

Abstract
The 8-oxo-guanine (8-oxo-G) lesion is the most abundant and mutagenic oxidative DNA damage existing in the genome. Due to its dual coding nature, 8-oxo-G causes most DNA polymerases to misincorporate adenine. Human Y-family DNA polymerase iota (polι) preferentially incorporates the correct cytosine nucleotide opposite 8-oxo-G. This unique specificity may contribute to polι’s biological role in cellular protection against oxidative stress. However, the structural basis of this preferential cytosine incorporation is currently unknown. Here we present four crystal structures of polι in complex with DNA containing an 8-oxo-G lesion, paired with correct dCTP or incorrect dATP, dGTP, and dTTP nucleotides. An exceptionally narrow polι active site restricts the purine bases in a syn conformation, which prevents the dual coding properties of 8-oxo-G by inhibiting syn/anti conformational equilibrium. More importantly, the 8-oxo-G base in a syn conformation is not mutagenic in polι because its Hoogsteen edge does not form a stable base pair with dATP in the narrow active site. Instead, the syn 8-oxo-G template base forms the most stable replicating base pair with correct dCTP due to its small pyrimidine base size and enhanced hydrogen bonding with the Hoogsteen edge of 8-oxo-G. In combination with site directed mutagenesis, we show that Gln59 in the finger domain specifically interacts with the additional O8 atom of the lesion base, which influences nucleotide selection, enzymatic efficiency, and replication stalling at the lesion site. Our work provides the structural mechanism of high-fidelity 8-oxo-G replication by a human DNA polymerase.
Keywords: DNA replication, Y family polymerase, translesion DNA synthesis, oxidative lesion, DNA mutagenesis
The existence of all animals on our planet depends on oxygen. However, this essential molecule also represents a toxic precursor and a potent mutagen due to its conversion to oxygen radicals by aerobic respiration. Abundant oxygen radicals threaten the well-being of organisms, due to their high reactivity with many biological compounds, in particular, DNA. One of the most abundant and mutagenic oxidative DNA lesions, 7, 8-dihydro-8-oxo-guanine (8-oxo-G), forms when an oxygen radical covalently attaches to the C8 atom of a guanine base (Fig. 1A). Approximately 1,000 of these oxidative DNA lesions arise daily in every cell of our body (1).
Fig. 1.

Activity of 8-oxo-guanine and human Y-family DNA polymerases. (A) Structural changes of guanine to 8-oxo-G. Bracket emphasizes the Hoogsteen edge and gray circles represent sites of modification on 8-oxo-G. The mismatched OG∶dATP base pair is shown on the right. (B) Primer extension assays show differences in nucleotide incorporation by polκ, polη, polι, and polιQ59A mutant for undamaged G (Upper) and 8-oxo-G (Lower) bases. The DNA substrate used for replication assays is shown at the top with G* representing the site of modification. Vertical black arrows indicate nucleotide insertion preference, whereas horizontal black arrows indicate replication stalling. Enzymes were incubated with DNA in the absence of nucleotides (0), presence of all four nucleotides (N), or individual nucleotides (A, T, C, G). (C) Steady-state kinetic parameters for dCTP incorporation opposite undamaged G (dG) and 8-oxo-G by wild-type polι and the polιQ59A mutant. The relative incorporation efficiency between damaged and undamaged DNA is reported as frel = (kcat/Km)damaged/(kcat/Km)undamaged.
The 8-oxo-G lesion is highly mutagenic because of its dual coding properties and the ability of high-fidelity replicative polymerases to replicate through the lesion. Structural studies demonstrate that the 8-oxo-G lesion can adopt two alternate conformations (anti or syn) in the active site of DNA polymerases (2–5). The anti conformation allows correct base pairing with an incoming cytosine (C) nucleotide, whereas the syn conformation forms a stable mispairing with an incoming adenine (A) nucleotide in a normal anti conformation (Fig. 1A). These alternate conformations of 8-oxo-G promote high dA misincorporation rates for most DNA polymerases (6). Consequently, the 8-oxo-G lesion can induce a high frequency of guanine (G) to thymine (T) transversions. Although the mutagenic potential of 8-oxo-G has been well characterized at the structural level, the structural basis of preferential correct C incorporation by eukaryotic DNA polymerases still remains elusive. Given the sheer volume and the dual coding nature of 8-oxo-G, this lesion represents a major hurdle for cells to overcome in maintaining genome integrity.
Organisms evolved repair mechanisms to deal with the abundant 8-oxo-G∶A mispairs produced in the genome. In the base excision repair (BER) pathway, an adenine glycosylase (MutY) removes the mismatched A base, and the subsequent gap can be filled by a specialized polymerase that strongly prefers the correct C incorporation opposite 8-oxo-G (7). Although the identity of this DNA polymerase remains unknown, Y-family DNA polymerases, which specialize in replicating through DNA lesions, likely play a role. Y-family DNA polymerases have a similar domain organization to that of replicative polymerases, consisting of palm, finger, and thumb domains with an additional fourth domain, referred to as the little finger or polymerase associated domain (8). Smaller finger and thumb domains in Y-family polymerases produce a spacious and solvent accessible active site, which enables the accommodation and bypass of bulky and distorted DNA lesions. The process of lesion bypass is referred to as translesion DNA synthesis (TLS).
The human Y-family DNA polymerase iota (polι) appears to play an important role in cellular protection against oxidative stress. Polι is recruited to chromatin after cells are exposed to oxidative DNA damage, and down-regulating polι greatly increases the sensitivity of cells to oxidizing agents (9). In addition, polι displays BER activity in vitro and in vivo and functionally interacts with the BER scaffold protein XRCC1 (9–11). Furthermore, polι displays 5′ deoxyribose phosphate (dRP) lyase activity, a characteristic of BER polymerases (11). Lastly, polι is one of only a few DNA polymerases that preferentially incorporate the correct dC nucleotide opposite 8-oxo-G (12). Other human Y-family polymerases, such as polη and polκ, have dA misincorporation rates of 45–60% (13, 14).
In the present study, we report four crystal structures of polι in complex with DNA containing an 8-oxo-G lesion at the template position, paired with the correct dCTP nucleotide, or incorrect dATP, dTTP, and dGTP nucleotides. Our results demonstrate how a specialized human DNA polymerase, polι, preferentially incorporates the correct C nucleotide opposite an 8-oxo-G lesion, with high specificity over the mismatched A, T, and G nucleotides.
Results and Discussion
Replication Fidelity on 8-Oxo-Guanine .
Primer extension assays were carried out with three different human Y-family polymerases (polκ, polη, and polι) to assess their 8-oxo-G bypass activity and fidelity (Fig. 1B). Opposite undamaged dG, all three polymerases insert the correct dC nucleotide with high specificity (Fig. 1B, vertical arrows). In the presence of 8-oxo-G, only polι maintains a dC incorporation preference over the mismatched nucleotides. Both polκ and polη demonstrate equal or greater A misincorporation rates over C opposite 8-oxo-G with little to no decrease in replication efficiency compared to undamaged DNA (Fig. 1B). In contrast, polι displays significantly reduced replication efficiency opposite 8-oxo-G in spite of preferential dC incorporation (Fig. 1B). Steady-state kinetic analysis of polι reveals a 3.8-fold decrease in dCTP incorporation efficiency opposite 8-oxo-G compared to undamaged G (Fig. 1C). In contrast, steady-state kinetics for polη have revealed no change in replication efficiency between undamaged G and 8-oxo-G templates (15). The replication assay results are consistent with previously published 8-oxo-G fidelities for polι, polκ, and polη (12–14) and indicate that polι is a specialized human Y-family polymerase that replicates the 8-oxo-G base in an error-free manner, albeit with reduced incorporation efficiency.
Polι/8-Oxo-G/dNTP Ternary Complexes.
In order to elucidate how polι selects the correct dC nucleotide opposite 8-oxo-G, we crystallized polι in ternary complex with the 8-oxo-G lesion. The DNA substrate used for crystallization was a self-annealing oligonucleotide, forming a double-stranded DNA substrate with two primer–template junctions (Fig. S1A). Both junctions contain the 8-oxo-G lesion at the first template base position ready for dNTP incorporation. The DNA substrate also contains 2′,3′-dideoxy 3′ ends in order to inhibit DNA polymerization and thus trap ternary complexes for crystallization. The DNA oligo sequence and modification were confirmed by mass spectrometry. The DNA substrate was incubated with polι and cocrystallized with each incoming nucleotide (dCTP, dATP, dTTP, dGTP) separately. The resulting four crystal structures are denoted as OG∶dCTP, OG∶dATP, OG∶dTTP, and OG∶dGTP, according to the incoming nucleotide against the lesion in the active site. All four structures have the same crystal form (space group, P6522) and diffract to 1.95, 2.03, 2.09, and 2.45 Å resolutions, respectively (Table S1).
Polι in all four 8-oxo-G complexes is virtually identical, with rmsd within 0.2 Å between all Cα atoms (Fig. S1B). In addition, the Cα rmsd between the current 8-oxo-G structures and a previously solved polι ternary structure with undamaged G [Protein Data Bank (PDB) ID 2ALZ] (16) is within 0.3 Å (Fig. S1B). This result indicates that polι does not undergo significant conformational changes when replicating through the 8-oxo-G lesion. The significant difference between the current four structures lies in the replicating base pairs. The incoming nucleotides in all four ternary structures are in an active position with the α-phosphates within 4 Å to the putative 3′ end of the primer strands. One active site Mg2+ ion is present in the standard B-site positions of the active sites, whereas the ion density is not well defined for the A sites. Lack of ion coordination at the A site has been previously observed due to ion mobility at this location (8, 17, 18).
Template Base Positioning of 8-Oxo-G.
The 8-oxo-G template base is oriented in a syn conformation opposite the incoming nucleotides in all four structures (Fig. 2A). The syn conformation of 8-oxo-G presents the Hoogsteen edge exclusively for hydrogen bonding. Polι induces syn conformations on template purines due to an exceptionally narrow active site, which restricts the C1′–C1′distance to under 9 Å (16, 17). Accordingly, the C1′–C1′ distance of all four replicating base pairs in the current 8-oxo-G structures is less than 9 Å (Fig. 2A). Compared to undamaged dG in the polι active site, 8-oxo-G is pushed out toward the solvent exposed major groove by approximately 1 Å and tilted approximately 30° off the base stacking plane due to its O8 atom clashing with the OE1 atom of Gln59 from the finger domain (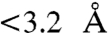 ) (Fig. 2B). The NE2 atom of Gln59 forms a hydrogen bond to the backbone carbonyl oxygen of Gly96 from the interior of the finger domain (Fig. 2B), which contributes to the structural integrity of the domain. Thus, alternate Gln59 conformations are likely unfavorable for the stability of the polι finger domain, which fixes the OE1 atom in its current orientation with a clashing interaction to the 8-oxo-G O8 atom. Consequently, the 8-oxo-G base is shifted away from its optimal hydrogen bonding position with the incoming nucleotides and further crowds the already narrow active site (Fig. 2). Interestingly, the position of the 8-oxo-G base is almost identical to that of undamaged T in the polι active site (PDB ID 3GV7) (Fig. S2A). Undamaged T also shifts into the major groove with tilting due to its exocyclic oxygen group (O2) approaching Gln59 in a similar manner to that of the O8 atom in 8-oxo-G (17). Thus, Gln59 appears to sterically position template bases containing exocyclic groups approaching its OE1 atom. In contrast, template G or A bases are not tilted in polι (19), as these bases lack exocyclic oxygen atoms that approach Gln59. The specific positioning of the 8-oxo-G base by Gln59 appears to contribute to incoming nucleotide stability by influencing hydrogen bonding potential of the lesion base.
) (Fig. 2B). The NE2 atom of Gln59 forms a hydrogen bond to the backbone carbonyl oxygen of Gly96 from the interior of the finger domain (Fig. 2B), which contributes to the structural integrity of the domain. Thus, alternate Gln59 conformations are likely unfavorable for the stability of the polι finger domain, which fixes the OE1 atom in its current orientation with a clashing interaction to the 8-oxo-G O8 atom. Consequently, the 8-oxo-G base is shifted away from its optimal hydrogen bonding position with the incoming nucleotides and further crowds the already narrow active site (Fig. 2). Interestingly, the position of the 8-oxo-G base is almost identical to that of undamaged T in the polι active site (PDB ID 3GV7) (Fig. S2A). Undamaged T also shifts into the major groove with tilting due to its exocyclic oxygen group (O2) approaching Gln59 in a similar manner to that of the O8 atom in 8-oxo-G (17). Thus, Gln59 appears to sterically position template bases containing exocyclic groups approaching its OE1 atom. In contrast, template G or A bases are not tilted in polι (19), as these bases lack exocyclic oxygen atoms that approach Gln59. The specific positioning of the 8-oxo-G base by Gln59 appears to contribute to incoming nucleotide stability by influencing hydrogen bonding potential of the lesion base.
Fig. 2.

Positioning of 8-oxo-G∶dNTP in the Polι/DNA/nucleotide ternary structures. (A) Superposition of replicating base pairs from all four polι/8-oxo-G/nucleotide ternary structures: OG∶dCTP (blue), OG∶dATP (cyan), OG∶dGTP (purple), and OG∶dTTP (pink). The oxygen modification on 8-oxo-G is colored red and the C1′–C1′ distances are indicated with black arrows. Sphere representation is shown for the incoming nucleotides. (B) Positioning (Left) and tilting (Right) of the 8-oxo-G bases by Gln59. The 8-oxo-G bases are superimposed with a previous template G polι structure (PDB ID 2ALZ, gray) with a black arrow indicating repulsion between the OE1 atom of Gln59 and the O8 atom of 8-oxo-G and a dashed line on the NE2 atom representing a hydrogen bond with a backbone carbonyl oxygen. The template bases are numbered 0 and the underlying bases are numbered -1. Gray dashed lines on the right indicate the planes of the guanine bases.
Noticeably, the electron density does not cover the 8-oxo-G base as well as observed in other polymerase structures (2–5, 20) (Fig. 3). The poor density likely results from the crowded active site in which the syn lesion base is squeezed by Gln59 and the incoming dNTP (Fig. 3). Deformation of the density indicates certain structural mobility of the lesion base, which has higher B factors than neighboring bases. The density appears slightly shrunken along the long dimension of the purine base and elongated through the short dimension, according to the syn orientation (Fig. 3, Left), which connects the electron density between the lesion base and the incoming nucleotides (Fig. 3). Alternate conformations of anti/syn 8-oxo-G would account for this deformation of electron density. To reflect this observation, we modeled the majority of the lesion bases (approximately 80%) in the syn conformation and approximately 20% in an anti conformation, which satisfies the electron density better than syn conformations alone (Fig. 3). However, the anti conformation excludes stable binding of incoming dNTPs (Fig. 3, Right) and thus forms nonproductive complexes. This structural observation is consistent with the reduced efficiency observed for polι opposite 8-oxo-G compared to undamaged G (12) (Fig. 1B). The Gln59 clashing with the extra O8 atom of 8-oxo-G makes the syn conformation less stable than that of the undamaged G base in polι and induces the nonproductive 8-oxo-G anti conformation. Accordingly, it has been predicted by molecular simulation work that large major-groove lesions prefer the anti conformation in the polι active site (21). However, the 8-oxo-G lesion is the smallest major-groove adduct with only one atom attachment (O8). Thus, the 8-oxo-G syn conformation can still be accommodated in the polι active site with minor disturbances in stability, which explains why only a small portion of anti conformations are observed in our structures.
Fig. 3.

Replicating base pairs in polι/8-oxo-G ternary structures: (A) OG∶dCTP, (B) OG∶dATP, (C) OG∶dGTP, and (D) OG∶dTTP. For the syn 8-oxo-G structures (Left), double-sided arrows indicate steric repulsion from Gln59 and between crowded bases, and dashed lines indicate hydrogen bonding with bonding and repulsion distances indicated. Divalent metal ions (green) and water molecules (red) are shown as spheres. For the anti conformation structures (Right), the replicating base pairs in syn form are shown in light gray as reference. The replicating base pairs are shown with 2Fo - Fc electron density maps contoured at 1.0σ level.
Replicating Base Pairs.
The nucleotide bases approach the syn 8-oxo-G base in different conformations (Fig. 3). The smaller pyrimidine dCTP and dTTP nucleotides are in anti conformations, whereas the larger purine-based dATP and dGTP are in syn conformations in order to fit in the narrow active site of polι. In the OG∶dCTP structure, the anti dCTP base forms the strongest hydrogen bonding network with the 8-oxo-G Hoogsteen edge among all four replication base pairs (Fig. 3). Two bonds occur between the N3 and N4 atoms of dCTP and the N7 and O6 atoms of the 8-oxo-G base. In addition to hydrogen bonding, the small pyrimidine base of dCTP opposite 8-oxo-G conforms to the narrow polι active site. In contrast, the syn dATP base forms only one hydrogen bond with the 8-oxo-G Hoogsteen edge in the OG∶dATP structure. Thus the dATP nucleotide would be less favorable than dCTP in terms of H-bonding potential. Furthermore, syn purine bases are energetically less favored than the anti conformations in solution (22, 23). Thus, the narrow polι active site forces dATP to adopt an energetically unfavorable conformation, which also increases the entropy cost of incorporation. These effects together enable polι to discriminate against purine–purine mismatched base pairs and favor a less strained purine–pyrimidine pair.
In the OG∶dTTP structure, dTTP has loose H bonding to the lesion template with two potential nonlinear H bonds between its N3 and O4 atoms and the O8 and N7 atoms of 8-oxo-G (Fig. 3C). The dGTP base in OG∶dGTP structure is in an unfavorable syn conformation and forms one hydrogen bond between its O6 atom and the N7 atom of 8-oxo-G (Fig. 3D). Thus, both dTTP and dGTP nucleotides would be less favorable than dCTP for replication opposite 8-oxo-G by polι. Overall, dCTP is the most favorable incoming nucleotide opposite 8-oxo-G in terms of nucleotide conformation and H bonding to the syn lesion base. The structural analysis fits the biochemical observations that dCTP is preferentially incorporated opposite 8-oxo-G by polι.
Glutamine 59 and Polι 8-Oxo-G Activity.
From our structural observations, we postulated that Gln59 from the polι finger domain would influence base-pair selection and bypass efficiency opposite 8-oxo-G by approaching the O8 atom. To test this hypothesis, we mutated polι Gln59 to Ala (Q59A mutant) and assayed the enzyme for incorporation specificity and activity. The Q59A mutant increases dATP misincorporation against 8-oxo-G compared to wild-type polι (Fig. 1B). The increased dA insertion by the Q59A mutant shifts the 8-oxo-G fidelity of polι toward that of polη and polκ. In addition, the bypass efficiency opposite 8-oxo-G has increased for the mutant. Steady-state kinetics demonstrates only a 1.2-fold decrease in dCTP incorporation efficiency when the Q59A mutant replicates opposite the 8-oxo-G lesion (Fig. 1C). Thus, the Q59A mutation has less replication efficiency loss compared to wild-type polι when replicating opposite 8-oxo-G, which is consistent with our structural observations, and which demonstrates the Q59 residue in polι reduces productive 8-oxo-G ternary complexes. It is likely that the Q59A mutation creates a less-restrictive polι active site, which alleviates the 8-oxo-G steric clash. The less-restrictive active site would reduce nonproductive 8-oxo-G anti conformations and possibly allow the accommodation of a mismatched anti dATP nucleotide.
Opposite the undamaged and lesion templates, polι displays replication inhibition as shown by stalling bands in the presence of all four nucleotides (horizontal black arrows) (Fig. 1B). The stalling band in 8-oxo-G DNA is caused by the lesion, and the undamaged DNA stalling is the result of a T base inhibition (17). Interestingly, the stalling bands disappear in the Q59A mutant assays for both undamaged and damaged DNA substrates (horizontal dashed arrows) (Fig. 1B), indicating Gln59 may contribute to stalling opposite 8-oxo-G in a similar manner to T bases. The similarity in replication stalling is consistent with the structural observations that both T and 8-oxo-G bases have an exocyclic oxygen atom clashing with Gln59, causing tilting and projection out of the regular template position (Fig. S2A). The stalling at 8-oxo-G implies that polι may require other DNA polymerases to extend the replication beyond the lesion site in a process known as multiple polymerase translesion replication (24).
Structural Comparison of 8-Oxo-G Replication.
It has been demonstrated that active site flexibility around the template base allows conformational alternation of 8-oxo-G in DNA polymerases with standard active site dimensions (25). This template flexibility allows the syn 8-oxo-G base to form a potent base pair with mismatched anti dATP, causing A misincorporations by virtually all classes of DNA polymerases (2, 4, 26) (Fig. 4A). Such mutagenic base pairing has been observed for Bacillus fragment (BF pol I) and T7 polymerase (T7) from the high-fidelity A family (4, 5), polβ from the X family (26, 27), and polκ and Dpo4 from the Y family (2, 28) (Fig. 4A). However, for polι, this mutagenic syn-OG∶anti-dATP mismatch is too wide to be accommodated in its narrow active site (Fig. 4A). Thus, polι appears to be the only known DNA polymerase that can replicate the 8-oxo-G base in a syn conformation without inducing dA misincorporations.
Fig. 4.

Comparison of 8-oxo-G base pairs in polι and other DNA polymerases. (A) Superposition of the 8-oxo-G∶dA base pair within polι (OG∶dATP, blue) and other DNA polymerases with syn 8-oxo-G (gray). For clarity, only the bases were shown for the dA nucleotides. From left to right, the gray base pairs correspond to pol kappa (PDB ID 3IN5), BF pol I (PDB ID 1U49), and T7 pol (PDB ID 1TK8). The narrow polι C1′–C1′ distance is indicated with black lines on top, whereas the standard C1′–C1′ distance of the other polymerases is indicated with gray lines on bottom. (B) Superposition of the 8-oxo-G∶dC base pair in polι (OG∶dCTP, blue) and other DNA polymerases with anti 8-oxo-G (gray). From left to right, the gray base pairs correspond to Dpo4 (PDB ID 2C2E), BF pol I (PDB ID 1U47), and T7 pol (PDB ID 1TKD).
Interestingly, it has been shown that the 8-oxo-G syn conformation is more favorable than the anti conformation in high-fidelity polymerases due to the O8 atom of anti 8-oxo-G clashing with the DNA backbone phosphates (4, 5). The clashing interaction results in a decreased stability for the correct anti-OG∶anti-dCTP Watson–Crick base pair. In polι, the O8 atom of anti 8-oxo-G also clashes with the DNA backbone. However, due to the crowded nature of the polι active site, the anti 8-oxo-G is mainly destabilized by clashing with the incoming nucleotides instead of the DNA backbone phosphates (Fig. 3, Right). Although the syn 8-oxo-G conformation appears more favorable than anti, most DNA polymerases can replicate the lesion in both syn and anti orientations with standard C1′-C1′ distances (Fig. 4B). In addition, Dpo4 is able to alter the syn/anti equilibrium by specifically interacting with the O8 atom of anti 8-oxo-G (29). In contrast, polι replicates the 8-oxo-G lesion exclusively using the mutagenic Hoogsteen edge (Fig. 4). Remarkably, polι is able to achieve high-fidelity 8-oxo-G replication opposite the Hoogsteen edge by creating a unique base-pairing environment, which stabilizes incoming dCTP and destabilizes incoming dATP.
Implication of Polι Function in the Repair of Oxidative DNA Damage.
Polι has a strong preference for inserting C against the 8-oxo-G lesion compared to other polymerases, which generally have A misincorporation preferences (4). However, polι may also be specialized for functioning over a broad range of oxidative damage rather than being solely specific for 8-oxo-G. Indeed, other BER DNA polymerases such as polλ have also been implicated in accurate 8-oxo-G replication in vivo (30). Interestingly, polι is also associated with another highly abundant oxidative lesion resulting from the oxidative deamination of cytosine: 5-hydroxy-uracil (5-OH-U). Unlike most DNA polymerases that incorporate A opposite the 5-OH-U base, polι incorporates a G nucleotide (31). Polι also preferentially inserts G opposite template T, which structurally resembles the 5-OH-U lesion (17, 32, 33) (Fig. S2B). Thus, similar to 8-oxo-G replication, polι could restore the correct incorporation opposite cytosine lesions during DNA replication or oxidative DNA damage repair.
During DNA replication, a high-frequency of 8-oxo-G bases will be mismatched with dA nucleotides by high-fidelity replicative polymerases. These mutagenic base pairs must be repaired using a specialized form of BER, where excision of the dA mismatch occurs, followed by error-free TLS opposite the 8-oxo-G base. This process allows restoration of the original genomic sequence. Polι likely contributes to this specialized TLS/BER pathway (9), which greatly reduces the mutational burden of 8-oxo-G∶A mismatches. Once the correct sequence has been restored, the 8-oxo-G base can be repaired using the standard BER pathway. In this manner, polι could greatly reduce mutagenesis induced by oxidative damage, which suggests possible roles for this enzyme in cellular protection against oxidizing agents.
Conclusions
This work describes the structural mechanism of preferential C incorporation opposite the highly mutagenic 8-oxo-G lesion by a human DNA polymerase. There are two unique features of the polι active site which allow high-fidelity 8-oxo-G replication: (i) The narrow size fixes purine bases in syn conformations, which destabilizes mismatched A and favors correct C; and (ii) the conserved Gln59 residue from the finger domain positions the syn 8-oxo-G base, which optimizes H bonding for the dCTP nucleotide. The unique polι active site may be specialized for accommodating a variety of mutagenic oxidative lesions and is likely involved in restoring the genomic sequence of mismatched oxidative base pairs via a specialized BER/TLS pathway.
Methods
Protein Preparation.
Polι used for crystallization (amino acids 1–420) was purified as previously described (17). Proteins used for replication assays (polι 1–430, polη 1–445, and polκ 19–523) were cloned from full length cDNA into the “His-parrallel” vector (34), overexpressed in BL21(DE3) cells and purified by nickel affinity and ion exchange chromatography. The Q59A point mutation within polι (1–430) was created by Quickchange mutagenesis using primers A (5′-GACAAACCTTTAGGGGTTCAAGCTAAATATTTGGTGGTTACCTGTAAC-3′) and B (5′-GTTGCAGGTAACCACCAAATATTTAGCTTGAACCCCTAAAGGTTTGTC-3′). The resulting vector was sequenced to confirm the mutation.
DNA Preparation.
The 8-oxo-G substrate used for crystallization was purchased from Keck Oligo, Inc. The purified oligonucleotide containing a 2′,3′-dideoxy 3′ cytosine (Cdd) and an 8-oxo-G (G*) lesion (5′-TCAG∗GGGTCCTAGGACCCdd-3′) was confirmed by mass spectrometry and annealed with itself to give a DNA substrate with two replicative ends. Oligonucleotides used for primer extension assays were purchased from Sigma Aldrich. A 12-nt primer (5′-GGGGGAAGGAC-3′) was annealed to either an 18-nt 8-oxo-G (G*) template (5′-TTCATTG*GTCCTTCCCCC-3′) or an undamaged 18-nt G template (5′-TTCATTGGTCCTTCCCCC-3′). The primer was 5′-end labeled using [γ-32P]ATP and T4 polynucleotide kinase.
Primer Extension Assays.
DNA substrates (10 nM) were incubated with polι, polη, polκ, or polι Q59A (10 nM) and 100 uM of all four dNTPs or individual dNTPs at 37 °C for 2 min. The polι reactions were carried out with 80 uM dNTP. The reaction buffer contained 40 mM Tris (pH 8.0), 5 mM MgCl2, 250 ug/mL BSA, 10 mM DTT, and 2.5% glycerol. Reactions were terminated with loading buffer (95% formamide, 20 mM EDTA, 0.025% xylene, 0.025% bromophenol blue) and resolved on a 20% polyacrylamide gel containing 7 M urea. Gels were visualized using a PhosphorImager.
Steady-State Kinetic Assays.
Standing-start primer extension assays for steady-state kinetic parameters were carried out with 50 nM DNA substrate, 2 nM protein, and various concentrations of dCTP in a range appropriate for single-hit conditions (35). Reactions were performed in replicates of three and were carried out at 37 °C for 2 min and resolved on a 20% polyacrylamide/7M urea gel. Products were quantified using a PhosphorImager and ImageQuant software (Molecular Dynamics). The relationship between the rate of dNTP incorporation (product formation per unit time) and dNTP concentration were fit to the Michaelis–Menten equation and the values for kcat (Vmax normalized to enzyme concentration) and Km were determined using nonlinear regression (GraphPad Prism). The relative incorporation efficiency between damaged and undamaged DNA is reported as frel = (kcat/Km)damaged/(kcat/Km)undamaged.
Crystallization and Structure Determination.
Ternary complexes were formed for OG∶dATP, OG∶dCTP, OG∶dTTP, and OG∶dGTP by incubating protein (0.2 mM) and DNA in a 1∶1.2 ratio with dNTP (2 mM) and MgCl2 (5 mM). Crystals of all four complexes were obtained in 15% PEG 5000 monomethyl ether, 0.2 M NH4SO4, 2.5% glycerol, and 0.1 M MES, pH 6.5. Extensive seeding was performed to increase the diffraction quality of the crystals. Crystals were flash-frozen in liquid nitrogen directly from dehydrated crystallization drops to prevent crystal cracking. X-ray diffraction data were collected on beamlines 24-ID-C and 24-ID-E at the Advanced Photon Source in Argonne National Laboratory. The data were processed and scaled using HKL2000 (36).
All four structures were solved by molecular replacement using PHASER (37) with a previously solved ternary complex (PDB ID 3GV5) as a search model. Structural refinement was performed using PHENIX (38), starting with rigid body refinement, followed by positional and B-factor refinement, and lastly translation/libration/screw motion (TLS) refinement (39). Model building and inspection were done using the program COOT (40). All structures have good stereochemistry with over 95% of the residues in the most favored region of the Ramachandran plot. Figures were created using PyMOL (41).
Supplementary Material
Acknowledgments.
We would like to thank beamline support at 24-ID of Advanced Photon Source in Argonne National Laboratory. This research was funded by the Canadian Institutes of Health Research (Operating Grant MOP 93590 to H.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. W.Y. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB), www.rcsb.org (PDB ID codes 3Q8Q, 3Q8P, 3Q8R, and 3Q8S for the structures OG∶dATP, OG∶dCTP, OG∶dGTP, and OG∶dTTP, respectively).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013909108/-/DCSupplemental.
References
- 1.Collins AR. Oxidative DNA damage, antioxidants, and cancer. Bioessays. 1999;21:238–246. doi: 10.1002/(SICI)1521-1878(199903)21:3<238::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Vasquez-Del Carpio R, et al. Structure of human DNA polymerase kappa inserting dATP opposite an 8-OxoG DNA lesion. PLoS One. 2009;4:e5766. doi: 10.1371/journal.pone.0005766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rechkoblit O, et al. Impact of conformational heterogeneity of OxoG lesions and their pairing partners on bypass fidelity by Y family polymerases. Structure. 2009;17:725–736. doi: 10.1016/j.str.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 5.Brieba LG, et al. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004;23:3452–3461. doi: 10.1038/sj.emboj.7600354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 7.Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine∶8-oxoguanine DNA glycosylase. Nucleic Acids Res. 1999;27:3638–3644. doi: 10.1093/nar/27.18.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 9.Petta TB, et al. Human DNA polymerase iota protects cells against oxidative stress. EMBO J. 2008;27:2883–2895. doi: 10.1038/emboj.2008.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasad R, et al. Localization of the deoxyribose phosphate lyase active site in human DNA polymerase iota by controlled proteolysis. J Biol Chem. 2003;278:29649–29654. doi: 10.1074/jbc.M305399200. [DOI] [PubMed] [Google Scholar]
- 11.Bebenek K, et al. 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science. 2001;291:2156–2159. doi: 10.1126/science.1058386. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Yuan F, Wu X, Taylor JS, Wang Z. Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 2001;29:928–935. doi: 10.1093/nar/29.4.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases delta and eta. Nucleic Acids Res. 2009;37:2830–2840. doi: 10.1093/nar/gkp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maga G, et al. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 16.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Human DNA polymerase iota incorporates dCTP opposite template G via a G.C + Hoogsteen base pair. Structure. 2005;13:1569–1577. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Kirouac KN, Ling H. Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase iota. EMBO J. 2009;28:1644–1654. doi: 10.1038/emboj.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong JH, Fiala KA, Suo Z, Ling H. Snapshots of a Y-family DNA polymerase in replication: Substrate-induced conformational transitions and implications for fidelity of Dpo4. J Mol Biol. 2008;379:317–330. doi: 10.1016/j.jmb.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 19.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-iota active site. Structure. 2006;14:749–755. doi: 10.1016/j.str.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Rechkoblit O, et al. Stepwise translocation of Dpo4 polymerase during error-free bypass of an oxoG lesion. PLoS Biol. 2006;4:e11. doi: 10.1371/journal.pbio.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donny-Clark K, Shapiro R, Broyde S. Accommodation of an N-(deoxyguanosin-8-yl)-2-acetylaminofluorene adduct in the active site of human DNA polymerase iota: Hoogsteen or Watson-Crick base pairing? Biochemistry. 2009;48:7–18. doi: 10.1021/bi801283d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stolarski R, Hagberg CE, Shugar D. Studies on the dynamic syn-anti equilibrium in purine nucleosides and nucleotides with the aid of 1H and 13C NMR spectroscopy. Eur J Biochem. 1984;138:187–192. doi: 10.1111/j.1432-1033.1984.tb07898.x. [DOI] [PubMed] [Google Scholar]
- 23.Salter E, Wierzbicki A, Sperl G, Thompson W. Quantum mechanical study of the Syn and Anti conformations of solvated cyclic GMP. Struct Chem. 2003;14:527–533. [Google Scholar]
- 24.Livneh Z, Ziv O, Shachar S. Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis. Cell Cycle. 2010;9:729–735. doi: 10.4161/cc.9.4.10727. [DOI] [PubMed] [Google Scholar]
- 25.Beard WA, Batra VK, Wilson SH. DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat Res. 2010;703:18–23. doi: 10.1016/j.mrgentox.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH. Structure of DNA polymerase beta with the mutagenic DNA lesion 8-oxodeoxyguanine reveals structural insights into its coding potential. Structure. 2003;11:121–127. doi: 10.1016/s0969-2126(02)00930-9. [DOI] [PubMed] [Google Scholar]
- 27.Batra VK, et al. Mutagenic conformation of 8-oxo-7,8-dihydro-2′-dGTP in the confines of a DNA polymerase active site. Nat Struct Mol Biol. 2010;17:889–890. doi: 10.1038/nsmb.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zang H, et al. Efficient and high fidelity incorporation of dCTP opposite 7,8-dihydro-8-oxodeoxyguanosine by Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2006;281:2358–2372. doi: 10.1074/jbc.M510889200. [DOI] [PubMed] [Google Scholar]
- 29.Eoff RL, Irimia A, Angel KC, Egli M, Guengerich FP. Hydrogen bonding of 7,8-dihydro-8-oxodeoxyguanosine with a charged residue in the little finger domain determines miscoding events in Sulfolobus solfataricus DNA polymerase Dpo4. J Biol Chem. 2007;282:19831–19843. doi: 10.1074/jbc.M702290200. [DOI] [PubMed] [Google Scholar]
- 30.van Loon B, Hubscher U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc Natl Acad Sci USA. 2009;106:18201–18206. doi: 10.1073/pnas.0907280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaisman A, Woodgate R. Unique misinsertion specificity of poliota may decrease the mutagenic potential of deaminated cytosines. EMBO J. 2001;20:6520–6529. doi: 10.1093/emboj/20.22.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tissier A, et al. Misinsertion and bypass of thymine-thymine dimers by human DNA polymerase iota. EMBO J. 2000;19:5259–5266. doi: 10.1093/emboj/19.19.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Yuan F, Wu X, Wang Z. Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase iota. Mol Cell Biol. 2000;20:7099–7108. doi: 10.1128/mcb.20.19.7099-7108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expression Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 35.Creighton S, Bloom LB, Goodman MF. Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.DeLano WL. San Carlos, CA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. Version 1.3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.