

Abstract
Purpose
We are in great need of specific biomarkers to detect pancreatic cancer (PDAC) at an early stage, ideally before invasion. Plectin-1 (Plec1) was recently identified as one such biomarker. However, its suitability as a specific biomarker for human pancreatic cancer, and its usability as an imaging target remain to be assessed.
Experimental Design
Specimens of human PDAC, chronic pancreatitis and normal pancreata were evaluated by IHC and Western blot analysis. To validate Plec1 as an imaging target, Plec1-targeting peptides (tPTP) were used as a contrast agent for single photon emission computed tomography in an orthotopic and liver metastasis murine model of PDAC.
Results
Plec1 expression was noted to be positive in all PDACs but negative in benign tissues. Plec1 expression increases during pancreatic carcinogenesis. It was found to be misexpressed in only 0–3.85% of early PDAC precursor lesions (PanINs-I/II) but in 60% of PanIN III lesions. Plec1 expression was further noted to be retained in all metastatic foci assayed and clearly highlighted these metastatic deposits in lymph nodes, liver and peritoneum. In vivo imaging using tPTP specifically highlighted the primary and metastatic tumors. Biodistribution studies performed after imaging demonstrate that the primary pancreatic tumors and liver metastasis retained 1.9–2.9 fold of tPTP over normal pancreas and 1.7-fold over normal liver.
Conclusions
Plec1 is the first biomarker to identify primary and metastatic PDAC by imaging and may also detect preinvasive PanIN III lesions. Strategies designed to image Plec1 could therefore improve detection and staging.
Keywords: Plectin, biomarker, imaging biomarker, pancreatic cancer, PanIN
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the 4th leading cause of cancer-related deaths in the U.S. and other industrialized countries (1). Despite many efforts, it remains a devastating disease with a 5-year survival rate of less than 5% and a median survival of less than 1 year (1). This grim prognosis is mostly due to the cancer’s aggressive biological behavior with early invasion and metastasis, leading to an initial diagnosis at an advanced incurable stage in more than 80% of patients (2). The only potentially curative treatment is radical surgical resection (3). Prognosis after resection depends on tumor size (<3 cm), lymph node involvement, and status of the resection margin (3).
Early diagnosis of small or even preinvasive cancers before the onset of metastasis is currently the only means to substantially improve resectability, prognosis after resection, and ultimately survival (4). However, all available diagnostic tools and biomarkers for PDAC fail to detect early or preinvasive cancer and suffer from low specificity and sensitivity (5, 6). The only clinically available serum biomarker for PDAC is CA 19–9, which is of limited use (7). Invasive endoscopic procedures (EUS and ERCP) suffer from potential for injury to the pancreas and are highly operator-dependent (5, 6, 8, 9). Cross-sectional abdominal imaging is not reliable enough to allow for screening of high-risk patient populations (10, 11) and fails to detect metastases in up to 30% of patients preoperatively (12, 13). All three screening modalities also cannot safely discriminate PDAC from chronic pancreatitis (CP), which is of particular importance since PDAC can arise in the background of chronic pancreatitis and chronic pancreatitis often mimics PDAC due to their similar clinical signs and symptoms (14, 15). This similarity makes the identification of biomarkers that distinguish PDAC from CP very challenging (16).
PDAC is believed to progress through precursor lesions termed pancreatic intraepithelial neoplasia (PanIN). Early PanIN I and II lesions are frequently observed in normal pancreata and chronic pancreatitis. PanIN III lesions are considered carcinoma in situ (preinvasive cancer) and possess many of the genetic aberrations of invasive cancer. The detection of PanIN III lesions has a direct impact on clinical treatment decisions (17–19). Thus, the ideal biomarker for PDAC should not only differentiate benign conditions (CP) from malignancy, but also be able to detect small cancers, ideally at the pre-invasive PanIN III phase. Novel biomarkers that satisfy these demands have been difficult to identify, and none of the potential candidate biomarkers discovered so far have been able to meet all of the above criteria (20–24). Based on findings in vitro and in a genetically engineered mouse model, Plectin-1 (Plec1) was recently suggested as a biomarker for PDAC (25), but its suitability as a biomarker for primary and metastatic human PDAC and its precursor lesions, as well as its capability to differentiate PDAC from CP, remain to be assessed.
This study demonstrates that Plec1 is not only a biomarker for human invasive and metastatic PDAC, but may also serve as a marker for preinvasive PanIN III lesions. Plec1 also distinguishes malignant pancreatic disease from chronic pancreatitis. In preclinical orthotopic mouse models of PDAC, Plec1 overexpression can be exploited for non-invasive imaging of PDAC and its metastases. This data suggests that Plec1 may indeed be an ideal biomarker for small and preinvasive cancers and that Plec1-targeted imaging of PDAC is feasible. Clinical use of Plec1-based imaging should permit the early diagnosis of small or even preinvasive cancers in addition to metastases, potentially leading to improved resectability rates and survival.
Materials & Methods
Tissue samples
All tissues and biologic samples were collected with the approval and in accordance with the requirements of the Institutional Review Board of the Massachusetts General Hospital, Boston, Massachusetts.
Paraffin-embedded tissue samples were obtained from the files of the Department of Pathology of the Massachusetts General Hospital, Boston, Massachusetts. All specimens had an established diagnosis at the time of assessment. A total of 4 normal pancreata, 15 chronic pancreatitis, 14 PanIN I, 26 PanIN II, 15 PanIN III and 41 PDAC, 8 liver metastasis, 11 lymph node metastasis, 10 with matching primary tumors, and 9 peritoneal metastasis were obtained. For the assessment of Plec1 expression in extra-pancreatic human cancer, a commercial tumor tissue microarray (MTU951, US Biomax, Rockville, MD) was used.
Mice & cell lines
All animal procedures were approved by the University of Virginia Animal Care and Use Committee and the Massachusetts General Hospital Subcommittee on Research Animal Care. Nude mice (nu/nu) were purchased from the National Cancer Institute (Fredericksburg, MD). FVB/NJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were maintained in a germ-free environment and had access to food and water available ad libitum.
The L3.6pl pancreatic cancer cell line was originally derived from a repeated cycle of injecting COLO-357 cells into the pancreas of nude mice, selecting for liver metastases, and re-injecting into the pancreas. AK134 cells were derived from spontaneous PDAC arising in Pft1-Cre; LSL-K-RasG12D; p53 ± mice in the background of an inbred FVB/NJ strain. The Panc1 pancreatic cancer cell line was obtained from ATCC. All cell lines were routinely verified by morphology and growth curve analysis, and tested for Mycoplasma.
Animal models
Three orthotopic mouse models and one mouse model of liver metastasis were employed to assess the suitability of Plec1 as an in vivo imaging biomarker. 1×106 L3.6pl (n=10) or 11×106 Panc1 cells (n=5) in 50 μl Hanks Buffered Sterile Saline (HBSS) were injected into the head of the pancreas of nu/nu mice. 2.5×105 AK134 were injected into the pancreas of FVB/NJ mice (n=8). To obtain liver metastasis, 2.5×105 AK134 cells (n=5) in 50 μL of HBSS were injected into the capsule of the spleen. Seven days (AK134), 10 days (L3.6pl), 4 weeks (Panc1) or 15 days (AK134 liver metastasis) after injection, animals were imaged, sacrificed, and tPTP biodistribution assessed. All animals underwent gross inspection of the abdominal cavity and liver for metastasis. Histology was used to confirm macroscopic findings. As control animals, FVB/NJ (n=2) or nu/nu (n=5) mice were injected with 50 μL of HBSS into the pancreas and spleen and then imaged 1–4 weeks after injection.
Western blot analysis
50 mg of pancreatic tissue obtained as snap-frozen surgical specimens was homogenized in RIPA buffer (50mM Trizma Base, pH 7.4, 1% Triton X-100, 0.25% sodium desoxycholate, 100mM EDTA 150mM NaCl) in combination with a protease inhibitor cocktail (0,001mg/ml aprotinin, bestatin, pepstatin, leupeptin and 0.005 mg/ml 20mM PMSF, Sigma-Aldrich, St. Louis, USA). The lysate was cleared by centrifugation. To ensure equal loading, a highly sensitive and precise copper-based assay was used to determine the protein concentration of each sample (2-D Quant Kit, Amersham Biosciences, NJ). 20 μg protein/lane were separated via SDS-PAGE and transferred onto a nitrocellulose membrane. Equal transfer was verified by Ponceau staining. Antigen detection was performed using a rabbit monoclonal antibody against human Plec1 (Abcam, Cambridge, MA). The secondary antibody was a HRP-coupled goat anti-rabbit polyclonal antibody (Sigma-Aldrich, St. Louis, USA). Bands were visualized with ECL. (control: rat brain lysate, Santa Cruz Biotechnology, La Jolla, CA).
Immunohistochemistry for Plectin-1
Paraffin-embedded sections were deparaffinized, hydrated with TBS and blocked with H2O2. Antigen retrieval was achieved by boiling tissue in Retrievit (BioGenex, San Ramon, CA). After blocking with avidin/biotin (Vector Laboratories, Burlingame, CA) and 5% goat serum in TBS, slides were incubated overnight at 4 C with 1:250 Plec1 antibody (Abcam). Sections were washed three times in TBST, followed by incubation with biotinylated anti-rabbit goat secondary antibody (Vector Laboratories, Burlingame, CA), than developed using DAB (Invitrogen, Carlsbad, CA) and counterstained with hematoxylin. Slides were evaluated using a Y-FL microscope (Nikon, Japan).
Expression of Plec1 in nerves within each slide was used as a staining control and reference for staining intensity. Nerves were noted to have a moderate staining intensity. Staining intensity was recorded by two independent observers, and in case of discrepant results, evaluated by a third observer. Plec1 staining was classified as negative if the staining intensity was weaker than nerves. It was classified as positive if the staining was as least as strong as nerves.
In-vitro competition assay
Tetramericplectin-1-targeted peptide (tPTP-4(βAKTLLPTPGGS(PEG5000))KKKDOTAβA-NH2)) was synthesized in a GMP grade facility (CS Bio Company, Menlo Park, CA). As a control, non-binding tetramer (ncPTP-4(βAKHVMSKQGGS(PEG5000))KKKDOTAβA-NH2)) was also synthesized. For Indium labeling, peptide (100 μg) was dissolved in 20 μl PBS, then diluted in 100 μl ammonium acetate buffer (0.1M, pH 4.5). Indium chloride (5mCi in water; Cardinal Health, VA) was mixed with the peptide and allowed to equilibrate with mixing at 40°C for 15 min. The reaction mixture was purified by size exclusion using a PD10 desalting column pre-equilibrated with DPBS. For in vitro peptide validation experiments, cells were incubated at room temperature for 1 hr with tPTP or ncPTP with concentrations ranging from 10−3 to 10−9 and 5 μCi tPTP-In111 in triplicate. After 1 hr the cells were washed and lysed with 100μL 1M NaOH for 5 min. The mixture was then transferred to tubes and activity analyzed on a gamma counter.
Imaging
Mice were injected with 1 mCi of 111In labeled tPTP, then imaged 4 hours post injection with a microSPECT/CT scanner designed and built at UVa. CT acquisition used 200 evenly spaced projections spanning 200 degrees over approximately 5 minutes. Pinhole SPECT scanning was then performed using two opposing gamma cameras simultaneously. The two cameras were fitted with 0.5 mm diameter tungsten pinholes. 60 evenly spaced projection views per camera were obtained over 180 degrees, for a total of 120 views at 3 degree increments over 360 degrees. The SPECT acquisition time was approximately 45 minutes. The reconstructed CT voxel size was 0.082×0.082×0.082 mm on a 640×640×768 image matrix. The reconstructed SPECT voxel size was 0.65×0.65×0.65 mm on an 80×80×80 image matrix. All SPECT images were corrected for radioactivity decay but not for gamma ray attenuation.
Biodistribution and blood half-life
After mice were imaged via SPECT/CT, animals were sacrificed and their organs harvested and placed into pre-weighed Eppendorf tubes. Each tube was then re-weighed to determine the weight of the organ and the radiation measured. Tubes containing organs were analyzed on a gamma counter. To determine the plasma lifetime of the probe, a mouse injected with the tPTP-Peg-111In was bled 0, 15, 30, 45, 60, and 120 min post injection, and the sample analyzed on a gamma counter. Tissue samples were then placed in histology cassettes and fixed for paraffin embedding. After the radioactivity in the tissue samples decayed, the blocks were sectioned on a microtome and evaluated by H&E.
Results
Plec1 expression intensity and pattern distinguish malignant from benign pancreatic disease
To determine whether Plec1 can be used as a marker for the detection of PDAC, IHC of human tissue sections was performed. Plec1 expression was scored as negative in all benign tissues (4/4 normal pancreata and 15/15 chronic pancreatitis). In contrast 41/41 PDAC stained strongly positive for Plec1 (Figure 1A, B). Similarly, Western blotting of pancreatic tissue lysates detected no Plec1 in normal pancreas or chronic pancreatitis whereas it is present in each lysate from PDAC (Figure 1C). Thus, Plec1 is identified in all PDACs and clearly distinguishes malignant from benign pancreatic disease (Figure 1).
Figure 1. Plectin-1 Immunohistochemistry and Western Blot.
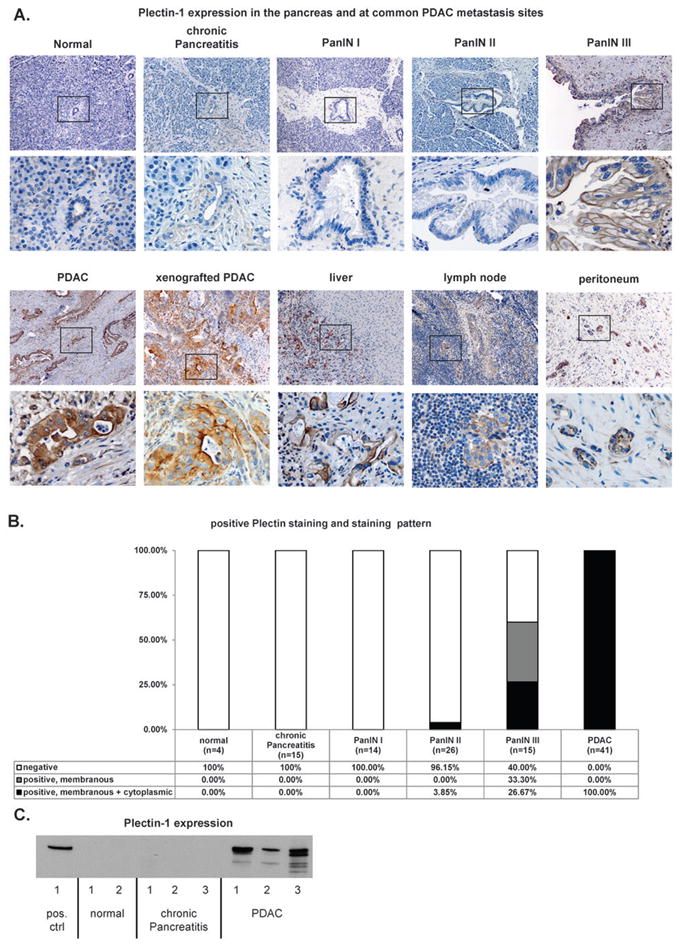
A) Representative images of the evaluated normal pancreata, chronic pancreatitis, PanIN, PDAC, xenografted PDAC and PDAC metastasis sites (liver, lymph node and peritoneum). Overview (upper panel) and detailed view of the black box (lower panel). Chronic pancreatitis and normal pancreas do not express Plectin-1. PanIN III has a membranous staining pattern. PDAC and PDAC xenograft tissue stain moderately to strongly cytoplasmic and membranous for Plectin-1. Common PDAC metastasis sites do not show significant Plectin-1 expression, while the tumor cells stain intensely for Plec1. B) Distribution of staining intensity and staining pattern in the specimens. All PDAC cases were Plec1-positive, whereas normal pancreas and chronic pancreatitis did not express Plectin-1. All PanIN I and most II lesions were Plec1-negative, while the majority of PanIN III lesions were Plec1-positive. The cellular localization of Plec1 also changes during carcinogenesis. The protein is found only in the membrane in 33% of PanIN III lesions, while 27% of PanIN III and all PDAC show membranous and cytoplasmic PLec1 expression. C) Quantitative Western Blot for Plec1 from 50 mg of pancreatic tissue (snap-frozen surgical specimens). No Plec1 was detected in the normal pancreas and CP, while it was present in each PDAC.
To determine whether Plec1 expression changes during carcinogenesis, tissue sections of PDAC precursor lesions, PanINs, were evaluated by IHC. While 0% PanIN I and only 3.85% PanIN II are Plec1-positive, 60% of PanIN III and 100% of invasive PDAC (Figure 1A, B) were Plec1-positive. Plec1 expression thus increases during pancreatic carcinogenesis and discriminates early stage PanIN I and II lesions from PanIN III and PDAC. During carcinogenesis, the cellular localization of Plec1 also changes. Plec1 was identified in the cytoplasm and/or on the cell membrane. While its expression was restricted to the cell membrane in 33% of PanIN III (5/13), a membranous and cytoplasmic expression of Plec1 was observed in 26.67% of PanIN III (4/13) and 100% invasive PDAC (41/41; Figure 1C). Sensitivity and specificity of Plec1 for differentiating PanIN III and PDAC from normal pancreata, chronic pancreatitis and lower grade PanIN lesions were 87% and 98%, respectively. Sensitivity for invasive cancers (PDAC) alone was 100%.
Plec1 is not expressed in most normal human tissue and is retained in PDAC metastasis
IHC of a human tissue microarray revealed that Plec1 is not expressed by most normal tissue, with the exception of the skin and genitourinary tract (Figure 2). Specifically, it is not expressed in the liver, lymph node, lung or peritoneum. PDAC has a propensity to metastasize early to these sites. To evaluate the suitability of Plec1 as a biomarker for metastatic disease, IHC of metastatic deposits was performed. All metastatic foci assayed retained their Plec1 expression, clearly identifying and highlighting metastatic deposits in the liver (8/8), lymph nodes (11/11), and peritoneum (9/9). The 10 lymph node metastases had the same pattern and staining intensity as their matched primary tumor (Figure 1A).
Figure 2. Plectin-1 expression in normal and malignant human tissue: Immunohistochemistry of a tissue microarray evaluated for Plectin-1 expression.

A) Most normal tissue shows only weak Plectin-1 expression. A clear difference in Plectin-1 expression distinguishing normal from malignant disease is observed in the pancreas, esophagus, stomach and lung. Common PDAC metastasis sites (lymph node, liver) do not express Plec1.
Plec1 is an ideal biomarker for detecting pancreatic cancer, but may also be an ideal biomarker for detecting other cancers, such as esophageal, stomach, and lung cancers, where a differential expression between normal and cancerous tissue was also noted in the tissue microarray. The discovery that Plec1 can be used to highlight tumors and their potential metastatic foci suggests that smart imaging agents targeting this marker could be used to improve diagnosis and staging (Figure 2).
Plec1 targeting probes can be used for non-invasive imaging of PDAC
To determine whether Plec1 can be used as an imaging biomarker to facilitate the detection of human PDAC in vivo, we employed Plec1-targeted peptides derived from a phage display screen (25) to synthesize a tetrameric synthetic peptide (tPTP) that functions as a clinically relevant imaging agent for single photon emission computed tomography (SPECT). In vitro validation of the specificity of the tPTP was performed by competition assay with labeled tPTP and nonrelated control PTP (nrPTP). The Ki (inhibition dissociation constant) for tPTP was 8.3 ×10−7 M vs. 2.86 × 10−6 M for nrPTP (Figure 3A). Animals bearing orthotopically injected human pancreatic cancer cells (L3.6pl or Panc1) were administered tPTP and imaged via SPECT/CT 4 hours after tPTP injection. In both L3.6pl and Panc1 animals, imaging illuminated the tumor in the pancreas (Fig. 3B and data not shown). Panc1 orthotopically injected animals did not form metastatic disease. Consistent with this finding, no tPTP uptake was identified outside the pancreas. However, 2 of 10 mice injected with L3.6pl at autopsy were found to have peritoneal metastases. tPTP SPECT/CT imaging was able to accurately detect the peritoneal metastases in these two animals (Figure 3B, L3.6pl). Likewise, in a syngeneic mouse model of PDAC, tPTP was able to highlight the primary tumor and associated metastases in the peritoneum (Figure 3B, AK134). In contrast, only the kidneys were visible in control animals (Figure 3B, null).
Figure 3. In vivo imaging of Plec1 in orthotopic PDAC.

A) In vitro validation of tPTP. L3.6pl cells were plated on a 96-well plate and incubated with 111In-tPTP and increasing log concentrations of unlabeled tPTP or negative control tetramer. B) Mice bearing tumors from orthotopically implanted L3.6pl, AK134 cells and mice without tumors (null) were injected with 111In-tPTP and imaged via SPECT/CT 4 hours post injection. Note the accumulation of tPTP in PDAC, allowing the in vivo imaging of tumor in the pancreas and in peritoneal metastases. Coronal (left) and axial (right) SPECT/CT slices through the tumor are presented. T-tumor, K-kidney, M-peritoneal metastasis. C) After SPECT/CT imaging, animals were sacrificed, organs harvested and gamma counts assessed. Null data is from both nu/nu and FVB/NJ animals that were injected in the pancreas with saline. D) Histology. Animals that had orthotopically implanted tumors or null animals were sacrificed and pancreas and regions of visible peritoneal metastases were removed, embedded, sectioned and stained with H&E (20x image). PT–primary tumor, PM–peritoneal metastasis
In order to validate and quantitate imaging results, biodistribution studies were performed to confirm tumor-specific tPTP accumulation. Biodistribution results demonstrated that the pancreatic tumors from all three cell lines had a statistically significant 1.9- to 2.9-fold higher uptake when compared with pancreata from control animals (Figure 3C, p<0.01). The probe was identified in the kidneys, which are its main route of elimination (Figure 3C). H&E staining and of sectioned pancreas and peritoneal metastases demonstrated the presence of Plec1-expressing tumors in the pancreas and peritoneum (Figure 3D). These data show that Plec1-targeted imaging using tPTP functions as a highly specific imaging tool for PDAC, clearly distinguishing PDAC and its metastases from their adjacent normal tissues.
To determine the ability of tPTP to highlight liver metastases, AK134 cells were used in a well-known model of liver metastases which form after intra-splenic injection. Animals injected with tPTP were imaged 4 hours later, followed by biodistribution measurements. Metastases in the liver were readily identified via tPTP-mediated SPECT/CT imaging (Figure 4A). Biodistribution analysis confirmed the imaging results, with a 1.7-fold increase in tPTP accumulation in livers that had metastases over livers from animals devoid of tumors (p<0.01; Figure 4B). H&E confirmed the presence of metastatic disease in livers which were positive via tPTP-mediated SPECT imaging (Figure 4C). These data show that Plec1-targeted imaging using tPTP functions as a highly specific imaging tool for PDAC, highlighting PDAC and its metastases.
Figure 4. Plec1 imaging allows non-invasive detection of liver metastases.
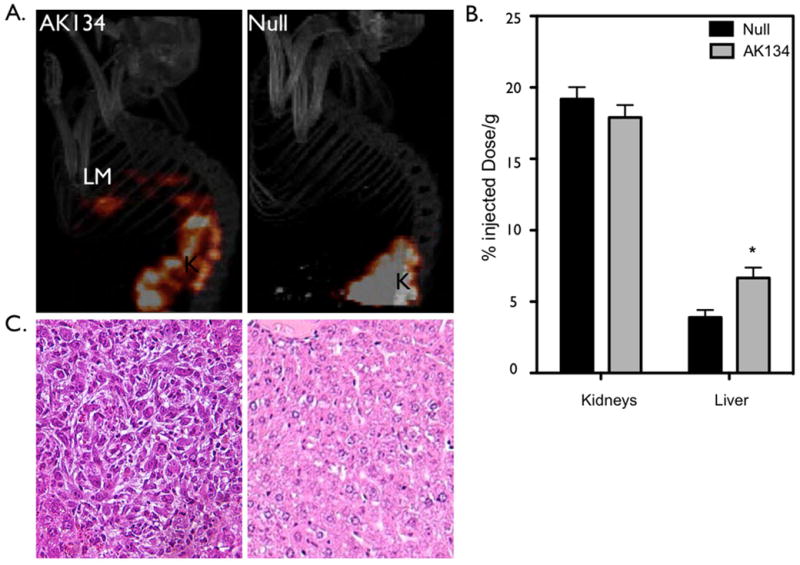
A) AK134 cells, or saline (null), were injected intrasplenically to produce liver metastases. Left panel: Mice bearing liver metastasis (LM) from AK134 injection. Right panel: Null animals without intrasplenic tumor cell injected. K-kidney B) Biodistribution studies were performed on indicated tissues subsequent to imaging experiments. * p<0.01. C) Histology: Left panel: Histologic confirmation of liver metastasis. Right panel: normal liver.
Discussion
In this study we demonstrate that Plec1 expression identifies preinvasive PanIN III lesions as well as primary and metastatic human PDAC. Plec1 expression intensity also clearly discriminates cancers from benign conditions, in particular chronic pancreatitis. In a preclinical orthotopic mouse model of PDAC, Plec1 targeted non-invasive imaging detects primary and metastatic PDAC. Taken together, this data suggests that Plec1 may indeed be an ideal biomarker for PDAC.
The use of Plec1 as a biomarker offers several advantages over current clinical diagnostic tools and markers. Unlike CA 19–9, which lacks sensitivity and specificity (7), Plec1 is a specific biomarker for invasive and preinvasive pancreatic cancer. In contrast to conventional cross-sectional abdominal imaging (10–13), Plec1-based imaging would highlight small, currently unidentifiable metastatic foci preoperatively, thus substantially improving preoperative staging. Due to its ability to potentially highlight preinvasive PDAC, it has the potential to be rapidly developed into present-day screening protocols for high risk patients with the promise of detecting PDAC prior to invasion.
Due to its unique properties, the use of Plec1 as a biomarker for preinvasive and invasive PDAC also holds promise to be superior to other recently described biomarkers. Lactose-binding protein, which is overexpressed in acinar cells that surround PDAC, is ideal to detect small cancers within the pancreas, but cannot detect small metastases (24). Neutrophil gelatinase-associated lipocalin delineates PanIN lesions and some invasive cancers, but does not identify poorly differentiated adenocarcinoma (23). MUC1 and MUC4 are overexpressed in a subset of invasive adenocarcinoma, but their expression does not distinguish early PanIN I/II from preinvasive (PanIN III) and invasive cancers. MUC4 is also aberrantly expressed in chronic pancreatitis (20, 21). In contrast, Plec1 detects all PDAC and their small metastatic foci as well as the majority of preinvasive cancers (PanIN III) in addition to discriminating PDAC from chronic pancreatitis.
To date, the role of Plec1 overexpression in pancreatic cancer is unknown. Plec1 itself is a cytolinker protein of the plakin family. Plakins connect intermediate filaments to desmosomes and hemidesmosomes, stabilize cells mechanically, regulate cytoskeleton dynamics, and serve as a scaffolding platform for signaling molecules. Plakins were first described as essential for skin and skeletal muscle integrity (26) and mutations of the Plec1 gene were therefore initially identified in skin disease, such as epidermolysis bullosa (27, 28). A recent study, in which Plec1 was found to interact with the breast cancer susceptibility gene 2 (BRCA2) (29), provides more insight into the protein’s potential role in cancer. BRCA2 mutations are associated with an increased risk of pancreatic cancer (30, 31). BRCA2 itself plays an important role in DNA damage repair and is mainly found in the cell nucleus, but has also been identified in the centrosome. The Plec1/BRCA2 interaction is involved in the regulation of centrosome localization and Plec1 misexpression leads to displacement of the centrosome. This may contribute to genomic instability and therefore cancer development (29). We found that Plec1 expression is acquired during the transition from PanIN II to PanIN III. As lesions progress, Plec1 is not retained at the cell membrane, but rather is ubiquitously expressed in the cancer cells. It thus appears that Plec1 overexpression and cytoplasmic localization begin at the stage of PanIN III and further increase as lesions progress to invasive cancer.
In summary, Plec1 may be the best novel biomarker for PDAC identified to date. Although further studies are needed to verify Plec1 as a target in humans, strategies designed to image Plec1 could substantially improve detection and staging, thus contributing to improved resectability, prognosis and ultimately survival in pancreatic cancer.
Clinical relevance.
Specific biomarkers for the detection of pancreatic cancer (PDAC) at an early or preinvasive stage are currently unavailable. Here we report on a pancreatic cancer biomarker, Plectin-1, that distinguishes PDAC from benign inflammatory diseases such as chronic pancreatitis. Plectin-1 is identified in 100% of tested PDAC tumors and 60% of preinvasive PanIN III lesions, and is retained in metastatic deposits; characteristics needed for an ideal imaging biomarker. In vivo imaging in orthotopic and liver metastases models of pancreatic cancer using a Plec1 targeted imaging agent for single photon emission/CT resulted in enhanced detection not only of the primary tumor but also of small peritoneal and liver metastases. This data suggests that Plectin-1 is a specific novel imaging biomarker for PDAC. Although further translational studies in humans will be needed, this study shows that it can be incorporated into present day imaging technologies used in humans and that this target may be used for improved early detection and staging.
Acknowledgments
We would like to gratefully acknowledge Nabeel Bardeesy for giving us the AK134 cells and the syngeneic mice. We also thank Sanford Feldman for help with the animals, Sara Adair and Dustin Walters for their help with the orthotopic tumor implantation, and Nancy Neyhard, Fred Reynolds, and Marc Seaman for technical assistance.
Abbreviations
- Plec1
Plectin-1
- tPTP
tetrameric Plectin1 targeting peptide
- SPECT
single photon emission computed tomography
- PDAC
pancreatic ductal adenocarcinoma
- PanIN
pancreatic intraepithelial neoplasia
- CP
chronic pancreatitis
- H&E
hematoxylin–eosin
- IHC
immunohistochemistry
- id
injected dose
- EUS
endoscopic ultrasound
- ERCP
endoscopic retrograde cholangiopancreatography
Footnotes
Financial Disclosure
Johnson & Johnson Corporate Office of Science and Technology, Wallace H. Coulter Foundation, National Institutes of Health (NIH) RO1 CA137071, American Association for Cancer Research (AACR) AACR-PAN-CAN, Lustgarten Foundation (K.A. Kelly)
National Institutes of Health (NIH) P01 5P01CA117969, 1P50CA127003 (S.P. Thayer)
Karin Grunebaum Cancer Research Foundation (D. Bausch).
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Koorstra JB, Hustinx SR, Offerhaus GJ, Maitra A. Pancreatic carcinogenesis. Pancreatology. 2008;8:110–25. doi: 10.1159/000123838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeo CJ, Cameron JL, Sohn TA, Lillemoe KD, Pitt HA, Talamini MA, et al. Six hundred fifty consecutive pancreaticoduodenectomies in the 1990s: pathology, complications, and outcomes. Annals of Surgery. 1997;226:248–57. doi: 10.1097/00000658-199709000-00004. discussion 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furukawa H, Okada S, Saisho H, Ariyama J, Karasawa E, Nakaizumi A, et al. Clinicopathologic features of small pancreatic adenocarcinoma. A collective study. Cancer. 1996;78:986–90. doi: 10.1002/(SICI)1097-0142(19960901)78:5<986::AID-CNCR7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 5.Krishna NB, Mehra M, Reddy AV, Agarwal B. EUS/EUS-FNA for suspected pancreatic cancer: influence of chronic pancreatitis and clinical presentation with or without obstructive jaundice on performance characteristics. Gastrointestinal Endoscopy. 2009;70:70–9. doi: 10.1016/j.gie.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 6.Varadarajulu S, Tamhane A, Eloubeidi MA. Yield of EUS-guided FNA of pancreatic masses in the presence or the absence of chronic pancreatitis. Gastrointestinal Endoscopy. 2005;62:728–36. doi: 10.1016/j.gie.2005.06.051. quiz 51, 53. [DOI] [PubMed] [Google Scholar]
- 7.Goggins M. Identifying molecular markers for the early detection of pancreatic neoplasia. Seminars in Oncology. 2007;34:303–10. doi: 10.1053/j.seminoncol.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad NA, Kochman ML, Brensinger C, Brugge WR, Faigel DO, Gress FG, et al. Interobserver agreement among endosonographers for the diagnosis of neoplastic versus non-neoplastic pancreatic cystic lesions. Gastrointestinal endoscopy. 2003;58:59–64. doi: 10.1067/mge.2003.298. [DOI] [PubMed] [Google Scholar]
- 9.Meining A, Rosch T, Wolf A, Lorenz R, Allescher HD, Kauer W, et al. High interobserver variability in endosonographic staging of upper gastrointestinal cancers. Zeitschrift fur Gastroenterologie. 2003;41:391–4. doi: 10.1055/s-2003-39422. [DOI] [PubMed] [Google Scholar]
- 10.Chari ST. Detecting early pancreatic cancer: problems and prospects. Seminars in Oncology. 2007;34:284–94. doi: 10.1053/j.seminoncol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelaez-Luna M, Takahashi N, Fletcher JG, Chari ST. Resectability of presymptomatic pancreatic cancer and its relationship to onset of diabetes: a retrospective review of CT scans and fasting glucose values prior to diagnosis. American Journal of Gastroenterology. 2007;102:2157–63. doi: 10.1111/j.1572-0241.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-del Castillo C, Rattner DW, Warshaw AL. Further experience with laparoscopy and peritoneal cytology in the staging of pancreatic cancer. Br J Surg. 1995;82:1127–9. doi: 10.1002/bjs.1800820839. [DOI] [PubMed] [Google Scholar]
- 13.John TG, Greig JD, Carter DC, Garden OJ. Carcinoma of the pancreatic head and periampullary region. Tumor staging with laparoscopy and laparoscopic ultrasonography. Ann Surg. 1995;221:156–64. doi: 10.1097/00000658-199502000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boll DT, Merkle EM. Differentiating a chronic hyperplastic mass from pancreatic cancer: a challenge remaining in multidetector CT of the pancreas. European Radiology. 2003;13 (Suppl 5):M42–9. doi: 10.1007/s00330-003-2100-8. [DOI] [PubMed] [Google Scholar]
- 15.Oto A, Eltorky MA, Dave A, Ernst RD, Chen K, Rampy B, et al. Mimicks of pancreatic malignancy in patients with chronic pancreatitis: correlation of computed tomography imaging features with histopathologic findings. Current problems in diagnostic radiology. 2006;35:199–205. doi: 10.1067/j.cpradiol.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Kloppel G, Adsay NV. Chronic pancreatitis and the differential diagnosis versus pancreatic cancer. Archives of Pathology & Laboratory Medicine. 2009;133:382–7. doi: 10.5858/133.3.382. [DOI] [PubMed] [Google Scholar]
- 17.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–72. [PubMed] [Google Scholar]
- 18.Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Advances in Anatomic Pathology. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 19.Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. American Journal of Surgical Pathology. 2004;28:977–87. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 20.Swartz MJ, Batra SK, Varshney GC, Hollingsworth MA, Yeo CJ, Cameron JL, et al. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am J Clin Pathol. 2002;117:791–6. doi: 10.1309/7Y7N-M1WM-R0YK-M2VA. [DOI] [PubMed] [Google Scholar]
- 21.Gold DV, Karanjawala Z, Modrak DE, Goldenberg DM, Hruban RH. PAM4-reactive MUC1 is a biomarker for early pancreatic adenocarcinoma. Clin Cancer Res. 2007;13:7380–7. doi: 10.1158/1078-0432.CCR-07-1488. [DOI] [PubMed] [Google Scholar]
- 22.Karanjawala ZE, Illei PB, Ashfaq R, Infante JR, Murphy K, Pandey A, et al. New markers of pancreatic cancer identified through differential gene expression analyses: claudin 18 and annexin A8. American Journal of Surgical Pathology. 2008;32:188–96. doi: 10.1097/PAS.0b013e31815701f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, et al. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540–7. doi: 10.1038/sj.bjc.6604329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flores LG, Bertolini S, Yeh HH, Young D, Mukhopadhyay U, Pal A, et al. Detection of pancreatic carcinomas by imaging lactose-binding protein expression in peritumoral pancreas using [18F]fluoroethyl-deoxylactose PET/CT. PloS One. 2009;4:e7977. doi: 10.1371/journal.pone.0007977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly KA, Bardeesy N, Anbazhagan R, Gurumurthy S, Berger J, Alencar H, et al. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS medicine. 2008;5:e85. doi: 10.1371/journal.pmed.0050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonnenberg A, Liem RK. Plakins in development and disease. Experimental Cell Research. 2007;313:2189–203. doi: 10.1016/j.yexcr.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 27.Pfendner E, Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. Journal of Investigative Dermatology. 2005;124:111–5. doi: 10.1111/j.0022-202X.2004.23564.x. [DOI] [PubMed] [Google Scholar]
- 28.Pulkkinen L, Smith FJ, Shimizu H, Murata S, Yaoita H, Hachisuka H, et al. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Human Molecular Genetics. 1996;5:1539–46. doi: 10.1093/hmg/5.10.1539. [DOI] [PubMed] [Google Scholar]
- 29.Niwa T, Saito H, Imajoh-Ohmi S, Kaminishi M, Seto Y, Miki Y, et al. BRCA2 interacts with the cytoskeletal linker protein plectin to form a complex controlling centrosome localization. Cancer Science. 2009 doi: 10.1111/j.1349-7006.2009.01282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Research. 1996;56:5360–4. [PubMed] [Google Scholar]
- 31.Naderi A, Couch FJ. BRCA2 and pancreatic cancer. International Journal of Gastrointestinal Cancer. 2002;31:99–106. doi: 10.1385/IJGC:31:1-3:99. [DOI] [PubMed] [Google Scholar]