 2006;188:305–316. doi: 10.1128/JB.188.1.305-316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hou S, et al. Prolonged control of patterned biofilm formation by bio-inert surface chemistry. Chem Commun. 2009:1207–1209. doi: 10.1039/b822197a. [DOI] [PubMed] [Google Scholar]
- 74.Lee J, et al. Indole and 7-hydoxyindole diminish Pseudomonas aeruginosa virulence. Microb Biotechnol. 2009;2:75–90. doi: 10.1111/j.1751-7915.2008.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ozsolak F, et al. Direct RNA sequencing. Nature. 2009;461:814–818. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- 76.Lenz AP, et al. Localized gene expression in Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2008;74:4463–4471. doi: 10.1128/AEM.00710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sintim HO, et al. Paradigm shift in discovering next-generation anti-infective agents: targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med Chem. 2010;2:1005–1035. doi: 10.4155/fmc.10.185. [DOI] [PubMed] [Google Scholar]
- 78.Ren D, et al. Inhibition of biofilm formation and swarming of Escherichia coli by (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2-(5H)-furanone. Environ Microbiol. 2001;3:731–736. doi: 10.1046/j.1462-2920.2001.00249.x. [DOI] [PubMed] [Google Scholar]
- 79.Barraud N, et al. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic diguanosine-5′-monophosphate levels and enhanced dispersal. J Bacteriol. 2009;191:7333–7342. doi: 10.1128/JB.00975-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolodkin-Gal I, et al. d-amino acids trigger biofilm disassembly. Science. 2010;328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]