

Abstract
Communication between the vasculature and nervous system is important during embryogenesis but the molecular mechanisms mediating this are ill-defined. We evaluated the molecular mechanisms by which Nerve Growth Factor (NGF) and Brain-derived neurotrophic factor (BDNF) regulate VEGF production. NGF activation of TrkA causes a marked increase in VEGF secretion by neuronal cells. The NGF induced increase in VEGF is accompanied by an increase in HIF-1α. Pharmacologic inhibitors of the Trk tyrosine kinase, PI-3 kinase and mTOR paths prevent NGF stimulated increases in HIF-1α and VEGF. NGF induced increase in VEGF transcription is dependent on a hypoxia response element (HRE) in the VEGF promoter. Mutation of the HRE or siRNA mediated silencing of HIF-1α expression blocks NGF induced increases in VEGF transcription. In primary cultures of TrkA expressing neurons from dorsal root ganglion, NGF induces VEGF expression that is accompanied by increases in HIF-1α but not HIF-2α expression. In CGN neurons, BDNF induces VEGF that is dependent on induction of HIF-1α. Our study indicates that neurotrophin activation of Trk stimulates an increase in VEGF transcription that is mediated by induction of HIF-1α.
Keywords: NGF/TrkA, BDNF/TrkB, VEGF, HIF-1α, dorsal root ganglion neurons, cerebellar granule neurons, neuroblastoma
Introduction
During embryogenesis neural-vascular communication is important for the appropriate development of the vasculature. In vivo sensory neurons determine the pattern of arterial differentiation and blood vessel formation. Moreover, during development Phox2b mutant mice lack peripheral autonomic nerves resulting in a disorganized neural network yet the alignment of the arteries with the misrouted axons is maintained (Mukouyama et al, 2002). Recently, a role for neurotrophins during angiogenesis and in response to vascular injury has emerged (Nico et al, 2007; Kermani and Hempstead, 2007). Neurotrophic factors (NTs) are family of structurally and functionally related growth factors that include NGF, BDNF, NT-3 and NT-4/5 that stimulate the survival, differentiation and function of neural cells via selective activation of tyrosine kinase receptors (TrkA, TrkB, and TrkC) in the absence or presence of p75NTR (Smeyne et al, 1994).
Studies indicate that NTs may act directly on Trk expressing vascular precursor cells or indirectly via induction of pro-angiogenic factors. NGF induces proliferation of TrkA and p75 expressing endothelial cells (Moser et al, 2004) while BDNF is an endothelial survival factor (Donavan et al, 2000), stimulating angiogenesis (Kermani et al, 2005) and increasing levels of VEGFR in TrkB expressing endothelial cells (Kim, et al 2004). Nerve growth factor (NGF) increases the levels of VEGF in normal neural cells (Calza et al 2001) and stimulates angiogenesis in animal models of ischemia (Turrini et al, 2002). VEGF is a key pro-angiogenic growth factors and may have neurotrophic properties under certain conditions. VEGF under hypoxic conditions is predominantly regulated by inhibition of the proteolytic degradation of HIF-1α and HIF-2α that typically occurs under normoxic conditions. This leads to increased levels of HIF-1α and HIF-2α which dimerize with constitutively expressed HIF-1β and bind to specific regions on the VEGF 5′ proximal region to increase VEGF transcription (Forsythe et al, 1996). Studies indicate that growth factors such as IGFs and TGFβ cytokines increase VEGF transcription under normoxic conditions via both HIF-1α dependent and independent mechanisms. (Loureiro and D’Amore, 2005). We have reported that BDNF activation of TrkB induces an increase in VEGF production in neuronal cell lines and the increased production of VEGF is dependent on the concomitant induction of HIF-1α (Nakamura et al, 2006).
In this study we examine the molecular mechanisms by which NGF activation of the TrkA signal transduction pathway stimulates increases in VEGF expression in neuronal cell lines and cultures of primary neurons.
Materials and Methods
Primary cells culture and neuronal cell lines
Dorsal root ganglia (DRG) tissues were removed from postnatal day 7(P7) mice pups and then incubated with 0.05mg/mml trypsin (Sigma) and 1.0mg/ml collagenase type I (Worthington) and 0.1mg/ml DNAseI for 30min at 37°C (DeBellard et al 1996; Cai et al 1999). Trypsinization was stopped by adding appropriate Dulbecco’s Modified Eagle Medium (DMEM, GIBCO) containing 10% FCS. The digested tissues were centrifuged at 800rpm for 5min and DRG neurons were resuspended in neurobasal medium and cultured in 10ng/ml NGF for 10–14 days. Cells were shifted to serum-free conditions for 6 hours prior to treatment with NGF 100ng/ml. Cerebellar granule neurons (CGNs) were prepared as described by Levi et al. (1984) and Romero et al (2003). Cerebella were removed from P7 mice pups, transferred into a Petri dish containing cold Hank’s Balanced Salt Solution (GIBCO, HBSS). Meninges and blood vessels of the cerebella were peeled off and discarded to ensure minimal contamination from endothelial cells. Cerebella were then minced into fine pieces (<0.5 mm) and incubated in HBSS containing 0.025% trypsin for 15 min at 37°C. DMEM containing 10%FBS was added to stop trypsinization of the cerebella. The digested tissues were centrifuged at 1000rpm for 5min, and further treated with HBSS with 0.05% DNAse I. Pasteur pipettes were used to disperse the tissues into a homogeneous suspension. The suspension was filtered through a 40um mesh and centrifuged for 5 min at 1000 rpm. Cerebellar granule neurons were re-suspended in neurobasal medium. Ninety-five percent of the CGN culture expressed βIII-tubulin, while 2.5% expressed Glial-fibillary protein, consistent with neural cells and the remaining 2.5% were assumed to be contaminating fibroblasts. Isolated CGNs were cultured for 2 days and treated with 100ng/ml NGF or BDNF and cells and conditioned media harvested after 24hrs. In a pilot experiment, we found that 10 or 100ng/ml of NGF in DRG neurons or similar concentrations of BDNF CGNs stimulated similar increase in VEGF protein secretion.
The tetracycline regulated TrkA expressing NB cells (TA6 and TA25) have been previously described (Kim, 1999). Although originally reported to be SY5Y cells, genetic analyses indicates these cells are the neuroblastoma cell line CHP-234 (unpublished data). Cells were transfected with the tetracycline-regulated vector, pBPSTR1, as a control (E2). 15N-TrkA cells (G3) and 15N-control cells (C16) have been previously described (Woo, 2004).
Immunocytochemistry
DRG neurons were plated onto the laminin and poly-L-lysine -coated 24 well plates (with coverslips) at a density of 3000/well. CGNs were plated onto poly-L-lysine -coated 24 well plates at a density of 60,000/well. DRG neurons or CGNs were cultured with either NGF 100ng/ml or BDNF 100ng/ml for 48hr. The supernatant media were collected and assayed for VEGF by ELISA (R & D Systems). The cells were fixed with 4% paraformaldehyde, rinsed in PBS and incubated with the appropriate primary antibody (TrkA sc-118, TrkB sc-8316 from Santa Cruz Biotechnology; HIF-1α NB 100–105, HIF-2α NB 100–132 from NOVUS; β-III tubulin T 8660 from Sigma; Neurofilament AB9568 from Chemicon) which was diluted by 1:100 in PBS containing 0.1% Triton and O.5% Goat Serum for 2h. Fluorescein-conjugated (TRITC and FITC) secondary antibodies (goat anti-rabbit secondary antibodies for TrkA, TrkB and Neurofilament; Goat anti-mouse secondary antibodies for HIF-1α, HIF-2α and β-III tubulin) were added to the cells by a 1:100 dilution in PBS containing 0.1% Triton and 0.5% Goat Serum for 1h. 4,6-diamindion-2-phenylindole (DAPI) was used for nuclear staining. For immunofluorescence image analysis, 5 fields of CGNs and 15 fields of DRG neurons were randomly selected in order to count at least 100 cells and evaluate immunofluorescence. For HIF-1α immunoflurescence staining, the cells were classified into negative cells (no staining), positive cells with low staining, and positive cells with high staining. The results are quantified by scoring the percentage of cells with high expression divided by total cells. The immunoflurescence staining was assessed using a Nikon ECLIPSE TE300 microscope equipped with an epi-fluorescence illuminator and fluorescent filter set to visualize the fluorochrome.
Cell Culture and Reagents
One million NB cells (E2, TA6, TA25, and 15N-TrkA cells) per well were cultured into 6-well plates in RPMI-1640 containing 10% fetal bovine serum for 8 or 16h. NB cells were shifted into serum-free RPMI medium for 6 h, and then treated with control media, NGF (100 ng/ml; Upstate, Lake Placid, NY) or BDNF (100 ng/ml; PeproTech, Inc., Rocky Hill, NJ) for 16 h. Cells and media were collected and stored as previously described (Beppu et al, 2005). Cells were treated with 100 ng/ml NGF for the indicated times with or without a 0.5 h pretreatment of the pharmacological inhibitors. TrkA inhibitor K252a (Calbiochem, San Diego, CA), PI3K inhibitor LY294002 (Sigma, St. Louis, MO), MEK inhibitor PD98059 (Sigma), and mTOR inhibitor rapamycin (Cell Signaling Technology, Beverly, MA) were obtained and reconstituted according to manufacturer’s specifications.
Protein assays
Western blotting and immunoprecipitation were performed as described previously (Jaboin et al, 2002; Beppu et al, 2005). Antibodies utilized were: anti-phospho-tyrosine antibody (PY-99; Santa Cruz Biotechnology, Santa Cruz, CA), anti-pan-Trk antibody (C-14; Santa Cruz Biotechnology), anti-TrkA antibody (763; Santa Cruz Biotechnology), anti-human HIF-1α antibody (BD Transduction laboratories, San Jose, CA), anti-mouse HIF-1α antibody (Novus Biologicals, Littleton, CO), anti-HIF-2α antibody (Novus Biologicals), anti-HIF-1β antibody (Novus Biologicals). The concentration of VEGF protein was measured using ELISA kits with a mouse monoclonal antibody against human and mouse VEGF (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Real-time quantitative PCR
Total RNA was isolated using a Qiagen Extraction procedure. Equal amounts of total RNA (1μg) were reverse-transcribed using the SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen, Carlsbad, CA), and the resulting first strand cDNA was diluted and used as template in the real-time quantitative-PCR analysis. All measurements were performed in triplicate. Actin served as internal control and was used to normalize the variances in input cDNA. The following VEGF primer pair was purchased from RealTimePrimers. com: Forward Primer 5′-TTCCAGGAGTACCCTGATGA -3′; Reverse Primer 5′-TGAGGTTTGATCCGCATAAT -3′. Detection of VEGF expression was performed with SYBR® GreenER™ qPCR SuperMix for ABI PRISM (Invitrogen) and an ABI PRISM 7700 Sequence Detection System (Applied Biosystems), using the relative standard curve method. Results presented represent the mean of 2 independent experiments.
Plasmids
The series of VEGF reporter construct, pVEGF-KpnI (from −2274 to +379) (Forsythe, 1996), P11w (from −985 to −939: CCACAGTGCATACGTGGGCTCCAACAGGTCCTCTTCCCTCCCATGCA) (Forsythe, 1996), and P11m (from −985 to −939: CCACAGTGCATAAAAGGGCTCCAACAGGTCCTCTTCCCTCCCATGCA) (Forsythe, 1996), were purchased from ATCC (ATCC, Manassas, VA). The pGL2TKHRE plasmid containing three copies of the HRE (5′-GTGACTACGTGCTGCCTAG-3′) was a generous gift from Dr. Giovanni Melillo (Rapisarda et al, 2002).
Transient Transfections
DNA plasmids were prepared using a commercially available kit (Endofree Maxi-Prep; Qiagen). Transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After 16 h, the protein lysates were made and luciferase assays were performed in 96-well optiplates (Packard Instrument, Inc., Meriden, CT) as previously described (Matsuo et al, 2000). The HIF-1α small interference (si) RNA (AAAGGACAAGTCACCACAGGA) to target HIF-1α and a nonspecific siRNA control (AATTCTCCGAACGTGTCACGT) (Qiagen) were utilized and targets both human and murine HIF-1α. One million TA25 cells were co-transfected with KpnI, P11w, P11m or pGL2TKHRE and HIF-1α siRNA using Lipotectamine 2000 (Invitrogen) according to manufacture’s the instructions. After 24 h, the cell supernatants were collected and evaluated for VEGF protein expression by ELISA while the cells were lysed with buffer containing 1% NP-40 and protein expression evaluated by Western blot analysis. Luciferase assays were performed in 96-well optiplates (Packard Instrument, Inc.).
Results
Mechanism of NGF/TrkA regulation of VEGF
To study NGF regulation of VEGF, we utilized the TA6 and TA25 cell lines in which a neuroblastoma cell line has been transfected with a tetracycline (TET)-regulated TrkA expression plasmid (pBPSTR1-TrkA) and E2 cells transfected with the empty vector plasmid (pBPSTR1) were used as controls (Kim, 1999). Both TA6 and TA25 cells express a 3- to 5-fold increase in TrkA levels when cultured in the absence of TET, and NGF induced an almost 4-fold increase in phospho-Trk levels under TrkA high-expressing condition (TET−) in both TA6 and TA25 cells (Fig. 1A). Conversely, in E2 cells, the basal level of TrkA does not change in either the TET+ or TET− conditions and there is no detectable phosphorylation of Trk after NGF stimulation.
Fig. 1. Nerve growth factor regulates VEGF levels in TrkA expressing NB cells.
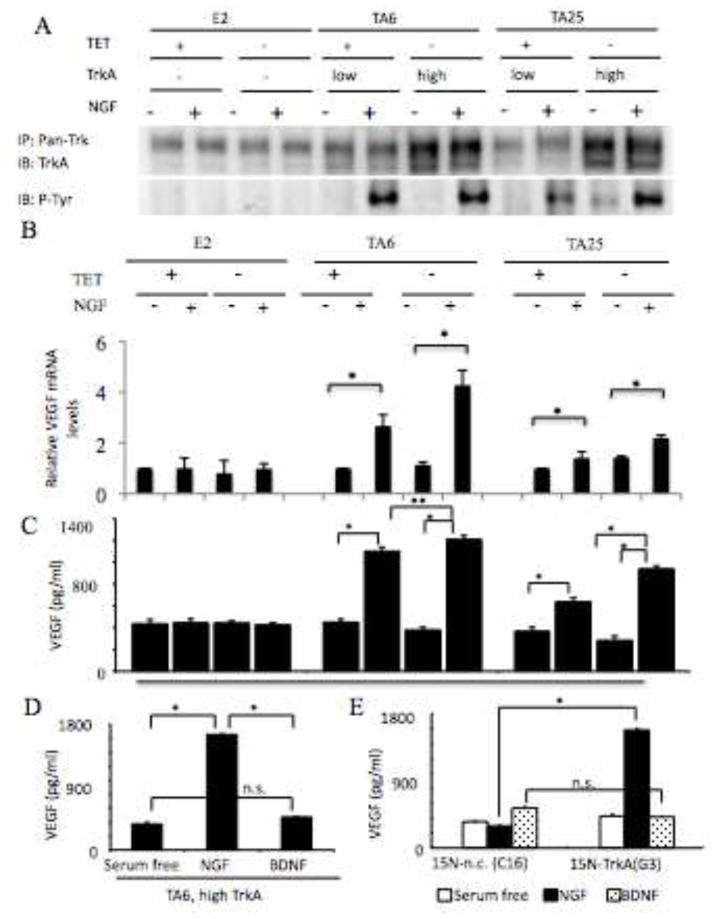
Panel A, TrkA pathway. Protein lysates (500 μg) from E2, TA6 and TA25 treated with or without TET or NGF (100 ng/ml) were immunoprecipitated with rabbit polyclonal anti-pan Trk antibody (C-14) and subjected to Western analysis (as described in Materials and Methods) for evaluation of phosphorylation of TrkA and total TrkA levels. Panel B. E2, TA6 and TA25 cells (1 × 106) treated with or without TET or NGF (100 ng/ml) for 16h were analyzed. VEGFmRNA was assessed using Q-PCR as detailed in Materials and Methods. For each cell line, the normalized VEGF/Actin expression level in each sample was normalized to the level of VEGF/actin in the TET+ condition. Results represent mean +/− standard deviation of triplicate values for each condition for 2 independent experiments. (*; p<.05). PanelC. Conditioned media (CM) from E2, TA6 and TA25 cells (1 × 106) treated with or without TET or NGF (100 ng/ml) for 16h was analyzed by ELISA for VEGF expression. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01, **: p < 0.05). Panel D. CM from TA6 cells (1 × 106) expressing high TrkA (TET−) treated with or without NGF (100 ng/ml) or BDNF (100 ng/ml) for 16h was analyzed by ELISA for VEGF expression. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01). Panel E. CM from 15N-negative control and 15N-TrkA cells (1 × 106) expressing high TrkA (TET−) treated with or without NGF (100 ng/ml) or BDNF (100 ng/ml) for 16h was analyzed by ELISA for VEGF expression. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01).
To examine whether NGF stimulates VEGF expression, we evaluated the levels of VEGF mRNA (Fig. 1B) and protein secretion (Fig. 1C) in E2, TA6 and TA25 cells. NGF stimulates a 2.7 and a 4.2 fold increase in VEGF mRNA in TA6 cells expressing low (TET+ ) and high (TET−) levels of TrkA, respectively after 16hrs (Fig. 1B). In TA25 cells expressing low TrkA (TET+), NGF stimulates only a 1.3 fold increase in VEGF mRNA in the low TrkA(TET+) cells and a 2.2 fold increase in the high TrkA(TET−) cells (Fig. 1B). NGF stimulates a 2.6-fold in VEGF secretion protein in TA6 cells expressing low TrkA(TET+) and a 2.8-fold increase in cells expressing relatively higher (TET−) levels of TrkA, after 16h (Fig. 1C). NGF stimulates an almost 1.5-fold in VEGF secretion protein in TA25 cells expressing low TrkA (TET+) and a 3.3-fold increase in cells with higher (TET−) levels of TrkA after 16h (Fig. 1C). However, NGF does not stimulate VEGF protein secretion in E2 cells (Fig. 1C). These data indicate that the levels of endogenous TrkA are not sufficient to stimulate VEGF protein secretion in the E2 cells or are below the levels detectable in our system. However, when cells are treated with retinoic acid (RA) to induce endogenous TrkA (Kaplan et al, 1993), NGFstimulates an induction of VEGF protein secretion in KCNR neuroblastoma cells (data not shown). NGF binds TrkA and the p75 neurotrophin receptor, a member of the death receptor family that binds all NGF family neurotrophins. To examine whether activation of p75 alone stimulates an increase in VEGF, we treated TA6 cells expressing high levels of TrkA (TET−) with BDNF (100 ng/ml), which is also a ligand for p75. NGF increases VEGF protein expression (*: p < 0.05) but BDNF does not cause of significant change in VEGF levels (Fig. 1D). To extend these findings to an additional neuronal cell lines expressing TrkA, we utilized 15N-TrkA cells (G3) and control vector-transfected 15N NB cells (C16). NGF stimulates a 4-fold increase in VEGF protein secretion compared to media-treated cells in G3 cells (Fig. 1E) but neither neurotrophin stimulates VEGF protein secretion in C16 (Fig. 1E). These data indicate that activation of p75 alone is not sufficient to induce VEGF secretion.
Induction of HIF-1α protein by NGF
Previously, we have shown that the ability of BDNF to induce VEGF in TrkB expressing cells was dependent on induction of HIF-1α (Beppu et al, 2005). To determine whether NGF stimulates HIF-1α expression, we treated TA6 and TA25 cells cultured in the absence or presence of TET with or without NGF. NGF stimulates HIF-1α protein levels in both TA6 and TA25 cells expressing low (TET+) and high (TET−) levels of TrkA (Fig. 2A). We treated the high TrkA-expressing TA6 and TA25 cells (TET −) with NGF or BDNF and found that NGF, but not BDNF, induces HIF-1α protein (Fig. 2B). However, under these conditions the levels of HIF-2α and HIF-1β are unchanged (Fig. 2B). Similarly, NGF but not BDNF induces HIF-1α but not HIF-2α (data not shown) in 15N-TrkA expressing cells (Fig 2C). Thus, in neuronal cell lines expressing TrkA, NGF stimulates HIF-1α expression while the levels of HIF-2α and HIF-1β are unchanged.
Fig. 2. Nerve growth factor regulates HIF-1 α levels in TrkA expressing NB cells.
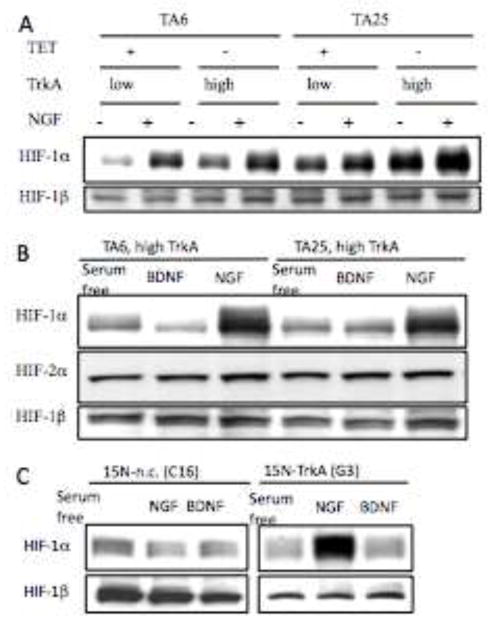
Panel A. Western analysis of total protein lysates (30 μg) from indicated TA6 and TA25 cells treated for 16 h with or without TET or NGF (100 ng/ml) for HIF-1α and HIF-1β levels. Panel B. Total protein lysates from indicated TA6 and TA25 cells expressing high TrkA (TET−) treated for 16 h with or without NGF (100 ng/ml) or BDNF (100 ng/ml) for HIF-1α, HIF-2α and HIF-1β levels. Panel C. Western analysis of 30 μg of HIF-1α levels in protein lysates from indicated 15N-necative control (C16) and 15N-TrkA (G3) cells treated for 16 h with or without NGF (100 ng/ml) or BDNF (100 ng/ml) for HIF-1α and HIF-1β levels.
Signal transduction pathway of NGF/TrkA
To evaluate the signal transduction pathway that contribute to the NGF-induced HIF-1α and VEGF expression in TrkA expressing NB cells, we used pharmacologic inhibitors of the TrkA (K252a), MAPK pathway (PD98059), PI3K (LY294002) and mTOR (rapamycin). When TA25 cells were treated with NGF following pretreatment with K252a, LY294002 or rapamycin, NGF-induced HIF-1α expression was markedly inhibited (Fig. 3A). Also, pretreatment with PD98059, the MAPK inhibitor, partially inhibited the NGF-induced HIF-1α expression (Fig. 3A). Consistent with inhibition of HIF-1α protein, K252a, LY294002, and rapamycin blocked the NGF-induced VEGF protein secretion from TA25 cells. PD98059 partially inhibited the VEGF secretion (*: p < 0.01, **: p < 0.05, Fig. 3B). These findings indicate that the PI3K/mTOR pathway markedly decreases expression of HIF-1α, which leads to decreases in VEGF expression.
Fig. 3. NGF regulates HIF-1 α and VEGF levels via PI3K/mTOR and MAPK pathways.
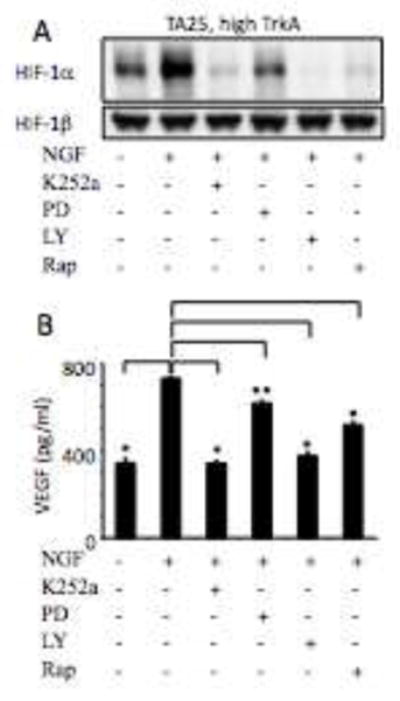
Panel A. Western analysis of 30 μg of HIF-1α levels in protein lysates from the serum-deprived (6h) high TrkA-expressing TA25 cells (TET−) pretreated with control solvent or indicated inhibitor for 1 h followed by stimulation with NGF for 8 h. HIF-1β is shown as a loading control. Panel B. Conditioned media from the high TrkA-expressing TA25 cells (TET−) (1 × 106) incubated with serum-free media for 6 h following a 1 h pre-incubation with control solvent or indicated inhibitor was analyzed by ELISA for VEGF expression. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01, **: p < 0.05).
VEGF transcription by HIF-1 α
In order to directly evaluate whether the NGF induction of HIF-1α plays a role in stimulating VEGF transcription, we transfected an artificial hypoxia response element (HRE) promoter containing three copies of HRE (pGL2TKHRE) into the high TrkA-expressing TA25 cells (TET−) and cultured for 16 h in the absence of serum (Serum-free), with NGF (100 ng/ml), or with BDNF (100 ng/ml). NGF stimulated a 2.7-fold increase in HIF-1α transcriptional activity in TA25 cells expressing high levels of TrkA, but BDNF did not (Fig. 4A). The increase in HIF-1α transcriptional activity may also be regulated by the level of TrkA expression, since NGF stimulated a 2.7-fold increase in TA25 cells expressing high levels of TrkA (TET−) compared to a 2-fold increase in TA25 cells expressing low levels of TrkA (TET+) (Fig. 4B).
Fig. 4. HIF-1α induced by NGF mediates VEGF protein expression.
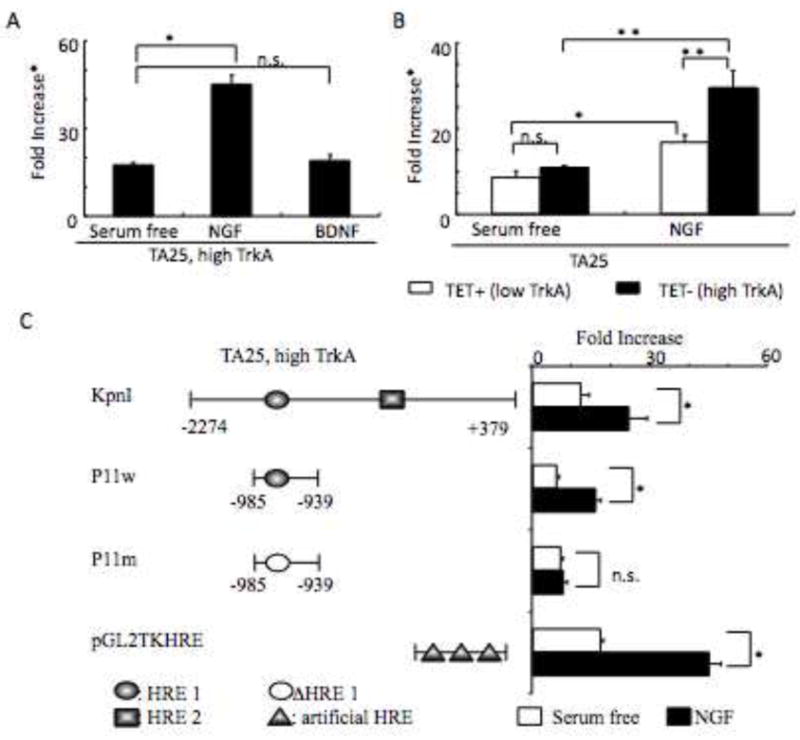
Panel A. Luciferase activity from TA25 cells (1 × 106) at TrkA-high expressing conditions (TET−) treated with or without NGF (100 ng/ml) or BDNF (100 ng/ml) for 16 h was analyzed by luciferase assay. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01). *Fold increase, relative fold increase of normalized luciferase activity. Panel B. Luciferase activity from TA25 cells (1 × 106) at both high and low TrkA (TET− and TET+) treated with or without NGF (100 ng/ml) for 16 h was analyzed by luciferase assay. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.05, **: p < 0.01). Panel C. Luciferase activity from the high TrkA-expressing TA25 cells (TET−) (1 × 106) transfected with KpnI, P11w, P11m, or pGL2TKHRE treated with or without NGF (100 ng/ml) for 16 h was analyzed by luciferase assay. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01).
In order to evaluate NGF regulation of the natural VEGF promoter, we transfected TA25 cells expressing high levels of TrkA with a luciferase reporter plasmid containing sequence −2272 to +379 of the VEGF promoter (KpnI) (Forsythe et al, 1996). NGF induced a 2- to 2.6-fold increase in transcriptional activity in TA25 cells expressing high levels of TrkA (TET−) that were transfected with KpnI or with P11w, a luciferase reporter construct containing just the HRE of the natural VEGF promoter (Fig. 4C). NGF did not stimulate transcriptional activity in P11m, a luciferase reporter construct in which the HRE was mutated and non-functional (Fig. 4C). These data indicate that NGF activation of the TrkA signal transduction pathway via HIF-1α drives VEGF transcription.
siRNA knockdown of HIF-1α decreases VEGF protein expression
To directly evaluate whether HIF-1α plays a role in NGF/TrkA-stimulated VEGF protein expression, we transfected a HIF-1α siRNA or a control siRNA into the high TrkA-expressing TA25 cells (TET−) and cultured for 24 h in NGF (100ng/ml). Western blot analysis indicated that HIF-1α protein expression decreased in cells transfected with the HIF-1α siRNA compared to cells transfected with the control siRNA treated with NGF (Fig. 5A). Furthermore, HIF-1α siRNA knockdown caused a 42%, 46%, and 48% decrease in NGF induced transcriptional activity in the high TrkA expressing TA25 cells (TET−) transfected with KpnI, P11w, and pGL2TKHRE, respectively (Fig. 5B). When VEGF protein levels were measured in the supernatants from these same samples, the NGF stimulated VEGF protein levels decreased approximately 40% in the HIF-1α siRNA-transfected samples compared to the control siRNA-transfected samples (Fig. 5C). These results indicate that a large portion of the NGF-induced VEGF expression is regulated by HIF-1α in this system.
Fig. 5. HIF-1α siRNA knockdown of VEGF protein expression.
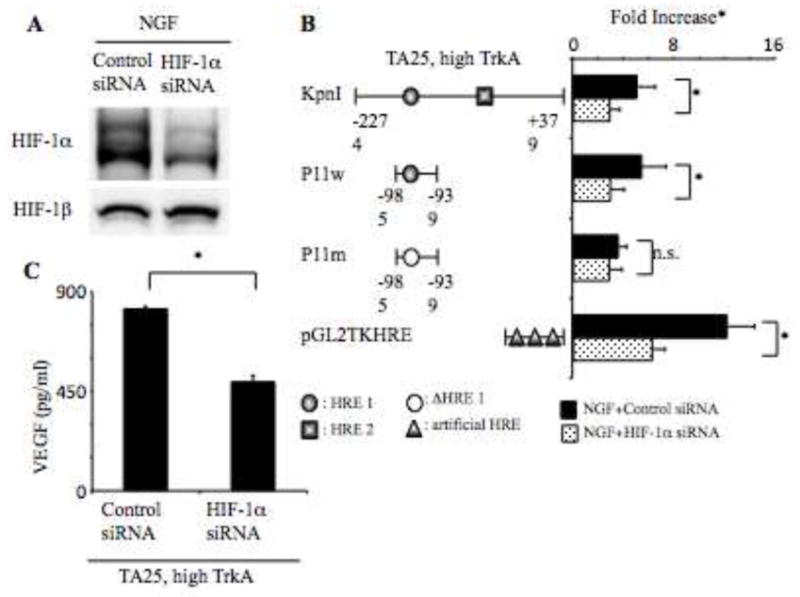
Panel A. Western analysis of total protein lysates (30 μg) from indicated TA25 cells expressing high TrkB (TET−) for 6 h in serum-free RPMI medium transfected HIF-1α siRNA with NGF after 24 h for HIF-1α and HIF-1β levels. Panel B. Luciferase activity from the high TrkA-expressing TA25 cells (TET−) (1 × 106) co-transfected with KpnI, P11w, P11m, or pGL2TKHRE with control siRNA or HIF-1α siRNA treated with NGF (100 ng/ml) for 24 h was analyzed by luciferase assay. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01). Panel C. VEGF protein levels from the same samples as Fig. 4B were analyzed by ELISA. Results represent mean + standard deviation of triplicate values for each condition (*: p < 0.01). *Fold increase, relative fold increase of normalized luciferase activity.
Neurotrophin regulation of VEGF production in primary culture of normal neurons
To evaluate the effects of neurotrophins on primary neuronal cells, we isolated dorsal root ganglion (DRG) neurons as a model for TrkA expressing normal neurons and cerebellar granule neurons (CGNs) as a model for TrkB expressing normal neurons. To evaluate whether NGF activation of TrkA would induce VEGF expression in, we cultured DRG neurons from P7 murine pups and found 87% of the neurons expressed TrkA with no neurons expressing TrkB (Fig 6A). After NGF (100ng/ml) treatment, 73% of DRG neurons had increased expression of HIF-1α with only 5% of cells having increased immunoreactivity for HIF-1α after BDNF treatment. The intensity of HIF-2α staining did not change in DRG neurons after treatment with either NGF or BDNF (Fig. 6B, C). Consistent with this, Western analysis showed NGF induced an increase in HIF-1α while levels of HIF-2α and HIF-1β were relatively unchanged (Fig 6D). Due to the difficulty in obtaining sufficient protein, only 1 Western analysis was performed. In the DRG cultures, there was a 1.6 fold increase in secreted VEGF in cells treated with NGF and no change in VEGF levels in cultures treated with BDNF (Fig. 6D). These data indicate that NGF activation of TrkA induced increases in VEGF levels that are associated with increases in HIF-1α.
Fig. 6. NGF regulates VEGF protein secretion via HIF-1α in murine DRG neurons.
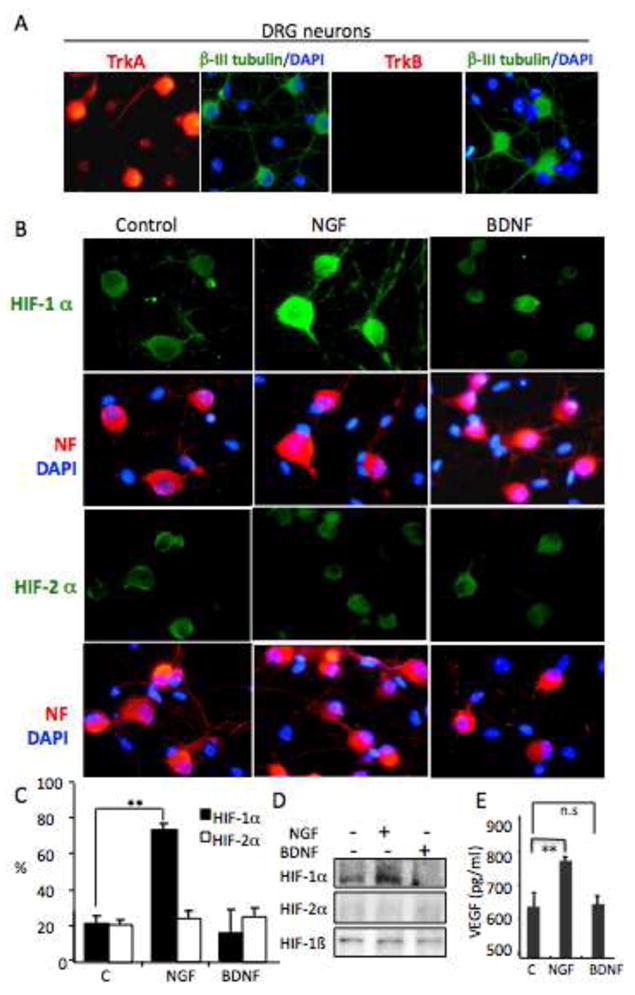
Panel A. DRG neurons express TrkA, but not TrkB. DRG neurons were fixed and stained with anti-β III tubulin (FITC-green) and DAPI (blue) or anit-TrkA or anti-TrkB (TRITC-red), relative levels of protein expression are assayed by immunocytochemistry (as described in Materials and Methods). Panel B. NGF, not BDNF, induces increases in expression of HIF-1α, but not HIF-2α, in DRG neurons. DRG neurons were treated with NGF (100ng/ml) or BDNF (100ng/ml) for 24hrs, then fixed and stained for Neurofilament ((NF), TRITC-red) and DAPI (blue), or NF and double staining with HIF-1α or HIF-2α(FITC-green), relative levels of protein expression HIF-1α (FITC-green), HIF-2α (FITC-green) are assayed by immunocytochemistry (as described in Materials and Methods). Panel C. DRG neurons are treated with NGF (100ng/ml) or BDNF (100ng/ml) for 24hrs, then fixed and stained with HIF-1α or HIF-2α. The number of cells with increases in HIF-1α or HIF-2α were enumerated and graphed as % of total counted cells. The statistic differences between treated cells (NGF or BDNF) and untreated control cells were performed. ** P<0.001. Panel D. Western analysis of 30ug of total protein from DRG neurons treated with NGF (100ng/ml) or BDNF (100ng/ml) for 24hrs and evaluated for HIF-1α, HIF-2α and HIF-1β levels. Panel E. NGF, not BDNF, induces an increase in VEGF secretion in DRG neurons. DRG neurons were treated with NGF (100ng/ml) or BDNF(100ng/ml) for 24hrs, and the conditioned media was harvested and the VEGF secreted into the media was analyzed by ELISA. The statistic differences between treated cells (NGF or BDNF) and untreated control cells were performed. Results were shown as Mean ± SD, ** P<0.001.
Our previous study indicated that BDNF activation of TrkB induced an increase in secretion of VEGF in neuronal cell lines that was dependent on induction of HIF-1α and HIF-1α dependent transcription from the VEGF promoter (Nakmura et al, 2005). To evaluate BDNF regulation of VEGF in normal neurons we cultured CGNs in which the majority of neurons express TrkB (Fig. 7A). CGNs were cultured for 2 days with control media, BDNF (100ng/ml) or NGF (100ng/ml). Culture supernatants were harvested and analyzed to determine levels of VEGF production and cells were evaluated for HIF expression by immunostaining. After BDNF treatment, 75% of CGNs had increased HIF-1α expression, while only a few cells had increased levels of HIF-1α after NGF treatment (Fig 7B, C). Immunoreactivity for HIF-2α did not change after BDNF or NGF treatment. Western analysis indicated BDNF induced an increase in HIF-1α (2.1-fold p<. 05) compared to NGF (0.8-fold p=ns) when normalized to the levels of HIF-1β while the levels of HIF-2α were variable yet not reproducibly significantly different from controls (Fig. 7D). There was a 1.8 fold increase in VEGF production in BDNF treated cells compared to controls, while the levels of VEGF in NGF treated cells were unchanged after 48hrs (Fig. 7E). To evaluate the dependency of BDNF induced VEGF production on HIF-1α we transfected CGNs with control siRNA or HIF-1α siRNA and compared the relative ability of BDNF to induce VEGF under these conditions. We found that CGNs treated with BDNF and transfected with siRNA targeted to HIF-1α failed to stimulate VEGF levels to the same extent as detected in CGNs transfected with a control siRNA (Fig. 7E). Thus, the relative BDNF induced increase in VEGF levels was attenuated in CGNs transfected with HIF-1α siRNA compared to the control siRNA transfected cells. These data indicate that the BDNF induced increase in VEGF in CGNs is dependent on increases in HIF-1α expression.
Fig. 7. BDNF regulates VEGF protein secretion via HIF-1 α in mice cerebellum granule neurons.
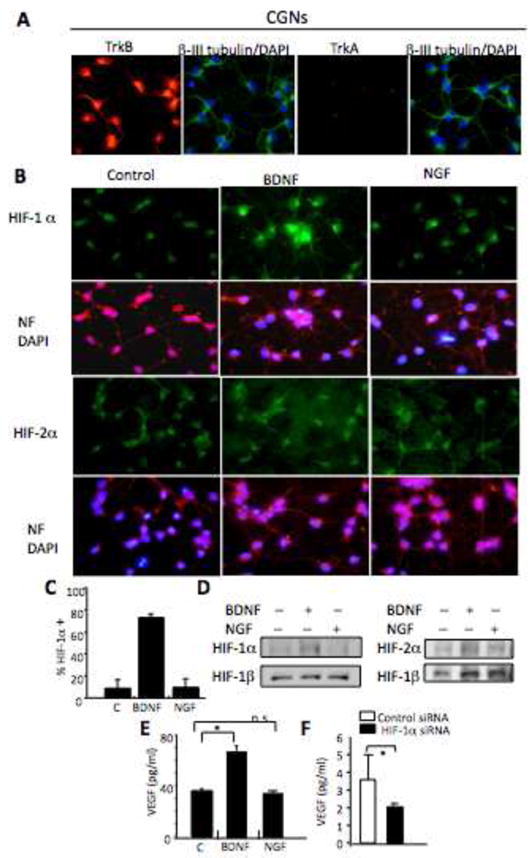
Panel A. CGNs express TrkB, but not TrkA. CGNs were fixed and stained with β III tubulin (FITC-green) and DAPI (blue), double staining with TrkB or TrkA (TRITC-red), relative levels of protein expression are assayed by immunocytochemistry (as described in Materials and Methods). Panel B. BDNF, not NGF, induces increases in expression of HIF-1 α, but not HIF-2α, in CGNs. CGNs were treated with NGF (100ng/ml) or BDNF (100ng/ml) for 24hrs, then fixed and stained for Neurofilament ((NF), TRITC-red) and DAPI (blue), or NF and double staining with HIF-1α or HIF-2 α (FITC-green), relative levels of protein expression HIF-1α (FITC-green), HIF-2 α (FITC-green) are assayed by immunocytochemistry (as described in Materials and Methods). Panel C. CGNs treated with BDNF (100ng/ml) or NGF (100ng/ml) for 24hrs were fixed and stained with anti-HIF-1α or anti-HIF-2α. The number of cells with increased HIF-1α expression with increased staining were counted and graphed as % of total counted cells. There was no change in HIF-2α staining in CGN neurons (data not shown). The statistic differences between treated cells (BDNF or NGF) and untreated control cells were performed. ** P<0.001. Panel D. Western analysis of 30ug of total protein from CGNs treated with NGF (100ng/ml) or BDNF (100ng/ml) for 24hrs and evaluated for HIF-1α, HIF-2α and HIF-1β levels. A representative Western blot from 2 independent experiments is presented. Panel E. BDNF, not NGF, induces an increase in VEGF secretion in CGNs. CGNs were treated with BDNF (100ng/ml) or NGF (100ng/ml) for 24hrs, conditioned media was harvested and level of secreted VEGF analyzed by ELISA. The differences between treated cells (BDNF or NGF) and untreated control cells were analyzed and results were shown as Mean ± SD, * * P<0.001. Panel F. CGN neurons were electroporated with control siRNAs or siRNA targeting HIF-1α and then treated with BDNF (100ng/ml) for 24hrs. The conditioned media was harvested and secreted VEGF levels analyzed by ELISA. The statistic differences between control siRNA and HIF-1α siRNA transfected cells were performed. Results were shown as Mean ± SD, * P<0.05.
Discussion
In this study we detail in neural cell lines and primary neural cultures, mechanisms of neurotrophin-mediated induction of VEGF. In TrkA expressing normal neurons, NGF activation of TrkA stimulates increases in VEGF production that is accompanied by increases in HIF-1α but not HIF-1β or HIF-2α. Moreover, pharmacologic inhibition of the Trk TK, PI-3kinase or mTOR pathways blocks NGF induced increases in HIF-1α and VEGF production. These data are consistent with our previous findings in which BDNF activation of a TrkB/PI-3kinase/AKT/mTOR pathway in a similar neuronal cell line model increases VEGF production via induction of HIF-1α dependent increases in VEGF promoter activity (Nakamura et al, 2005).
Our results in the neuroblastoma neuronal cultures are consistent with studies, which showed increases in VEGF production in PC12 cells only after treatment with high dose NGF (10–100ng/ml) (Middeke et al, 2002). While another report failed to detect NGF stimulated increases in VEGF in PC12 cells this may have been due to suboptimal concentrations of NGF (Claffey, 1992). The E2 cells express a level of endogenous TrkA that is similar at the protein level to the level of TrkA in TA25 yet fail to activate TrkA when treated with NGF. It is not clear whether endogenous TrkA signaling is impaired under steady-state culture conditions in neuroblastoma, however under these conditions only NGF treatment of TrkA transfected neuroblastoma cells activate TrkA and induce VEGF production. A previous report in neuroblastoma indicated that increased levels of TrkA in the absence of ligand decreased VEGF production in NB cells (Eggert et al 2002). Similarly we find a small but significant decrease in VEGF (p <0.05 comparing VEGF secretion in TA6 Tet+ vs Tet− and in TA25 Tet+ vs Tet−) secretion in the absence of ligand when cells express increased levels of TrkA (Fig 1C) despite increased HIF-1α levels (Fig. 2A) and a small increase in VEGFmRNA in the TA25 but not TA6 cell lines (Fig. 1B). The mechanism of TrkA reduction of VEGF expression is not known and whether other recently described TrkA interactors such as CCM2 (Haret et al 2009) are involved remains to be determined.
Furthermore our studies are consistent with reports in which NGF treatment of rat superior cervical ganglion (scg) cultures induced VEGF and eNOS expression in SCG neurons (Calza et al, 2001), and NGF treatment of normal rat dorsal root ganglion showed increases in VEGF expression (Sammi A, et al, 1999). We extend these studies to show that the increases in VEGF production are accompanied by increases in HIF-1α expression in neuronal cells. There are reports that NGF treated PC-12 cells decrease levels of HIF-2α (Naranjo-Suarez et al, 2002) or HIF-1α (Charlier et al, 2002) but differences in culture conditions and cell context may account for the discrepancies. Our findings in both primary cultures of murine DRG or CGN neurons that activation of Trk by cognate neurotrophin stimulated increases in VEGF production that were accompanied by increases in HIF-1α are consistent with our studies in neuronal cell lines. In the models we tested we see increased expression of HIF-1α and not HIF-2α upon NGF activation of TrkA. However it is possible that in other neural models there may be induction of HIF-2α over HIF-1α, since a PHD3-HIF-2α pathway is implicated in sympathoadrenal but not sensory neuron development (Bishop et al 2008). In neuroblastoma tumor cell lines transient induction of HIF-1α and sustained induction of HIF-2α has been reported (Holmquist-Mengelbier et al, 2006) but this may also be cell line dependent since we have seen sustained HIF-1α under hypoxic condtions in different neuroblastoma cell lines (unpublished results).
An important aspect of our study is the finding that the neurotrophin mediated increases in VEGF are dependent on increases in HIF-1α. While these studies have been performed in normoxia (21%O2), we have also found that in TrkB expressing cells BDNF enhances increases HIF-1α and VEGF expression even under hypoxic conditions (1%O2) (unpublished data). The importance of HIF-1α in this process is demonstrated by the finding that VEGF transcription is attenuated if the VEGF promoter contains the mutation in the HRE known to disrupt HIF -1α transcription factor binding. Furthermore, a siRNA targeted to HIF-1α blocks the ability of NGF to stimulate increases in VEGF transcription and causes a reduction in VEGF production in the neuronal cells. Pharmacologic inhibition of Trk (K252a), the PI3-kinase/mTOR and to a lesser extent the MAPK pathway decreased HIF-1α expression to a level lower than basal HIF-1α levels yet only prevented NGF mediated increases in VEGF production. What controls the basal level of VEGF is currently under investigation. The NGF/TrkA mediated increase in HIF-1α may be via increased translation of HIF-1α mRNA as BDNF induced increases in HIF-1α protein were not accompanied by an increases in HIF-1α mRNA (our unpublished data). Increased or stabilized HIF-1α and VEGF expression is found in neural stem/progenitor cells cultured under normoxic conditions (21% O2) and has been found to protect endothelial cells against ischemia associated cell death (Roitbak et al, 2008).
Recent studies also indicate that there is a complex interaction among neurotrophins, endothelial and neural cells during development and during ischemic alterations in the brain. NGF promotes endothelial vessel growth (Turrini et al, 2002; Cantarella et al, 2002; Emanueli et al, 2002; Hattori et al, 2002) and increases in arteriole length density (Turrini et al, 2002). NGF also stimulates VEGF expression in normal superior cervical ganglia neurons (Calza et al 2001). BDNF also promotes angiogenesis in the developing embryonic myocardium (Donavan et al 2000), recruits brain- (Kim et al, 2004) and bone marrow-derived TrkB-expressing endothelial precursor cells from the bone marrow (Kermani et al, 2005) and increases VEGFR on endothelial cells (Kim et al, 2004). BDNF secretion by cerebral endothelial cells is neuroprotective to cortical neurons (Guo S et al, 2008). Our findings that cognate neurotrophin activation of TrkB or TrkA stimulates VEGF production supports models of reciprocal paracrine survival or growth stimulatory interactions between the neural and vascular systems.
Acknowledgments
The authors are grateful to Dr. Giovanni Melillo and Dr. Annamaria Rapisarda for insightful discussions. We’d like to thank the members of the Cell and Molecular Biology Section for their encouragement. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beppu K, Nakamura K, Linehan WM, Rapisarda A, Thiele CJ. Topotecan blocks hypoxia-inducible factor-1α and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res. 2005;65:4775–81. doi: 10.1158/0008-5472.CAN-04-3332. [DOI] [PubMed] [Google Scholar]
- Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, Grosfeld A, Aragones J, Schneider M, van Geyte K, Teixeira D, Diez-Juan A, Lopez-Barneo J, Channon KM, Maxwell PH, Pugh CW, Davies AM, Carmeliet P, Ratcliffe PJ. Abnormal Sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol. 2008;28:3386–400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Calza L, Giardino L, Giuliani A, Aloe L, Levi-Montalcini R. Nerve growth factor control of neuronal expression of angiogenetic and vasoactive factors. Proc Natl Acad Sci USA. 2001;98:4160–5. doi: 10.1073/pnas.051626998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarella G, Lempereur L, Presta M, Ribatti D, Lombardo G, Lazarovici P, Zappalà G, Pafumi C, Bernardini R. Nerve growth factor-endothelial cell interaction leads to angiogenesis in vitro and in vivo. FASEB J. 2002 Aug;16(10):1307–9. doi: 10.1096/fj.01-1000fje. [DOI] [PubMed] [Google Scholar]
- Charlier N, Leclere N, Felderhoff U, Heldt J, Kietzmann T, Obladen M, Gross J. Hypoxia-induced cell death and changes in hypoxia-inducible factor-1 activity in PC12 cells upon exposure to nerve growth factor. Brain Res Mol Brain Res. 2002 Jul 15;104(1):21–30. doi: 10.1016/s0169-328x(02)00198-5. [DOI] [PubMed] [Google Scholar]
- Claffey KP, Wilkison WO, Spiegleman BM. Vascular Endothelial Growth Factor; Regulation by cell differentiation and activated second messenger pathways. J Biol Chem. 1992;267:16317–16322. [PubMed] [Google Scholar]
- DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT. Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci. 1996;7(2):89–101. doi: 10.1006/mcne.1996.0007. [DOI] [PubMed] [Google Scholar]
- Donavan MJ, Lin MI, Wiegn P, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531–40. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- Eggert A, Grotzer MA, Ikegaki N, Liu XG, Evans AE, Brodeur GM. Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Res. 2002;62:1802–8. [PubMed] [Google Scholar]
- Emanueli C, Salis MB, Pinna A, Graiani G, Manni L, Madeddu P. Nerve growth factor promotes angiogenesis and arteriogenesis in ischemic hindlimbs. Circulation. 2002 Oct 22;106(17):2257–62. doi: 10.1161/01.cir.0000033971.56802.c5. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996 Sep;16(9):4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008 May 27;105(21):7582–7. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel L, Costa B, Tcherpakov M, Zapatka M, Oberthuer A, Hansford LM, Vojvodic M, Levy Z, Chen ZY, Lee FS, Avigad S, Yaniv I, Shi L, Eils R, Fischer M, Brors B, Kaplan DR, Fainzilber M. CCM2 mediates death signaling by the TrkA recptor tyrosine kinase. Neuron. 2009;63:585–91. doi: 10.1016/j.neuron.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, Hicklin DJ, Zhu Z, Bohlen P, Witte L, Hendrikx J, Hackett NR, Crystal RG, Moore MA, Werb Z, Lyden D, Rafii S. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med. 2002 Aug;8(8):841–9. doi: 10.1038/nm740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10 :413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3′-kinase pathway. Cancer Res. 2002;62:6756–63. [PubMed] [Google Scholar]
- Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Neuron. 1993;11:321–31. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- Kermani P, Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007 May;17(4):140–3. doi: 10.1016/j.tcm.2007.03.002. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermani P, Rafii D, Jin DK, et al. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653–63. doi: 10.1172/JCI200522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CJ, Matsuo T, Lee KH, Thiele CJ. Up-regulation of insulin-like growth factor-II expression is a feature of TrkA but not TrkB activation in SH-SY5Y neuroblastoma cells. Am J Pathol. 1999;155:1661–70. doi: 10.1016/S0002-9440(10)65481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Li Q, Hempstead BL, Madri JA. Paracrine and autocrine functions of brain-derived neurotrophic factor and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem. 2004;279:33538–46. doi: 10.1074/jbc.M404115200. [DOI] [PubMed] [Google Scholar]
- Levi G, Aloisi F, Ciotti MT, Gallo V. Autoradiographic localization and depolarization-induced release of acidic amino acids in differentiating cerebellar granule cell cultures. Brain Res. 1984;290(1):77–86. doi: 10.1016/0006-8993(84)90737-6. [DOI] [PubMed] [Google Scholar]
- Loureiro RMB, D’Amore PA. Transcirptional regulation of Vascular edothelial growth factor in cancer. Cytokine & Growth Factor Reviews. 2005;16:77–89. doi: 10.1016/j.cytogfr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Stauffer JK, Walker RL, Meltzer P, Thiele CJ. Structure and promoter analysis of the human unc-33-like phosphoprotein gene. J Biol Chem. 2000;275:16560–8. doi: 10.1074/jbc.M001312200. [DOI] [PubMed] [Google Scholar]
- Middeke M, Hoffmann S, Hassan I, Wunderlich A, Hofbauer LC. Zielke ANGF stimulates VEGF and neoangiogenesis in PC12 cells. Exp Clin Endocrinol Diabetes. 2002 Nov;110(8):386–92. doi: 10.1055/s-2002-36424. [DOI] [PubMed] [Google Scholar]
- Moser KV, Reindl M, Blasig I, Humpel C. Brain capillary endothelial cells proliferate in response to NGF, express NGF receptors and secrete NGF after inflammation. Brain Res. 2004;1017:53–60. doi: 10.1016/j.brainres.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Mukouyama YS, Shin D, Britsch S, Taniguchi M, Anderson DJ. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002 Jun 14;109(6):693–705. doi: 10.1016/s0092-8674(02)00757-2. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1alpha in neuroblastoma cells. Cancer Res. 2006 Apr 15;66(8):4249–55. doi: 10.1158/0008-5472.CAN-05-2789. [DOI] [PubMed] [Google Scholar]
- Naranjo-Suarez S, Castellanos MC, Alvarez-Tejado M, Vara A, Landazuri MO, del Peso L. Down-regulation of hypoxia-inducible factor-2 in PC12 cells by nerve growth factor stimulation. J Biol Chem. 2003 Aug 22;278(34):31895–901. doi: 10.1074/jbc.M304079200. Epub 2003 Jun 12. [DOI] [PubMed] [Google Scholar]
- Nico B, Mangieri D, Benagiano V, Crivellato E, Ribatti D. Nerve growth factor as an angiogenic factor. Microvasc Res. 2008 Mar;75(2):135–41. doi: 10.1016/j.mvr.2007.07.004. Epub 2007 Jul 21. Review. [DOI] [PubMed] [Google Scholar]
- Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–24. [PubMed] [Google Scholar]
- Roitbak T, Li L, Cunningham LA. Neural stem/progenitor cells promote endothelial cell morphogenesis and protect endothelial cells against ischemia via HIF-1α-regulated VEGF signaling. J Cerebral Blood Flow& Metabol. 2008;28:1530–1542. doi: 10.1038/jcbfm.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero AA, Gross SR, Cheng KY, Goldsmith NK, Geller HM. An age-related increase in resistance to DNA damage-induced apoptotic cell death is associated with development of DNA repair mechanisms. J Neurochem. 2003;84:1275–87. doi: 10.1046/j.1471-4159.2003.01629.x. [DOI] [PubMed] [Google Scholar]
- Sammi A, Unger J, Lange W. NGF induces VEGF expression in normal sciatic and drg but to a lesser degree in neurons from diabetic animals. Neurosci Lett. 1999 Mar 12;262(3):159–62. doi: 10.1016/s0304-3940(99)00064-6. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994 Mar 17;368(6468):246–9. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Turrini P, Gaetano C, Antonelli A, Capogrossi MC, Aloe L. Nerve growth factor induces angiogenic activity in a mouse model of hindlimb ischemia. Neurosci Lett. 2002;323:102–12. doi: 10.1016/s0304-3940(02)00090-3. [DOI] [PubMed] [Google Scholar]
- Woo CW, Lucarelli E, Thiele CJ. NGF activation of TrkA decreases N-myc expression via MAPK path leading to a decrease in neuroblastoma cell number. Oncogene. 2004;23:1522–1530. doi: 10.1038/sj.onc.1207267. [DOI] [PubMed] [Google Scholar]