

SUMMARY
To define alterations of neuronal connectivity that occur during motor neuron degeneration, we characterized the function and structure of spinal circuitry in spinal muscular atrophy (SMA) model mice. SMA motor neurons show reduced proprioceptive reflexes that correlate with decreased number and function of synapses on motor neuron somata and proximal dendrites. These abnormalities occur at an early stage of disease in motor neurons innervating proximal hindlimb muscles and medial motor neurons innervating axial muscles, but only at end-stage disease in motor neurons innervating distal hindlimb muscles. Motor neuron loss follows afferent synapse loss with the same temporal and topographical pattern. Trichostatin A, which improves motor behavior and survival of SMA mice, partially restores spinal reflexes illustrating the reversibility of these synaptic defects. De-afferentation of motor neurons is an early event in SMA and may be a primary cause of motor dysfunction that is amenable to therapeutic intervention.
INTRODUCTION
In neurodegenerative diseases, abnormalities of synaptic connectivity are thought to account for early clinical deficits (Day et al, 2006; Shankar et al, 2008). Dysfunction of specific, vulnerable neuronal populations may precipitate secondary changes in neural circuits that could exacerbate neuronal dysfunction (Palop et al., 2006; Chen et al., 2002, Small et al., 2001). However, in many disease models, the primary targets and the precise sequence of functional and cellular changes that initiate the disease process remain unclear.
In the human motor neuron diseases amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA), motor neurons show altered function and many of them eventually die. Because of this, most research on ALS and SMA is focused on the motor neuron itself and on its synapse with skeletal muscle, the neuromuscular junction. However, in the spinal cord, motor neurons receive inputs from local spinal networks, descending pathways, and sensory neurons. Little is known about the response of these inputs to the factors that trigger disease. There is some evidence that spinal circuit abnormalities may occur in ALS (Soliven and Maselli, 1992; Schutz 2005; Jiang et al, 2009). In addition, Renault et al., (1983) described reduced or absent H-reflexes in Type I Human SMA, which they attributed to the severe muscle weakness due to concurrent motor neuron loss rather than proprioceptive abnormalities.
SMA is the most common inherited cause of infant death (Pearn, 1978). It is caused by mutation of the SMN1 gene and insufficient expression of the SMN protein (Lefebvre et al, 1995). Patients with severe SMA have profound muscle weakness in a stereotyped pattern (Crawford, 2004; Swoboda et al, 2005). Proximal muscles of the limbs are more affected than distal muscles, with the legs more affected than the arms (Montes et al, 2009). Muscles of the trunk are also severely affected leading to respiratory insufficiency and severe scoliosis. This characteristic pattern of selective muscle weakness suggests that particular subsets of motor neurons, or the circuits that control their activity, are particularly vulnerable in SMA. However, no studies have yet reported selective deficits among different motor pools in SMA.
The most commonly studied SMA mouse model, the SMA-Δ7 mouse (Le et al., 2005), recapitulates many features of the human disease and exhibits early impairments of motor behavior. Mice die at approximately 2 weeks (for review see Burghes and Beattie, 2009). Motor neuron cell loss has not been detected before postnatal day 4 (P4) (Le et al., 2005) and is modest even at end-stage (P13) (Le et al., 2005; Avila et al., 2007; Kariya et al., 2008) suggesting that the first behavioral abnormalities are due to impairments in motor neuron function rather than neuronal loss. Several recent studies have examined whether abnormalities of the neuromuscular junction (NMJ) synapse can account for the motor behavioral deficits of SMA mice (Murray et al., 2008, Kariya et al., 2008; Kong et al., 2009, Ruiz et al. 2010). Surprisingly, motor neurons in SMA are structurally well connected to their target muscles but functionally, NMJs in SMN-Δ7 mice exhibit a 50% reduction in quantal content indicating reduced synaptic vesicle release from motor neuron terminals in response to evoked stimulation (Kong et al., 2009, Ruiz et al., 2010). However, given the high safety factor for neuromuscular transmission (for review see Wood and Slater, 2001), the consequences of a 2-fold reduction in neurotransmitter levels would be expected to be mild, and it is unlikely that these abnormalities can alone account for the severe impairment of motor behavior evident in SMA.
These observations raise the possibility that the muscle weakness and motor dysfunction in SMA mice, and by inference in the human disease, might be the result of alterations in spinal circuit function. Here, we focused on the synaptic connections between motor neurons and muscle spindle afferents (the stretch reflex) because of their experimental accessibility and their importance in many aspects of motor function. We demonstrate here that the strength of monosynaptic connections between primary afferents and motor neurons is greatly reduced early in the disease, before substantial motor neuron cell loss can be detected. This loss of function is mediated in part by the loss of primary afferent boutons on motor neuron somata and proximal dendrites in SMA mice. These abnormalities are more severe in synapses formed upon motor neurons projecting to proximal muscles. Collectively, our findings suggest that spinal circuit dysfunction is one of the earliest and most pronounced pathological features of the disease and therefore may contribute significantly to the loss of motor function that characterizes both mouse models and human SMA patients.
RESULTS
Reduced synaptic responses of SMA motor neurons following proprioceptive fiber activation
We observed severe motor dysfunction in SMA mice as demonstrated by their inability to right at postnatal day 1 (P1) until death at approximately P14 (Figure 1A), as has been previously reported (Le et al, 2005; Avila et al 2007). The righting reflex is a complex motor function that is mediated by vestibular pathways, spinal interneurons, proprioceptive afferents, and motor neurons (Bignall, 1974; Bose et al, 1998). In order to examine the functional status of the sensory-motor spinal reflex circuitry in SMA mice at an early (P4) and a late stage (P13) of the disease, we used an in vitro preparation of the isolated intact spinal cord in which connectivity of spinal circuits can be investigated. We first examined motor neurons in the lumbar segment 1 (L1), which innervate axial and proximal hindlimb muscles such as the iliopsoas (Gerrits et al, 1997) which is severely affected in SMA. We recorded the synaptic responses evoked in the entire population of motor axons emerging from the L1 ventral root in response to stimulation of sensory axons in the L1 dorsal root in wild type (WT) and SMA spinal cords (Figure 1C). Stimulation of dorsal root sensory fibers at supra-maximal intensities (5×Threshold) resulted in a robust monosynaptic reflex in WT mice. In contrast, the reflex was significantly reduced in SMA spinal cords (Figure 1C) even at high stimulation intensities (10×T; data not shown). On average, there was an approximately 85% reduction in the peak amplitude of the monosynaptic response from SMA L1 motor neurons at P4 (Figure 1C). There was no difference between WT and SMA mice in the latency of onset of the monosynaptic reflex (Figure 1D1), suggesting that conduction velocities and synaptic delays were not significantly altered in SMA mice. At P13, the monosynaptic reflex remained significantly depressed (Figure 1D2).
Figure 1. SMA monosynaptic responses are markedly reduced following proprioceptive fiber stimulation.

(A) Time to right for WT (blue line, n=6) and SMA mice (red line, n=9) at P1–P12. Subsequent experiments were performed at P4 and P13 as indicated by the shaded rectangles. (B) Experimental setup for stimulation of L1 dorsal root (DR) and extracellular recording from L1 ventral root (VR). (C) VR responses to supra-maximal stimulation (5×T) of L1 DR for WT and SMA spinal cords at P4 and P13. (D1–D2) Average amplitude and latency of the monosynaptic reflex at P4 (D1) and P13 (D2) for WT and SMA mice. * p<0.01, t-test. Arrowheads indicate the stimulus artifact and the red arrows indicate the monosynaptic response. Numbers within the bars indicate the numbers of experiments. Scale bars: C (P4) = 0.4mV, 20ms; (P13) = 0.25mV, 20ms.
To test if the reduced amplitude of the monosynaptic reflex in the SMA spinal cords was simply the result of motor neuron loss, we obtained whole cell intracellular recordings from antidromically identified L1 motor neurons (see Experimental Procedures). Similar to the extracellular recordings, WT motor neurons responded robustly to supra-maximal stimulation of the dorsal root L1 (Figure 2B1) while all SMA motor neurons responded weakly and evoked fewer action potentials (Figure 2C1). We previously reported that monosynaptic excitatory post-synaptic potentials (EPSPs) due to primary afferent stimulation can be analyzed quantitatively by comparing the peak amplitude of the response 3ms after its onset (Shneider et al, 2009a). The resulting measurements are similar to other reports (Mears and Frank, 1997; Wang et al, 2008). SMA L1 motor neurons at P4 evoked smaller monosynaptic EPSPs compared to WT following supra-maximal stimulation of the L1 dorsal root (Figure 2C2). On average, there was a significant 70% reduction in the peak EPSP amplitude in SMA mice (Figure 2D). The latency of the monosynaptic EPSP was not significantly different between WT and SMA potentials consistent with the ventral root recordings. Therefore, the ability of proprioceptive afferents to excite motor neurons is severely depressed at early stages in the SMA disease process, at both the single cell and population level.
Figure 2. Monosynaptic potentials in individual SMA motor neurons are reduced.

(A) Setup for stimulation of L1 DR and whole-cell recording from individual motor neurons. (B1–C1) Intracellular responses to supra-maximal stimulation of the L1 dorsal root in a WT (B1) and SMA (C1) L1 motor neuron at P4. The monosynaptic part of the EPSP is shown on an expanded time scale in B2 (WT) and in C2 (SMA). (D) The average EPSP peak amplitude and latency for WT and SMA mice. Number of experiments in bars. * p<0.01, t-test. Arrowheads show the stimulus artifact. Scale bars: B1, C1 = 20mV, 50ms; B2, C2 = 10mV, 2ms.
SMA motor neurons are hyperexcitable
One possible explanation for the reduced responses to dorsal root stimulation in SMA mice could be that SMA motor neurons have lower input resistance and are hypoexcitable. To test this hypothesis, we performed intracellular recordings from identified SMA motor neurons and compared their intrinsic membrane properties with those from age-matched WT motor neurons. We analyzed the active and passive membrane properties of 6 wild type and 6 SMA L1 motor neurons at P3. Neurons were identified as motor neurons by the presence of an all-or-none antidromic action potential (AP) following stimulation of the ventral root (Figure 3A). Antidromically-evoked action potentials recorded from SMA motor neurons had a significantly greater amplitude and faster rate of rise compared to WT motor neurons (Figures 3A and 3B). Correspondingly, the time to peak of the AP was significantly shorter in SMA compared to WT motor neurons (SMA:1.0±0.06ms; WT: 2.3±0.2ms; p<0.001, t-test). The mean resting potential (corrected for a 6mV liquid junction potential) was not significantly different between WT (−56.8±2.5mV) and SMA motor neurons (−54.6±2.1mV; p=0.51; t-test).
Figure 3. SMA L1 motor neurons are hyperexcitable.

(A) Antidromically evoked action potentials recorded in a WT and a SMA L1 motor neuron. The superimposed traces shown on an expanded time scale reveal the greater amplitude and rate of rise in the SMA (red) motor neuron. (B) The average amplitude and rate of rise of the antidromic action potential in SMA and WT motor neurons. (C) Membrane responses following current injection in a WT and a SMA motor neuron. (D) Current/voltage relationships for the two neurons shown in C. (E) The average input resistance, rheobase current and spike threshold for WT and SMA motor neurons. * p<0.01, t-test. Arrowheads show the stimulus artifact. Scale bars: A = 20mV, 10ms; superimposed traces = 20mV, 2ms; C = 20mV, 20ms, current: 400pA (WT), 200pA (SMA).
To calculate the input resistance of the motor neurons, we injected progressively increasing hyperpolarizing or depolarizing current steps (Figure 3C). The slope of the linear current-to-voltage relationship defines the input resistance (Figure 3D). The input resistance (Figure 3E) of L1 motor neurons was approximately four-fold higher in SMA compared to WT motor neurons (p<0.05, t-test).To address whether changes in input resistance could be attributed to changes in soma size (input resistance is inversely proportional to soma size, Fulton and Walton, 1986; Mentis et al, 2007), we measured the maximum cross-sectional area of ChAT+ L1 motor neurons and found no differences between WT and SMA motor neurons at P4 (Supplemental Figures 1A and 1B; WT:298.7±26.8µm2; SMA:290.7±17.3µm2, p=0.83, t-test) or at P13 (Supplemental Figures 1C and 1D; WT:308.9±15.2µm2; SMA:271.1±13.5µm2, p=0.09, t-test). These results suggest that the increased input resistance of SMA motor neurons is due either to a reduction in the extent of the dendritic tree or an increase in the specific membrane resistivity of SMA motor neurons. In addition, SMA motor neurons had a significantly lower spike threshold (the membrane potential at which an action potential is evoked) than did WT motor neurons (p<0.05, t-test, Figure 3E). As a result of the increase in input resistance and the reduced threshold for eliciting action potentials, the minimum current necessary to elicit an action potential (rheobase) was significantly reduced in L1 SMA motor neurons (p<0.01, Mann-Whitney test, Figure 3E). These results indicate that the reduced amplitude of afferent-evoked monosynaptic potentials and the limited evoked firing cannot be explained by motor neuron hypoexcitability; instead SMA neurons are paradoxically hyperexcitable.
Reduction of proprioceptive synapses on the somata and dendrites of SMA motor neurons
The observed marked increase in input resistance of SMA motor neurons would be predicted to increase the amplitude of synaptic potentials evoked when stimulating the Ia afferents. Therefore, the reduction in the amplitude of primary afferent-evoked synaptic potentials in SMA motor neurons actually reflects a much greater reduction (~22 fold; for calculation details see Experimental Procedures) in the amplitude of these synaptic potentials. One possibility to account for this abnormality is diminished connectivity between primary afferents and motor neurons. This was examined by filling the dorsal roots with fluorescent dextran and immunostaining their synapses using antibodies against the vesicular glutamate transporter 1 (VGluT1), a known synaptic marker for proprioceptive primary afferents (Todd et al, 2003; Alvarez et al, 2004) (Figures 4A1–A2 and 4C1–C2). Fewer ventrally directed sensory collaterals (n=4) and fewer VGluT1+ boutons were observed in the ventral horns of SMA spinal cords compared to WT (Figures 4B1–B2 and 4D1–D2). We examined whether a reduction in the number of primary afferent neurons or an abnormality of the muscle spindles could account for this marked reduction of sensory afferent projections to the ventral horn. Quantification of the number of DRG neurons expressing parvalbumin, a marker for proprioceptive neurons, in the L1 segment at P13 revealed no reduction in SMA mice (Figures 4E–4G). In addition, the morphology and innervation by primary and secondary afferent endings in the proximal iliopsoas and the distal tibialis anterior muscles, were normal (Figures 4H–4I). We analyzed at P13, 75 WT muscle spindles (51 in the iliopsoas muscle and 25 in the tibialis anterior muscle; n=3 animals) and 35 in SMA muscles (23 in the iliopsoas and 12 in tibialis anterior muscle; n=3 animals) immunostained with PGP9.5 and VGluT1. No structural or immunoreactivity changes were observed in SMA spindles with respect to WT in either muscle.
Figure 4. Reduction of sensory fibers in the ventral horn in SMA.

(A1–A2) Transverse section from L1 segment in a WT spinal cord showing DR fibers filled with fluorescent dextran (A1) and stained for VGluT1-immunoreactivity. (B1–B3) Higher magnification z-stack projection showing sensory fibers in the ventral horn (B1), the location VGluT1+ synapses (B2) and their co-localization in the merged image (B3). (C1–C2) Transverse section from an SMA spinal cord as in A1–A2. (D1–D3) Labeled sensory fibers (D1) and VGluT1+ synapses (D2) in the ventral horn (D3 is a merged image). The dotted shape indicates the location of the motor neuron nucleus (MN). (E, F) Confocal images of parvalbumin+ (Pv+) L1 dorsal root ganglion (DRG) neurons in a P13 wild type (E), and an age-matched SMA (F) mouse. (G) Comparison of the total number of Pv+ DRG-L1 neurons between WT and SMA mice. Number of experiments in bars. (H, I) Confocal images of muscle spindles labeled with PGP9.5 (green) and VGluT1 (blue) from iliopsoas muscles in a P13 WT (H) and a SMA (I) mouse. Insets show the sensory VGluT1-immunoreactive components alone. In I, only half of the muscle spindle is shown. Total z-stack projection in series B1–B3 and D1–D3 = 40µm; in H and I=26µm. Scale bars: A1, A2, C1 and C2 = 200µm; B1, B2, B3, D1, D2, D3, E and F = 50µm; H and I = 100µm.
We next quantified the number of proprioceptive boutons on the somata and dendrites of SMA and WT motor neurons. WT and SMA L1 motor neurons were retrogradely filled from the ventral root with Cascade Blue dextran in the isolated spinal cord. The cords were then fixed and immunostained for VGluT1. Representative examples are shown in Figures 5A and 5B (P4) and Figures 5C and 5D (P13). We examined and analyzed 10 WT and 9 SMA motor neurons at P0, 22 WT and 26 SMA motor neurons at P4 and 18 WT and 15 SMA motor neurons at P13. The synaptic coverage on dendrites was expressed as the number of VGluT1+ synapses per 50 µm of dendritic length. We excluded small motor neurons (soma size less than 300 µm2) from our analysis at P4 and P13 because these are predominantly γ-motor neurons. Mouse γ-motor neurons older than 2 weeks of age have cross-sectional areas less than ~330µm2 and receive few or no VGluT1+ synapses (Friese et al, 2009; Shneider et al, 2009b).
Figure 5. Early reduction in the proprioceptive input to L1 motor neurons in SMA mice.

(A) Z-stack projection of confocal images from retrogradely labeled P4 motor neurons (blue) and VGluT1+ synaptic boutons (green) in a WT mouse. (B) Similar image projection for age-matched SMA motor neurons. Insets show areas in the dotted box at a higher magnification. (C–D) WT (C) and SMA motor neurons (D) and VGluT1+ immunoreactive boutons at P13. (E) The number of VGluT1+ boutons on the somata of WT and SMA motor neurons. (F–H) The density of VGuT1+ boutons on the dendrites of WT and SMA motor neurons at P0 (F), P4 (G) and P13 (H). The total distance in the z-axis for A and B was 5µm (20 optical sections at 0.2µm intervals) and 12µm (20 optical sections at 0.6µm intervals). * p<0.01, t-test. Scale bars: A, B = 10µm (insets: 2.5µm); C,D = 20µm (insets: 5µm). Numbers in the bars in E indicate the number of motor neurons analyzed.
At P0, the number of VGluT1+ terminals was similar on the soma and dendrites of WT and SMA motor neurons (Figure 5E and 5F). By contrast at P4, there was a significant reduction in the number of VGluT1+ synaptic terminals on the soma (Figure 5E; p<0.001, t-test) of SMA motor neurons with no significant change in the soma size (WT:323.3±14.3µm2; SMA:314.1±15.1µm2; t-test). We also found a significant reduction (p<0.001, t-test) in the linear density of proprioceptive synapses on the dendrites of SMA motor neurons, but only in the first two dendritic compartments (Figure 5F; 0–50µm and 50–100µm from soma). We detected no differences in the number of primary dendrites emanating from the soma of the sampled motor neurons (WT: 4.8±0.3; SMA:4.4±0.2, p=0.25, t-test). The loss of VGluT1+ contacts was not due to down-regulation of VGluT1 expression in proprioceptive terminals because we found a one-to-one correspondence between VGluT1+ staining and putative synaptic swellings in orthogradely labeled primary afferents (see Supplemental Figure 2).
Between P0 and P13, WT motor neurons showed a significant increase in the number and density of VGluT1+ proprioceptive synapses both on the soma as well as the dendrites (Figure 5E), consistent with the proliferation of proprioceptive VGluT1 synapses onto interneurons during the neonatal period (Mentis et al, 2006; Siembab et al, 2010). In contrast, there was a reduction in the density of proprioceptive boutons on the dendrites of SMA motor neurons between P0 and P13, although the number of contacts on the soma did increase during this period. The density of VGluT1+ contacts in the first two dendritic compartments (0–50 and 50–100µm) was further reduced at P13 compared to P4 in SMA (p<0.05 for 0–50µm and p<0.001 for 50–100µm, t-test).
These data indicate that the reduced afferent-evoked potentials in the SMA spinal cords is due, in part, to a loss of and/or reduced proliferation of proprioceptive synapses on motor neurons during the first two postnatal weeks. Moreover, it is known that, synaptic efficacy of Ia afferents correlates with bouton size and synaptic vesicle number (Pierce and Mendell, 1993). VGluT1+ bouton cluster sizes provide an estimate of the size of the vesicle pools. Comparing ventral horn VGluT1+ clusters in the L1 segment of WT and SMA P13 mice revealed that SMA proprioceptive synapses were slightly, but significantly smaller than WT synapses (WT: 1.70±0.04µm; SMA: 1.52±0.05µm; p<0.01, One Way ANOVA). This finding is consistent with the idea that the remaining VGluT1+ terminals on SMA motor neurons have impaired function.
To test whether the reduction of synapses on motor neurons was unique to proprioceptive projections, we investigated the somatic coverage of inhibitory synapses on L1 WT and SMA motor neurons at P13. For this purpose, we immunostained sections containing ChAT+ motor neurons with the neuronal glycinergic transporter 2 (GlyT2). We found no significant difference in the number of GlyT2+ synapses between WT and SMA motor neurons (p=0.52, t-test; Supplemental Figure 3).
Selective vulnerability of synapses onto medial motor neurons in SMA
In SMA patients, the most severely affected muscle groups are proximal and axial. To test if medial motor neurons innervating axial muscles are more affected than lateral motor neurons innervating distal limb muscles in SMA mice, we examined motor neurons in the L5 spinal segment where the medial and lateral pools can be easily distinguished by their location (see Figure 7F). We analyzed the monosynaptic reflex in the L5 spinal segment. As lateral motor neurons make up ~95% of motor neurons in this spinal segment, the monosynaptic reflex predominantly reflects the function of these neurons. We found that the peak amplitude of the reflex at P4 was similar to that measured in WT animals (Figure 6A and 6B1). At P13, however, there was a significant reduction in the amplitude in SMA animals (Figure 6A and 6B2). The onset latency of the monosynaptic response was similar at both ages tested.
Figure 7. Time course of motor neuron loss in L1 and L5 lumbar spinal segments in SMA.
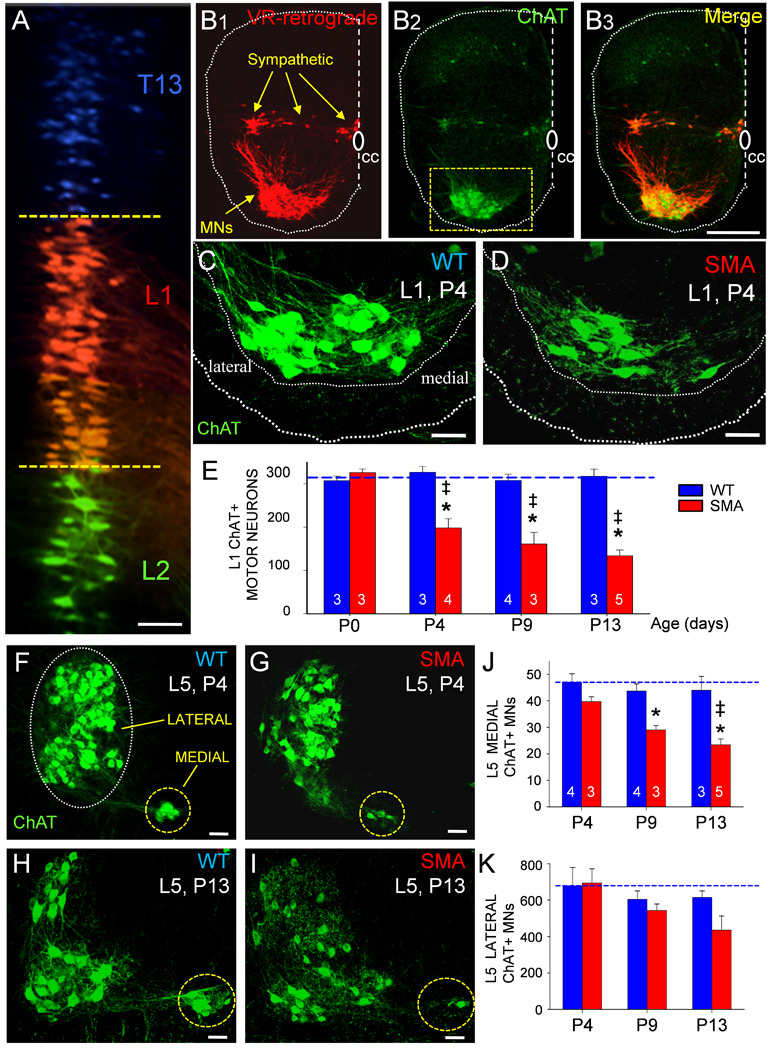
(A) Motor neurons in the 13th thoracic (T13, in blue), 1st lumbar (L1, in red) and 2nd lumbar (L2, in green) segment labeled with different fluorophores by retrograde transport from the respective VRs. Dotted lines indicate the rostral and caudal borders of the L1 motor neuron column. (B1–B3) the L1 segment of a wild type P4 hemisected spinal cord, showing the location of retrogradely filled motor and sympathetic neurons (B1), ChAT-immunoreactivity (B2) and both signals superimposed (B3). (C) Higher magnification of WT L1 ChAT+ P4 motor neurons (from boxed area in B2) and (D) from an aged-matched SMA mouse showing a significant loss of ChAT+ motor neurons. (E) Time course of L1 motor neuron loss in SMA (red bars) and WT mice (blue bars). Numbers of experiments in bars. (* p<0.05 vs. P0 SMA motor neurons, One Way ANOVA; ‡ p<0.05 vs. WT age-matched motor neurons, One Way ANOVA followed by Bonferroni-corrected t-test pair-wise comparisons).(F–I) Z-stack projection of confocal images from L5 ChAT+ motor neurons in P4 WT (F), P4 SMA (G), P13 WT (H) and P13 SMA (I) ventral horns. Dotted circles indicate the location of the medial and lateral motor neuron columns. (J) Change in the number of medial L5 motor neurons from P4 – P13 (* p<0.05 vs. WT, ‡ p<0.05 vs. P4-SMA; One Way ANOVA, Bonferroni t-tests as above). (K) The number of lateral L5 motor neurons at three ages in SMA and WT mice. Scale bars: A, C, D, F, G, H, I = 50µm, B1–B3= 200µm.
Figure 6. Medial motor neurons exhibit earlier and more severe pathology than lateral motor neurons in SMA.

(A, B1 and B2) Averaged L5 VR potentials evoked by supra-maximal stimulation of the L5 DR in P4 WT (top left), P4 SMA (top right), P13 WT (bottom left) and P13 SMA (bottom right) spinal cords. Red arrows indicate the maximum amplitude of the monosynaptic component. Black arrows indicate the stimulus artifact. Average amplitude and latency of the reflex at P4 (B1) and P13 (B2) for WT and SMA mice. Number of experiments in bars. * p<0.05, t-test. (C–F) Z-stack projections of confocal images in P4 WT (C), P4 SMA (D), P13 WT (E) and P13 SMA (F) showing retrogradely labelled medial motor neurons (blue) and VGluT1+ synaptic boutons (green). Insets show dendritic VGluT1+ synapses at higher magnification. (G) Comparison of the total number of VGluT1+ synaptic contacts on the somata (* p<0.05 vs. WT, t-test) and dendrites (* p<0.05 vs. WT, One Way ANOVA) of WT and SMA medial motor neurons at both ages. (H–K) Z-stack projections of confocal images from P13 WT (H), P4 SMA (I), P13 WT (J) and P13 SMA (K) lateral motor neurons (blue) and VGluT1+ synapses (green). (L) Comparison of the total number of VGluT1+ synapses in WT (blue bars) and SMA (red bars) lateral L5 motor neurons (* p<0.05 vs. wild type, t-test). Images in C, D, E and F are projection images for a total thickness of 5µm in the z-axis (at 0.2µm z-intervals) and in H, I, J and K are 7.2µm thick (at 0.6µm intervals). Number of experiments in bars. Scale bars: A: 10ms, (P4) = 2mV, (P13) = 0.2mV; C, D, H, I = 10µm; E, F, J, K = 20µm (insets: 5µm).
Examination of orthogradely labeled dorsal root fibers revealed that very few sensory collaterals travelled medially towards the medial motor column in SMA spinal cords, while the laterally projecting collaterals were less affected (Supplemental Figure 4). To quantify the number of proprioceptive synapses on the soma and dendrites of medial and lateral L5 motor neurons at P4 and P13, we counted VGluT1+ synapses on these two motor neuron populations in WT and SMA mice. We detected a substantial loss of VGluT1+ synapses on medial motor neurons in SMA mice compared to WT both at P4 (Figure 6C–6D) and at P13 (Figure 6E–6F). We estimated a 65% reduction of VGluT1+ synapses on the soma at P4 and 80% reduction at P13 (Figure 6G). The reduction in VGluT1+ contact density was statistically significant at both ages (p<0.05, t-test). Similar to the observations on L1 motor neurons, proximal dendrites also showed significant reductions of 67% and 74% loss at P4 and P13, respectively. The difference between P4 and P13 did not reach statistical significance (p=0.39 for 0–50µm and p=0.14 for 50–100µm, t-tests).
In striking contrast to the results for medial motor neurons, there was no loss of synaptic coverage on L5 lateral motor neurons (Figure 6H–6K) at P4 (Figure 6H and 6I), but there was a 50% reduction of VGluT1+ synapses on the soma (Figure 6G) and proximal dendritic compartments (Figure 6L) by P13. Taken together, these observations indicate that de-afferentation occurs earlier and is more severe in medial compared to lateral motor neurons in correspondence with the known progression of muscle weakness in diseased mice and humans.
Time course of motor neuron loss in lumbar motor neurons in SMA
The preceding data reveal significant and selective changes in synaptic transmission from proprioceptive afferents onto surviving motor neurons that may contribute to the disease phenotype in SMA mice. However, motor neuron degeneration is a hallmark of the disease so we were interested in establishing if sensory loss precedes or follows the loss of motor neurons. To investigate the relationship between impaired synaptic inputs and motor neuron loss, we quantified all motor neurons in the L1 and L5 segments throughout the course of disease. Motor neurons were retrogradely labeled from the ventral root by application of fluorescent dextran dyes (Texas Red dextran or Cascade Blue dextran). Ventral root retrograde fills demonstrated that motor neurons located in adjoining spinal segments do not overlap significantly in the rostrocaudal axis (Figure 7A). Based on this observation, we processed serial sections (75–80µm) for ChAT immunoreactivity. Using confocal microscopy, we counted every lamina IX ChAT+ motor neuron in all sections from each of the L1 and L5 segments defined as those, containing motor neurons retrogradely labeled from the corresponding ventral roots (Figure7B1–B3).
In the L1 segment, there was no significant loss of SMA motor neurons at birth (P0), but by P4 there was a significant (~40%) reduction in the number of ChAT+ motor neurons in SMA compared to WT mice. This value progressively increased to 50% and 60% at P9 and P13 respectively (p<0.05, vs. age-matched WT, One Way ANOVA) (Figures 7C–7E).
In the L5 segment, lateral and medial motor neurons (Figure 7F) exhibited a differential vulnerability during disease progression (Figures 7F–K). At P4, there was no statistically significant loss of L5 medial or lateral motor neurons (Figure 7F, 7G and 7K). By P9 and P13, however, the number of medial motor neurons was reduced by 35% and 50% respectively, in SMA mice as compared to WT (p<0.05, One Way ANOVA). In contrast, no significant loss of lateral motor neurons was detected, even at P13. These data demonstrate that the severity of de-afferentation and motor neuron loss are correlated within subsets of motor neurons. Furthermore, the loss of proprioceptive inputs precedes the loss of lateral L5 motor neurons.
Treatment with Trichostatin A (TSA) improves sensorimotor connectivity in SMA mice
The dramatic reductions in proprioceptive innervation and survival of specific motor neuron subsets we describe are likely to contribute significantly to the motor behavioral deficits in SMA mice. The histone deacetylase inhibitor, Trichostatin A (TSA), increases SMN expression and improves motor function, weight gain and survival of SMA-Δ7 mice (Avila et al., 2007; Narver et al., 2008). We therefore tested whether TSA could also ameliorate the loss of proprioceptive synapses and improve function of the monosynaptic reflex. SMA mice were treated with TSA 10mg/kg daily between P1–P12 as has been previously described (Narver et al, 2008). We assessed sensory-motor circuitry at P13 only in animals showing improved time to right (Figure 8A) and exhibiting weight gain (data not shown). Eighty percent of treated animals showed a therapeutic response to TSA treatment, similar to previous studies (Avila et al., 2007). Lack of response to TSA treatment might be due to failure of the drug to gain systemic access or because a weaker animal displaying a more advance stage of the disease. We found that the amplitude of the monosynaptic reflex recorded from the L1 ventral root improved significantly in the TSA-treated group compared to SMA animals treated with vehicle (p<0.05, One Way ANOVA, Figure 8B).
Figure 8. Treatment with Trichostatin A (TSA) partially improves synaptic function and rescues motor neurons in SMA mice.

(A) Time to right for WT (blue line), SMA (red line) and SMA+TSA treated mice (green line). (B) Average monosynaptic reflex amplitude in L1 VR for the three experimental groups. (* p<0.05 vs. WT; ‡ p<0.05 vs. SMA, One Way ANOVA). (C) Comparison of the somatic and dendritic VGluT1+ boutons for the 3 groups. (* p<0.05 vs. WT, ‡ p<0.05 vs. SMA, One Way ANOVA). (D–F) The average number of L1 (D) and L5 (E, medial; F, lateral) motor neurons at P13 for WT (blue bar), SMA (red bar) and SMA+TSA treated mice (green bar). (* p<0.05 vs. WT, ‡ p<0.05 vs. SMA, One Way ANOVA). Number of experiments in bars.
We next investigated whether this functional improvement was accompanied by structural improvements in the proprioceptive afferent synapses on L1 motor neurons at P13. TSA treatment resulted in a significant increase (~3 fold, p<0.05, One Way ANOVA) in the density of VGluT1+ terminals on the proximal dendrites (0–50µm and 50–100 µm) of SMA motor neurons (Figure 8C).
VGluT1+ synapses in TSA-treated animals were significantly larger than SMA vehicle-treated synapses, but not when compared to WT proprioceptive synapses (WT: 1.70±0.04µm; SMA: 1.52±0.05µm; SMA+TSA: 1.77±0.03µm; One Way ANOVA; posthoc pair comparisons: WT vs. SMA+TSA p=0.38; SMA vs. SMA+TSA, p<0.001, Bonferroni t-test). The measurements for all VGLUT1+ synapses on the somatodendritic surface of motor neurons were pooled because there was no difference in VGluT1-IR cluster size across the different locations (i.e. soma, 0–50µm and 50–100µm dendritic compartments; data not shown).
The increased function of motor neuron synaptic inputs was accompanied by an increase in the number of motor neurons in the L1 segment (Figure 8D) and in the medial and lateral motor nuclei of the L5 segment (Figures 8E and 8F) in TSA-treated compared to vehicle-treated SMA mice. These results suggest that the improved motor function of the TSA treated mice is likely to be the result of both improved synaptic function and a reversal of motor neuron loss.
DISCUSSION
Spinal muscular atrophy in human patients and mouse models is characterized by pronounced weakness of specific muscle groups. However, the relatively modest changes in transmission at the neuromuscular junction of SMA model mice seem unlikely to explain the severe phenotype. Here, we report that transmission in the monosynaptic reflex arc formed between proprioceptive afferents and spinal motor neurons, which is known to be important for motor function, shows massive and progressive failure early in the disease process. This functional deficit and the subsequent motor neuron loss are most pronounced in motor neurons innervating proximal hindlimb and axial muscles (Figure 9), mirroring the pattern of muscle weakness in SMA patients. Treatment with a drug known to improve motor function and increase survival of SMA model mice favorably affects all of these parameters, in support of the idea that they reflect novel therapeutic targets for this incurable disease.
Figure 9. Sensory-motor defects in SMA: temporal and spatial sequences.

A schematic showing the changes in the sensory-motor circuitry in SMA. L1 motor neurons innervate both proximal and axial muscles whereas medial L5 motor neurons innervate axial muscles and lateral L5 lateral motor neurons innervate distal hindlimb muscles. In SMA, soon after birth there is a significant de-afferentation (yellow synapses) which progresses with age (red dotted line). In the L5 segment, medial motor neurons are lost a few days (P9) after the loss of L1 motor neurons (P4). There is a significant de-afferentation of proprioceptive synapses before the loss of L5 medial motor neurons (P4). Lateral L5 motor neurons show signs of de-afferentation later (P13). Treatment with TSA, assessed after 12 days of daily treatment reverses motor neuron loss and partly restores proprioceptive synaptic coverage. The L1 spinal reflexes corresponding to the three conditions are shown on the bottom line.
Changes in sensory afferents in SMA mice
A central, unexpected finding of our study was the marked reduction (~70%) - probably an underestimate due to the increased input resistance of SMA motor neurons - in the strength of evoked synaptic transmission in the monosynaptic reflex circuit already apparent at P4. It is striking that this reduction was significantly greater than the morphological loss of VGluT1+ synapses (50%) at the same age. The mismatch suggests that at least part of the synaptic dysfunction in early SMA mice is not related to physical de-afferentation, but to more subtle functional alterations. This hypothesis is supported by our observation that the remaining VGluT1+ synapses were reduced in size, which has been reported to result in reduced synaptic efficacy at Ia afferent synapses (Pierce and Mendell, 1993).
Another surprising observation was the finding that the amplitude of the sensory-evoked ventral root potential in SMA mice was less attenuated (compared to the wild-type response) at P13 than at P4 (see Figures. 1C and D). Several factors could contribute to this apparently anomalous result. First, in wild type animals the input resistance of motor neurons is likely to fall significantly between P4 and P13 as the motor neurons mature and their dendritic tree expands. Although data are not available for mouse motor neurons, it has been reported that the input impedance of neonatal rat motor neurons drops by 50% between P3–P6 and P7–P9 (Mentis et al 2007). Second, if the abnormal intrinsic properties of SMA motor neurons - particularly the elevated input resistance - are progressive, then SMA motor neurons may have an even higher input resistance at P13 than at P4. Together, these two effects, progressively lower input resistance in wild type motor neurons but higher in SMA motor neurons with age will decrease the differences in response to a similarly decreased input in the older animals.
Several key questions about the mechanisms and sequence of the molecular and cellular events involved during disease progression are raised by these data. The first concerns the changes in sensory neurons of SMA mice between early (P4) and late (P13) stages of the disease. One model suggested by our data is that at a given proprioceptive synapse, SMN reduction first triggers shrinkage and dysfunction of the terminal and only later, perhaps as a secondary effect, leads to complete retraction. According to this model, the greater reduction in the number of VGluT1+ boutons at P13 as compared to P4 at L1 in SMA mice could reflect the fact that by late stages, more synapses have reached the stage of physical die-back. Such a sequence of functional loss followed by die-back has been considered in other neurodegenerative diseases (Selkoe, 2002; Yoshiyama et al, 2007), although in no case has the precise sequence of events been determined. Consistent with this idea, we found that that the number of VGluT1+ terminals on the dendrites of L1 motor neurons decreased between P0 and P13.
However, our data are also consistent with a second model in which the loss of synaptic function is due to a failure of postnatal maturation rather than neurodegeneration of the afferent pathway. Between P4 and P13, the number of VGlut1+ synapses on L1 motor neurons increased on average by 30/soma in wild type mice. It also increased in SMA mice but only by 5/soma, leading to a reduction in the number of proprioceptive synapses in SMA as compared to controls. A similar phenomenon was observed for lateral motor neurons at L5, and even on medial motor neurons at L5 there was only a modest loss of VGlut1+ boutons/soma over the same period. According to this model, the principal phenotype in SMA mice between P4 and P13 may be a failure to add additional Ia synapses at the normal rate as part of a maturational process, rather than a massive retraction of synapses.
Finally, it is also possible that some proprioceptive axons fail to reach their target motor neurons in SMA mice. In the L1 segment, this does not appear to occur up to P0 because the number of VGlut1+ terminals is similar on WT and SMA motor neurons at this time. However, it is not known to what extent the proliferation of VGlut1+ synapses on WT motor neurons between P0 and P14 (see Fig. 5) is due to the arrival of new proprioceptive axons or to proliferation by branching or sprouting of sensory axons that have already projected to motor neurons. As a result, the reduced and absent proliferation of VGluT1+ boutons on SMA motor neuron somata and dendrites respectively, could be due to a failure of sensory axon projection, sensory axon branching or to deficits in both processes.
A second question concerns the mechanisms through which SMN reduction leads to loss of function of the proprioceptive synapse. In principle, given that the SMN protein is expressed in motor neurons and DRG (see Suppl. Figure 5), changes in either or both cell types could contribute to the phenotype. The reduction of neurotransmission in this pathway is unlikely to be accounted for by the changes in the intrinsic motor neuron properties (increased input resistance) observed in the disease, which would tend to amplify rather than the depress the synaptic potentials. This makes it more likely that alterations in sensory afferents are responsible for the reduced synaptic strength. Since we found normal numbers of muscle spindles and proprioceptive neurons in the dorsal root ganglia at end-stage, the SMA phenotype is likely to involve the central axonal projections. In rodents, the connections between Ia afferents and motor neurons first form shortly before birth (E16) and progressively mature during the postnatal period (Kudo and Yamada, 1987; Mears and Frank 1997, Seebach and Ziskind-Conhaim 1994). Sensory neurons isolated from a severe mouse model of SMA show reduced capacity for axonal growth (Jablonka et al., 2006), so that some Ia synapses may fail to form, or do so late during embryogenesis. Even for those synapses that do form, reductions in SMN may create barriers to normal maturation, since deficits in axonal transport have been reported in SMA and it is known that primary afferents are exquisitely dependent on axonal transport for trophic interactions with their targets (Chen et al., 2007). In particular, muscle spindle-derived neurotrophin 3 (NT-3) is known to mediate the postnatal proliferation, maturation and maintenance of Ia afferent central synapses on postnatal motor neurons (Patel et al, 2003; Chen et al, 2002; Wang et al., 2007; Shneider et al., 2009a). In support of this idea, NT3 has been reported to enhance neurite growth in SMA sensory neuron cultures (Jablonka et al, 2006).
Thirdly, it is critical to ascertain whether the marked reduction of the monosynaptic reflex is sufficient to explain the severe motor phenotype of the SMA-Δ7 mice or that of human SMA patients. Ia afferents have been argued to constitute alone a small percentage of all synaptic inputs to mature motor neurons (Fyffe, 2001) and their exact contribution to synaptic modulation of motor neuron firing during early postnatal development is not yet fully understood. Nevertheless, in adults they do contribute a significant amount of synaptic drive that continually modulates motor output, muscle stiffness and tone which is particularly important for postural control (Davidoff, 1992; Dietz and Sinkjaer, 1997). Thus, in SMA mice, the loss of stretch reflex function may contribute to deficits in the righting during the disease.
Although few studies have examined the spinal reflex in human SMA patients, deep tendon reflexes and H-reflexes have been reported to be absent in a proportion of SMA patients (Iannaccone et al., 1993, Renault et al., 1983). The interpretation of this result is complicated by the presence of motor neuron cell loss and dysfunction which could account for the absent reflexes. However, when this finding is considered in the light of our current results, it raises the possibility that a loss of central Ia afferents may be a common characteristic of the disease in both human patients and animal models. We have not quantified the muscle afferent-evoked polysynaptic responses to motor neurons because these are labile and difficult to quantify reliably. Nevertheless, the afferent synapses on spinal interneurons might also be compromised leading to a reduction in the polysynaptic drive to motor neurons.
Changes in motor neurons in SMA mice
The second set of findings from our study concerns the motor neurons themselves. We have uncovered selectivity in the pattern of motor neuron loss that appears to be highly relevant to the clinical phenotype, and we have shown that surviving motor neurons exhibit marked changes in their intrinsic electrophysiological properties.
Although existing data on post mortem material from SMA patients point to extensive motor neuron death, especially in the severe Type 1 cases (Robertson et al, 1978; Soler-Botija et al, 2002), studies performed using mouse models have concluded that motor neuron loss is surprisingly modest. The values reported range from no loss (Kariya et al, 2008) to ~60% (Passini et al, 2010) at endstage and vary considerably between different studies (Le et al, 2005; Passini et al, 2010; Rose et al, 2009; Avila et al, 2007). Our data provide an explanation for these discrepancies and a novel insight into the specific motor neuron populations involved. Distal limb-innervating lateral motor neurons which constitute the majority of motor neurons in the lumbar enlargement are relatively spared, at least during the lifespan of the SMA-Δ7 mice. In contrast, rostral and medial motor neurons, which are less abundant and innervate axial muscles and proximal limb muscles, show a significant and progressive reduction in number. Clinical observations have long indicated a similar selectivity of muscle weakness in SMA, but it has not been clear to what extent this reflects differences in dysfunction among motor neuron populations or the muscles themselves. Our data suggest that selective motor neuron loss may contribute to the clinical presentation of symptoms and disease progression. Furthermore, the similarity between the pattern of muscle weakness in the human and mouse forms of the disease suggests that the selective vulnerability of medial motor neurons is not due to variations in the expression of the SMN2 transgene in medial and lateral motor neurons. However, it is possible that the expression of the SMN protein is differentially regulated in medial versus lateral motor neurons, since tissue-specific variations in the levels of full-length and truncated SMN protein have been reported (Soler-Botija et al, 2005; Vezain et al, 2007). Other functional and molecular differences between subsets of motor neurons that could increase their susceptibility to disease are only beginning to be defined (for review, see Kanning et al, 2010).
Motor neuron loss occurred after the early changes in proprioceptive afferent input. This is particularly clear in the L5 segment, where clear deficits in VGlut1+ synaptic coverage are apparent in medial and lateral populations before cell loss can be detected. Although the synaptic and motor neuron loss occur at different times, our data suggest that they are related, since the populations that show the greatest reduction in Ia innervation are those which subsequently show more pronounced motor neuron loss. This raises the question of whether motor neuron loss might be a direct result of deafferentation. Experiments in chicken embryos demonstrated enhanced motor neuron cell death after removal of the neural crest and all sensory afferents (Okado and Oppenheim, 1994). A number of studies in rodents however, argue against this possibility. Neonatal dorsal rhizotomy in rats has been reported to result in no loss of motor neurons (O’Hanlon and Lowrie, 1996) or a small but significant decrease of ~10% (Chatzisotiriou et al., 2005). Moreover, in several genetic mouse models the selective loss of proprioceptive afferents does not seem to be accompanied by significant cell death of alpha motor neurons (Tourtelotte and Milbrandt,1998; Chen et al., 2007; Shneider et al, 2009b). Nonetheless, it is possible that SMN-deficient motor neurons might be particularly vulnerable to the effects of de-afferentation.
We report that SMA motor neurons at the L1 level are hyperexcitable, which is paradoxical considering the marked reduction in their response to primary afferent input. This hyperexcitability may be directly related to SMN deficiency. In support of this, the marked (~4 fold) increase in input resistance does not reflect a change in soma size or the number of primary dendrites, factors that are known to govern input resistance. It is also unlikely that SMA motor neurons have shorter dendrites, since they extend at least 250µm from the soma and we did not observe any gross reduction in dendrite branching. Thus, the changes in input resistance are likely to be due to changes in the distribution of ion channels at the cell membrane (specific membrane resistivity). Moreover, the higher amplitude of the action potential in SMA neurons, the faster rate of rise and the lower threshold all point to a greater availability of voltage-gated Na channels (Miles et al., 2005). This could result either from higher expression levels, slower inactivation or faster recovery from inactivation.
Circuit properties and cell autonomy
The circuit and synaptic abnormalities that contribute to neurodegenerative diseases are complex. They comprise pathological changes that are secondary to the primary lesion and may be compensated for at the synaptic, circuit, and network levels. Such compensatory changes may in turn ameliorate or exacerbate the disease or initiate an irreversible cascade of events that leads to neuronal degeneration (Palop et al, 2006; Bezard et al, 2003). An example of toxic network interactions was reported in a mouse model of Alzheimer's disease, in which Aβ peptide triggers intermittent aberrant excitatory neuronal activity in the hippocampus. This results in compensatory remodeling of inhibitory circuits thereby increasing inhibition on granule cells and compromising the normal function of excitatory circuits (Palop et al, 2007).
Both cell types involved in the monosynaptic reflex arc (primary afferents and motor neurons) express SMN, and so it is possible that the phenotypes of sensory and motor neurons in SMA mice simply reflect cell-autonomous outcomes of SMN reduction. An alternative hypothesis is that SMN deficiency in motor neurons triggers changes in Ia afferents in a non-cell-autonomous manner and that these in turn lead to changes in motor neurons and other cell types. Testing this hypothesis rigorously will require the use of cell type-specific SMN mutants but it seems plausible that functional changes in motor neurons may be secondary to alterations in synaptic inputs. An imbalance between reduced excitatory and maintained inhibitory inputs will lead to a reduction in motor neuron activity, and motor neurons could react homeostatically by increasing their excitability to compensate for the decreased presynaptic activity. In support of this hypothesis, de-afferentation of chicken magnocellular neurons by surgical removal of the cochlea results in increased axonal INa and neuronal hyperexcitability (Kuba et al., 2010). In addition, training in a milder model of SMA (SmnΔ7+/+ Smn2) has beneficial effects on the motor phenotype (Biondi et al, 2008) which could be due to exercise-induced upregulation of NMDA-receptor signalling and a corresponding increase in SMN gene expression (Biondi et al., 2010).
Implications for therapy
One potential therapeutic target has already been identified in SMA: the SMN protein itself. Concordant data from human genetics and experimental mouse data suggest that if SMN levels can be upregulated early and strongly enough, there will be considerable therapeutic benefit (Sumner, 2006; Wirth et al, 2006; Oskoui and Kaufmann, 2008). This has most recently been demonstrated through the use of viral gene therapy vectors expressing full-length SMN in neonatal SMA-Δ7 mice (Foust et al, 2010; Passini et al, 2010; Valori et al, 2010). Other promising approaches include antisense oligonucleotides to promote correct splicing of full-length SMN, and drugs to induce increased transcription from the SMN2 gene (Williams et al, 2009; Hua et al, 2010). Despite the exciting progress of recent years, however, it remains uncertain what cell types should be targeted for possible SMN upregulation.
Our data are relevant to these concerns. First, they clearly identify neurotransmission at the proprioceptive afferent synapse as a parameter that needs to be corrected by treatments targeting SMN. Second, they suggest that correction of the sensory-motor transmission defects and the related motor neuron phenotypes may itself have significant therapeutic benefit, alone or in combination with SMN upregulation.
Our findings provide one approach for identifying the most critical parameters that need to be rescued, and for comparing the ability of different treatments to attain this goal. TSA is a small molecule that can cross the blood brain barrier and has proven therapeutic benefit in the SMA-Δ7 mouse (Avila et al, 2007; Narver et al, 2008) and we demonstrate here that it can significantly improve sensory-motor dysfunction. SMN protein levels in TSA-treated animals only reach ~50% of the level in wild type animals (Avila et al, 2007) which may explain the failure of TSA to completely reverse the sensory-motor abnormalities. In contrast to its effects on sensory motor function, TSA treatment prevented motor neuron cell loss suggesting that motor neuron survival may require a lower level of the SMN protein than that necessary to maintain sensory motor function. Collectively, these data provide a proof of principle for the therapeutic use of small molecules to improve synaptic function, which is likely to be a key factor in the restoration of normal motor function in this disease.
EXPERIMENTAL PROCEDURES
All surgical procedures were performed on postnatal mice in accordance with the National Institutes of Health Guidelines on the Care and Use of Animals and approved by the NINDS animal care and use committee, the Institutional Animal Care and Use Committee at Columbia University and Johns Hopkins University.
Animals
The original breeding pairs for the SMA mice used in our study (Smn+/− SMN2+/+SMNΔ7+/+) were provided by A. Burghes (Ohio State University, Columbus, Ohio, USA). The breeding colony was maintained by interbreeding Smn+/− SMN2+/+SMNΔ7+/+ mice, and offspring were genotyped using PCR assays on tail DNA. Precise genotyping protocols can be found in Avila et al. (2007).
In vitro electrophysiology
Eighty mice of 2 to 13 days of age (P2–P13; 30 wild type, 50 SMA-Δ7 with genotype: Smn−/−SMN2+/+SMNΔ7+/+) were used in electrophysiological experiments. Methods for recording from isolated spinal cord preparations have been described (Shneider et al., 2009). Further details provided in Supplemental Experimental Procedures. The cord was transferred to the stage of a 2-photon microscope (LSM510 Carl Zeiss) and the ventral and dorsal roots were placed into suction electrodes for stimulation or recording. Whole-cell recordings were obtained with patch electrodes as described previously (Mentis et al., 2005). Synaptic potentials were recorded from individual motor neurons (DC - 3 KHz, Multiclamp 700A, Molecular Devices) in response to orthodromic stimulation (A365, current stimulus isolator, WPI, Sarasota, FL) of a dorsal root (L1 or L5).
Tracing and Immunohistochemistry
Detailed protocols for tracing and immunohistochemistry used in our study have been reported (Mentis et al, 2006; Shneider et al, 2009a). Further details provided in Supplemental Experimental Procedures.
Confocal imaging
Sections were imaged using either a SP5 Leica or a LSM510 Carl Zeiss confocal microscope. Motor neurons were counted off-line from z-stack images (at 2–3µm intervals) collected for each section that contained a fluorescent signal from retrogradely labeled motor neurons (either in the L1 or L5 segment). Sections were scanned using a 40× objective. Only motor neurons (ChAT+) that contained the nucleus were counted in order to avoid double counting from adjoining sections. Further details of the quantitative analysis are provided in supplementary methods.
Trichostatin A treatment in vivo
We followed the same protocol for TSA administration as reported by Avila et al, 2008. Briefly, TSA was dissolved in sterile DMSO to a concentration of 2.5µg/µl.SMA mice and their WT littermates received intra-peritoneal daily injections (from P1 to P12) of 10 mg/kg. A group of control animals (WT and SMA) received equal volumes of the vehicle without TSA. For further details see supplementary methods.
Statistics
Results presented in this study are means ± standard error of the mean. Comparison was performed by either t-test or One Way ANOVA (post hoc pair-wise comparison methods indicated in the Results when necessary) using Sigmastat (v. 3.1, Jandel) software package.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Drs. T.M. Jessell, C.E. Henderson, S. Przedborski, L. Pellizzoni, N.A. Shneider and K. Fischbeck for comments on the manuscript and for the generous support of Dr K. Fischbeck. The technical assistance of the late Ms A.A. Taye (genotyping) and Mrs J. Sisco (muscle spindle staining) is also appreciated. Thanks to Dr D. Ladle for assistance in muscle dissection. The work was supported by the NINDS Intramural Program and the SMA Foundation (G.Z.M.). C.J.S. was supported by a NINDS grant (R01NS062869) and Howard Hughes Medical Institute Physician Scientist Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J. Comp. Neurol. 2004;472:257–280. doi: 10.1002/cne.20012. [DOI] [PubMed] [Google Scholar]
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Brotchie JM. Presymptomatic compensation in Parkinson's disease is not dopamine-mediated. Trends Neurosci. 2003;26:215–221. doi: 10.1016/S0166-2236(03)00038-9. [DOI] [PubMed] [Google Scholar]
- Bignall KE. Ontogeny of levels of neural organization: the righting reflex as a model. Exp. Neurol. 1974;42:566–573. doi: 10.1016/0014-4886(74)90079-x. [DOI] [PubMed] [Google Scholar]
- Biondi O, Grondard C, Lécolle S, Deforges S, Pariset C, Lopes P, Cifuentes-Diaz C, Li H, della Gaspera B, Chanoine C, Charbonnier F. Exercise-induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J. Neurosci. 2008;28:953–962. doi: 10.1523/JNEUROSCI.3237-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi O, Branchu J, Sanchez G, Lancelin C, Deforges S, Lopes P, Pariset C, Lécolle S, Côté J, Chanoine C, Charbonnier F. In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J. Neurosci. 2010;30:11288–11299. doi: 10.1523/JNEUROSCI.1764-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Fielding R, Ameis KM, Vacca-Galloway LL. A novel behavioral method to detect motoneuron disease in Wobbler mice aged three to seven days old. Brain Res. 1998;813:334–342. doi: 10.1016/s0006-8993(98)01033-6. [DOI] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzisotiriou AS, Kapoukranidou D, Gougoulias NE, Albani M. Effect of neonatal spinal transection and dorsal rhizotomy on hindlimb muscles. Brain Res. Dev. Brain Res. 2005;157:113–123. doi: 10.1016/j.devbrainres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Chen R, Cohen LG, Hallett M. Nervous system reorganization following injury. Neuroscience. 2002;111:761–773. doi: 10.1016/s0306-4522(02)00025-8. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J. Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TO. Concerns about the design of clinical trials for spinal muscular atrophy. Neuromuscul. Disord. 2004;14:456–460. doi: 10.1016/j.nmd.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Davidoff RA. Skeletal muscle tone and the misunderstood stretch reflex. Neurology. 1992;42:951–963. doi: 10.1212/wnl.42.5.951. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Dietz V, Sinkjaer T. Spastic movement disorder: impaired reflex function and altered muscle mechanics. Lancet Neurol. 2007;6:725–733. doi: 10.1016/S1474-4422(07)70193-X. [DOI] [PubMed] [Google Scholar]
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc. Natl. Acad. Sci. USA. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton BP, Walton KD. Electrophysiological properties of neonatal rat motoneurones studied in vitro. J. Physiol. 1986;370:651–678. doi: 10.1113/jphysiol.1986.sp015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe REW. Spinal motoneurons: synaptic inputs and receptor organization. In: Cope TC, editor. Motor Neurobiology of the Spinal Cord. CRC Press; 2001. pp. 21–46. [Google Scholar]
- Gerrits PO, Boers J, Holstege G. The lumbar cord location of the motoneurons innervating psoas and iliacus muscles: a single and double labeling study in the female Syrian golden hamster. Neurosci. Lett. 1997;237:125–128. doi: 10.1016/s0304-3940(97)00842-2. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannaccone ST, Browne RH, Samaha FJ, Buncher CR. Prospective study of spinal muscular atrophy before age 6 years. DCN/SMA Group. Pediatr. Neurol. 1993;9:187–193. doi: 10.1016/0887-8994(93)90082-n. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Karle K, Sandner B, Andreassi C, von Au K, Sendtner M. Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2006;15:511–518. doi: 10.1093/hmg/ddi467. [DOI] [PubMed] [Google Scholar]
- Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ. Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2009;29:15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Kudo N, Yamada T. Morphological and physiological studies of development of the monosynaptic reflex pathway in the rat lumbar spinal cord. J. Physiol. 1987;389:441–459. doi: 10.1113/jphysiol.1987.sp016665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Mears SC, Frank E. Formation of specific monosynaptic connections between muscle spindle afferents and motoneurons in the mouse. J. Neurosci. 1997;17:3128–3135. doi: 10.1523/JNEUROSCI.17-09-03128.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Siembab VC, Zerda R, O'Donovan MJ, Alvarez FJ. Primary afferent synapses on developing and adult Renshaw cells. J. Neurosci. 2006;26:13297–13310. doi: 10.1523/JNEUROSCI.2945-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Díaz E, Moran LB, Navarrete R. Early alterations in the electrophysiological properties of rat spinal motoneurones following neonatal axotomy. J. Physiol. 2007;582:1141–1161. doi: 10.1113/jphysiol.2007.133488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles GB, Dai Y, Brownstone RM. Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J. Physiol. 2005;566:519–532. doi: 10.1113/jphysiol.2005.086033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes J, Gordon AM, Pandya S, De Vivo DC, Kaufmann P. Clinical outcome measures in spinal muscular atrophy. J. Child Neurol. 2009;24:968–978. doi: 10.1177/0883073809332702. [DOI] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and postsynaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ. Sustained improvement of spinal muscular atrophy mice treated with trichostatinA plus nutrition. Ann. Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hanlon GM, Lowrie MB. The effects of neonatal dorsal root section on the survival and dendritic development of lumbar motoneurons in the rat. Eur. J. Neurosci. 1996;8:1072–1077. doi: 10.1111/j.1460-9568.1996.tb01274.x. [DOI] [PubMed] [Google Scholar]
- Okado N, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. IX. The loss of motoneurons following removal of afferent inputs. J. Neurosci. 1984;4:1639–1652. doi: 10.1523/JNEUROSCI.04-06-01639.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskoui M, Kaufmann P. Spinal muscular atrophy. Neurotherapeutics. 2008;5:499–506. doi: 10.1016/j.nurt.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O'Riordan CR, Klinger KW, Shihabuddin LS, Cheng SH. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TD, Kramer I, Kucera J, Niederkofler V, Jessell TM, Arber S, Snider WD. Peripheral NT3 signaling is required for ETS protein expression and central patterning of proprioceptive sensory afferents. Neuron. 2003;38:403–416. doi: 10.1016/s0896-6273(03)00261-7. [DOI] [PubMed] [Google Scholar]
- Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet. 1978;15:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JP, Mendell LM. Quantitative ultrastructure of Ia boutons in the ventral horn: scaling and positional relationships. J. Neurosci. 1993;13:4748–4763. doi: 10.1523/JNEUROSCI.13-11-04748.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Renault F, Raimbault J, Praud JP, Laget P. Electromyographic study of 50 cases of Werdnig-Hoffmann disease. Rev. Electroencephalogr. Neurophysiol. Clin. 1983;13:301–305. doi: 10.1016/s0370-4475(83)80042-2. [DOI] [PubMed] [Google Scholar]
- Robertson WC, Jr, Kawamura Y, Dyck PJ. Morphometric study of motoneurons in congenital nemaline myopathy and Werdnig-Hoffmann disease. Neurology. 1978;28:1057–1061. doi: 10.1212/wnl.28.10.1057. [DOI] [PubMed] [Google Scholar]
- Rose FF, Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R, Casañas JJ, Torres-Benito L, Cano R, Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz B. Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2005;20:131–140. doi: 10.1016/j.nbd.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Seebach BS, Ziskind-Conhaim L. Formation of transient inappropriate sensorimotor synapses in developing rat spinal cords. J. Neurosci. 1994;14:4520–4528. doi: 10.1523/JNEUROSCI.14-07-04520.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider NA, Mentis GZ, Schustak J, O’Donovan MJ. The importance of muscle spindles and spindle-derived NT3 in the specification and maintenance of synaptic connections between muscle spindle afferents and motor neurons. J. Neurosci. 2009a;29:4719–4735. doi: 10.1523/JNEUROSCI.5790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shneider NA, Brown MN, Smith CA, Pickel J, Alvarez FJ. Gamma motor neurons express distinct genetic markers at birth and require muscle spindle-derived GDNF for postnatal survival. Neural Dev. 2009b;2(4):42. doi: 10.1186/1749-8104-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siembab VC, Smith CA, Zagoraiou L, Berrocal MC, Mentis GZ, Alvarez FJ. Target selection of proprioceptive and motor axon synapses on neonatal V1-derived Ia inhibitory interneurons and Renshaw Cells. J. Comp. Neurol. 2010;518:4675–4701. doi: 10.1002/cne.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Botija C, Ferrer I, Gich I, Baiget M, Tizzano EF. Neuronal death is enhanced and begins during foetal development in type I spinal muscular atrophy spinal cord. Brain. 2002;125:1624–1634. doi: 10.1093/brain/awf155. [DOI] [PubMed] [Google Scholar]
- Soler-Botija C, Cusco I, Caselles L, Lopez E, Baiget M, Tizzano EF. Implication of fetal SMN2 expression in type I SMA pathogenesis: protection or pathological gain of function? J. Neuropathol. Exp. Neurol. 2005;64:215–223. doi: 10.1093/jnen/64.3.215. [DOI] [PubMed] [Google Scholar]
- Soliven B, Maselli RA. Single motor unit H-reflex in motor neuron disorders. Muscle Nerve. 1992;15:656–660. doi: 10.1002/mus.880150604. [DOI] [PubMed] [Google Scholar]
- Sumner CJ. Therapeutics development for spinal muscular atrophy. NeuroRx. 2006;3:235–245. doi: 10.1016/j.nurx.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda KJ, Prior TW, Scott CB, McNaught TP, Wride MC, Reyna SP, Bromberg MB. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgár E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur. J. Neurosci. 2003;17:13–27. doi: 10.1046/j.1460-9568.2003.02406.x. [DOI] [PubMed] [Google Scholar]
- Tourtellotte WG, Milbrandt J. Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat. Genet. 1998;20:87–91. doi: 10.1038/1757. [DOI] [PubMed] [Google Scholar]
- Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- Vezain M, Saugier-Veber P, Melki J, Toutain A, Bieth E, Husson M, Pedespan JM, Viollet L, Pénisson-Besnier I, Fehrenbach S, Bou J, Frébourg T, Tosi M. A sensitive assay for measuring SMN mRNA levels in peripheral blood and in muscle samples of patients affected with spinal muscular atrophy. Eur. J. Hum. Genet. 2007;15:1054–1062. doi: 10.1038/sj.ejhg.5201885. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li LY, Taylor MD, Wright DE, Frank E. Prenatal exposure to elevated NT3 disrupts synaptic selectivity in the spinal cord. J. Neurosci. 2007;27:3686–3694. doi: 10.1523/JNEUROSCI.0197-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li L, Goulding M, Frank E. Early postnatal development of reciprocal Ia inhibition in the murine spinal cord. J. Neurophysiol. 2008;100:185–196. doi: 10.1152/jn.90354.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy: from gene to therapy. Semin. Pediatr. Neurol. 2006;13:121–131. doi: 10.1016/j.spen.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog. Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.