

Abstract
Myelination is one of the fundamental biological processes in the development of vertebrate nervous system. Disturbance of myelination is found to be associated with progression in many neurological diseases such as multiple sclerosis. Tremendous efforts have been made to develop novel therapeutic agents that prevent demyelination and/or promote remyelination. These efforts need to be accompanied by the development of imaging tools that permit direct quantification of myelination in vivo. In this work, we describe a novel near-infrared fluorescence imaging technique that is capable of direct quantification of myelination in vivo. This technique is developed based on a near-infrared fluorescent probe, 3,3′-diethylthiatricarbocyanine iodide (DBT) that readily enters the brain and specifically binds to myelinated fibers. In vivo imaging studies were first conducted in two animal models of hypermyelination and hypomyelination followed by longitudinal studies in the cuprizone-induced demyelination/remyelination mouse model. Quantitative analysis suggests that DBT is a sensitive and specific imaging probe of myelination, which complements other current myelin-imaging modalities and is of low cost.
Introduction
Molecular imaging has entered a new era in which different imaging modalities have become available for in vivo studies of same disease-defining targets (Guy and Ffytche, 2005; Rudin, 2005). These imaging modalities includes positron emission tomography (PET), single photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and optical imaging, which complement each other in many ways (Fowler and Wolf, 1997; Caravan et al., 1999; Phelps, 2000; Merbach and Toth, 2001; Lowe, 2002; Bottrill et al., 2006; Caravan, 2006; Frullano and Meade, 2007). For example, PET provides high sensitivity but relative low resolution while MRI provides high resolution but relatively low sensitivity. PET provides high quantitative capacity compared with SPECT, but production of PET radiotracers is limited as it requires a cyclotron to produce positron-emitting carbon-11 or fluorine-18. Production of SPECT radiotracers is more affordable as it uses generators that are more readily available. PET, SPECT, and MRI are the primarily modalities in clinical settings. Optical imaging modalities such as bioluminescence imaging and fluorescent molecular tomography are primarily modalities in preclinical settings (Martin et al., 2008; Tan and Jiang, 2008; Weissleder and Pittet, 2008). Availability of different yet complementary modalities for imaging the same targets provides physicians and researchers a “tool box” with high flexibility. Multimodal imaging studies also permit cross-referencing to directly validate results (Jennings and Long, 2009; Lee and Chen, 2009).
One of the key targets that is ideal for multimodal imaging is myelin membranes. In the vertebrate nervous system, myelination is one of the fundamental biological processes that forms segmented sheaths around large-caliber axons to provide insulation and protection of neurons (Hildebrand et al., 1993; Morell and Quarles, 1999). Destruction or changes of myelination have been considered a causative event in numerous neurological diseases such as multiple sclerosis. We have developed a series of myelin-imaging agents (Stankoff et al., 2006; Wu et al., 2006, 2008; Wang et al., 2009) for PET studies. After radiolabeling, these agents were used as radiotracers for in vivo PET studies that were performed in various animal models. Our studies show that the PET modality not only enhances the specificity of myelin imaging but also allows for longitudinal studies of myelin changes (Wang et al., 2009).
In the course of our studies, we have also developed a series of myelin-imaging agents with different fluorescent properties. One type of these agents fluoresces in the near-infrared range with excitation and emission wavelengths >650 nm. We thus set out to explore the potential of these near-infrared agents for in vivo optical imaging of myelination. For this purpose, we thoroughly studied the myelin binding properties of 3,3′-diethylthiatricarbocyanine iodide (DBT). DBT readily enters the brain and selectively binds to myelin fibers. The imaging sensitivity and specificity based on this probe were demonstrated in two animal models with different myelin loads. Complementing the existing myelin imaging modalities, the near-infrared fluorescence (NIRF) imaging can be used as a powerful tool for in vivo studies in the preclinical screening and development of drugs.
Materials and Methods
Animal preparation.
SWR/J mice were obtained from Harlan Laboratory. C57BL/6 mice and C3Fe.SWV-Mbpshi/J shiverer mice were obtained from Jackson Laboratory, and the Plp-Akt-DD transgenic mice were generated as described previously (Flores et al., 2008; Narayanan et al., 2009). Briefly, the transgenic mice expressing constitutively active Akt (HAAkt308D473D, Akt-DD) driven by the Plp promoter (Wight et al., 1993) were generated and used as an hypermyelinated animal model. Akt cDNA was inserted into the AscI/PacI sites of the modified Plp promoter cassette, and hypermyelination was induced after the Plp promoter/Akt-DD insert was injected into SJL/SWR F1 mice. Positive founders were identified by PCR amplification of tail DNA using IntronSV40F (5′-GCAGTGGACCACGGTCAT-3′) and Akt lower (5′-CTGGCAACTAGAAGGCACAG-3′) primer sequences. Analyses were done from litter-matched mice in all developmental experiments, and where possible with older animals. In all three lines, Plp-Akt-DD9 was used for these studies. The cuprizone mouse model of demyelination was induced by feeding 6-week-old female C57BL/6 mice with a diet of milled mouse chow containing 0.2% of the copper chelator, cuprizone (Sigma-Aldrich) for 6 weeks for maximal demyelination. For remyelination, the mice were then fed normal milled mouse chow.
Immunohistochemistry.
For immunohistochemistry with free-floating sections, mice were deeply anesthetized with isoflurane and perfused with PBS followed by 4% paraformaldehyde (PFA) in PBS via the ascending aorta. Brains were excised, incubated for 24 h in 4% paraformaldehyde at 4°C, cryoprotected and sectioned (30 μm) with a sliding microtome. Free-floating sections were rinsed in 1× PBS (two times, 5 min), then incubated in 1% H2O2/10% Triton X-100, then washed with 1× PBS (three times, 5 min). After incubation with 3% normal goat serum for 60 min, the free-floating sections were immunostained overnight at 4°C with rabbit anti-MBP antibody (Millipore Bioscience Research Reagents) 1:2000 dilution in 3% normal goat serum in 1× PBS, which was followed by 1 h incubation at room temperature with IRDye 800CW goat anti-rabbit (LI-COR Biosciences) 1:5000 dilution, then rinsed in 1× PBS (two times, 5 min). The stained sections were covered with fluorescence mounting medium (Vectashield, Vector Laboratories). Images of the stained mouse brain sections were acquired on a LI-COR Odyssey infrared imaging system.
For immunohistochemistry with fresh frozen sections, mice were deeply anesthetized with isoflurane and perfused with 1× PBS followed by 4% paraformaldehyde in PBS. The brains were removed and fixed in 4% PFA overnight and then in 30% sucrose solution. The brains were then sectioned at 20 μm. After incubation with 3% normal goat serum in 1× PBS for 60 min, the fresh frozen sections were immunostained overnight at 4°C with purified mouse antibody monoclonal SMI99 (Covance) 1:500 dilution in 3% normal goat serum in 1× PBS, followed by 1 h incubation at room temperature with Fluorescein (FITC) AffiniPure goat anti-mouse IgG (H+L) (Jackson ImmunoResearch Laboratories) 1:500 dilution, then rinsed in 1× PBS (two times, 5 min). The stained sections were covered with fluorescence mounting medium (Vectashield, Vector Laboratories). Images of the stained mouse brain sections were acquired on a Leica DM5000 inverted microscope (L5 filter, BP 527/30).
For immunohistochemistry with neuronal marker, free-floating sections were incubated in 1% H2O2/10% Triton X-100, washed with 1× PBS (three times, 5 min), then immunostained overnight at 4°C with mouse anti-MAP2 (microtubule-associated protein 2) antibody (Sigma-Aldrich), 1:800 dilution in 3% normal goat serum in 1× PBS, followed by 1 h incubation at room temperature with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) 1:1000 dilution in 3% normal goat serum/1× PBS, then rinsed in 1× PBS (two times, 5 min). The stained sections were covered with fluorescence mounting medium (Vectashield, Vector Laboratories). Images of the stained mouse brain sections were acquired on a Leica DM5000 inverted microscope (L5 filter, BP 527/30).
DBT staining.
For DBT staining with free-floating sections, mice were deeply anesthetized with isoflurane and perfused with PBS followed by 4% paraformaldehyde in PBS via the ascending aorta. Brains were excised, incubated for 24 h in 4% paraformaldehyde at 4°C, cryoprotected and sectioned (30 μm) with a sliding microtome. Free-floating sections were incubated in 1% H2O2/Triton X-100 for 10 min, incubated in a solution of DBT (1 μm) in 1% DMSO/PBS for 30 min at room temperature, then washed three times with 1× PBS before coverslipping with fluorescence mounting medium (Vectashield, Vector Laboratories). Images were acquired on a LI-COR Odyssey infrared imaging system (LI-COR Biosciences).
For DBT staining with fresh frozen sections, mice were deeply anesthetized with isoflurane and perfused with 1× PBS followed by 4% paraformaldehyde in PBS. The brains were excised and fixed in 4% PFA overnight and then in 30% sucrose solution. The brains were then sectioned at 20 μm and fresh frozen sections with 20 μm thickness were incubated in 0.1% Triton X-100 in 1× PBS for 10 min, which was followed by incubation in a solution of DBT (1 mm) in 1% DMSO/1× PBS for 30 min at room temperature. The fresh frozen sections were then washed three times for 5 min each with 1× PBS before coverslipping with fluorescence mounting medium (Vectashield, Vector Laboratories). Images of the stained mouse brain sections were acquired on a Leica DMI6000 inverted microscope (Y5 filter, BP 700/75).
Coregistration of fluorescent and x-ray images.
Images were acquired with a KODAK In-Vivo Multispectral System FX (Carestream Health, Rochester, NY). The fur of the mouse was shaved to avoid autofluorescence artifacts. Twenty minutes after the intravenous injection of DBT (0.1 mg/kg), the mouse was anesthetized using isoflurane gas. The mouse was positioned on the center of the platen and imaged via fluorescence (excitation 760 nm, emission 790 nm) and x ray (vertical resolution, 885 ppi; horizontal resolution, 885 ppi).
Near-infrared fluorescence in vivo imaging.
The in vivo near-infrared fluorescence imaging was performed using a Xenogen IVIS Imaging System 200 (Caliper Life Sciences). To avoid the influence of autofluorescence and light scattering due to the fur, all the mice were shaved before the experiment. After the intravenous injection of DBT 0.1 mg/kg, the mouse was anesthetized using isoflurane gas, The mouse was then positioned on the center of the stage (working temperature 20−40°C) and near-infrared images were acquired by back-thinned, back-illuminated grade 1 CCD, which was thermoelectrically cooled to −90°C to ensure a low dark current and low noise. The fluorescent filter was set for excitation at 705–780 nm and for emission from 810 to 885 nm. The experiments were controlled with a Living Image 3.1 (Caliper Life Sciences) (lens, f/2; field of view, 6.5 × 6.5 cm; imaging pixels: 2048 × 2048; exposure time, 0.5 s for all of the images).
Quantification and statistical analysis.
The near-infrared images were evaluated with a Living Image 3.1 from Caliper Life Sciences. All the images used photon as the unit for the quantification, and the images were shown as an overlaid photograph with fluorescence. Statistical analysis was performed using a two-tailed t test with GraphPad Prism 5.
Results
Fluorescent properties of DBT
DBT is a highly conjugated dibenzothiazol derivative with a structure shown in Figure 1A. The fluorescent excitation and emission spectra of DBT were recorded using Varian fluorescent spectroscopy at a concentration of 100 nm in DMSO. As shown in Figure 1B, the excitation and emission maxima of DBT are 782 and 797 nm, respectively, which are located in the near-infrared region (650 nm-900 nm). Previous studies showed that the extinction coefficient of DBT at 780 nm is 43,000 (m cm)−1. The quantum yield of DBT is 0.034 with a lifetime of 1.18 ns (Sevick-Muraca et al., 1997).
Figure 1.

A, The structure of DBT. B, The excitation and emission spectra of DBT (100 nm in DMSO). Excitation spectra: emission at 797 nm (range 600–850 nm), maximal excitation wavelength at 782 nm. Emission spectra: excitation at 782 nm (range 600–850 nm), maximal emission wavelength at 797 nm. C, In vitro DBT staining of free-floating brain tissue sections of wild-type mice (1 μm). D, In vitro immunohistochemical staining of free-floating brain tissue sections of wild-type mice with MBP antibody.
In vitro binding studies
The myelin-binding property of DBT was first examined by fluorescent tissue staining of wild-type mouse brain sections. For comparison, immunohistochemical staining with myelin-specific MBP antibody was also conducted. At 1 μm concentration, DBT selectively labeled intact myelin sheaths present in the whole mouse brain, particularly in the corpus callosum and the caudate putamen (Fig. 1C). The pattern of myelin sheaths stained by DBT was virtually identical to the pattern stained by MBP antibody (Fig. 1D). These observations indicated that DBT binds specifically to myelin sheaths in vitro.
To determine the sensitivity of DBT binding to myelin, we examined tissue staining by DBT in different brain regions such as caudate putamen, cerebellum, and frontal cortex (Fig. 2, left panels). These regions were selected to represent different forms of myelin fibers in the brain. In the caudate putamen, DBT selectively stains compact myelin fibers. In the cerebellum, DBT stains both myelin tracks and surrounding granule layers. In the frontal cortex, DBT can also stain the small myelin fibers that are primarily located in the gray matter regions. These studies suggested that DBT not only binds to myelin fibers present in the white matter, but also to small bundles of myelin present in the gray matter.
Figure 2.

In vitro DBT staining of myelinated regions (left panels) and subsequent costaining of MBP antibodies (middle panels). A, DBT staining of compact myelin fibers present in caudate–putamen, which significantly blocks antibody staining of MBP in the same section. B, DBT staining of small myelin fibers present in frontal cortex, which also significantly blocks antibody staining of MBP in the same section. C, DBT staining of both myelin tracts and granule layers present in cerebellum, which significantly blocks antibody staining of MBP in the same section. Inhibition of immunohistochemical staining by DBT indicates that DBT stains myelin through specific binding to MBP. Scale bars: A, B, 50 μm; C, 200 μm.
To determine the exact binding sites of DBT, we compared both DBT and MBP staining conducted in the same brain tissue sections. Each section was first subjected to DBT staining followed by antibody staining of MBP (Fig. 2, middle panels). The antibody staining was significantly blocked by DBT in different regions including caudate putamen (Fig. 2A), cerebellum (Fig. 2B), and frontal cortex (Fig. 2C).
The results indicate that DBT binds specifically to MBP present in the CNS myelin.
As a further validation of the system, we conducted tissue staining of DBT in the hypermyelinated Akt and hypomyelinated shiverer mouse brains in comparison with wild-type mouse brains. The myelin staining of the corpus callosum region in each model is shown in Figure 3A–F. Compared with the corpus callosum present in the wild-type mouse brain (Fig. 3A,B), DBT staining of the same region showed significant myelin enrichment in the hypermyelinated Akt mouse brain (Fig. 3C,D) and diminishment in the shiverer mouse brain (Fig. 3E,F), which is consistent with their previously defined pathological findings.
Figure 3.

A–F, In vitro DBT staining and MBP antibody staining of corpus callosum in mouse brain tissue sections of wild-type (A, B), hypermyelinated Akt mouse mutant (C, D), and hypomyelinated shiverer mouse mutants (E, F). G–I, In vitro double staining of the corpus callosum and subcortical gray matter using MAP2 antibody (H) and DBT (G), with the merged images at 20× shown in I. J–L, Examination of the gray matter at high magnification (40×) to identify subcortical myelinated structures by DBT staining (J) and individual neurons (arrows) by MAP2 staining (K). Lack of DBT staining on some individual neurons is highlighted by arrows with the merged images shown in L. Scale bars: A–F, 300 μm; G–L, 50 μm.
To examine whether DBT binds to neurons, we conducted double staining for neuronal markers similar to that previously described for MBP. Thus, immunohistochemical labeling of floating mouse brain tissue sections for MAP2 followed DBT staining of the same sections. Images in both low and high magnification of the corpus callosum and subcortical regions were taken separately with the red and green channels. In the 20× images, as shown in Figure 3, G and H, the DBT staining was localized to both the corpus callosum and subcortical gray matter (Fig. 3G) while the MAP staining was largely confined to the subcortical region (Fig. 3H). When the subcortical region was examined at a higher magnification (40×) to identify individual neurons as shown in Figure 3J–L, individual neurons were clearly visualized by MAP2 staining (Fig. 3K), but were not labeled with DBT, which was localized only to the subcortical myelinated structures (Fig. 3J), suggesting that DBT selectively binds to myelin, but not to neurons. Further, no specific binding of DBT was detected on mouse forebrain dissociated cell cultures consistent with the binding specificity of DBT for myelin (results not shown).
Brain permeability
To determine the brain permeability of DBT, we administered DBT to wild-type mice through intravenous injection in the tail vein. At 20 min postinjection of DBT (0.1 mg/kg), both x-ray and fluorescent images of the mouse brain were acquired using a KODAK In-Vivo Multispectral System FX (Fig. 4). Coregistration of the high quality x-ray image and fluorescent image confirmed that the strong fluorescence was indeed emitted from inside the brain. The results indicate that DBT can readily penetrate the intact blood–brain barrier, making it a suitable near-infrared (NIR) probe for in vivo studies.
Figure 4.

A, B, The x-ray image (A) and fluorescent image (B) of a living wild-type mouse from Kodak multispectral imaging system. The precise overlay image without fiduciary markers is shown in C.
In vivo NIRF imaging of myelin
In vivo NIRF imaging was conducted in three mouse models that reflect different levels of myelination in the brain. Female 3-month old Plp-Akt-DD mice (n = 3) were used to represent hypermyelination in the brain. Age-matched Swiss Webster wild-type mice (n = 2) were used to represent intact, normal myelination in the brain. Shiverer mice (C3Fe.SWV-Mbpshi/J and n = 2) were used to represent deficient myelination in the brain.
In vivo NIRF imaging in a hypermyelinated mouse model
DBT (0.1 mg/kg) and vehicle (0.05% DMSO/PBS (V/V)) were first administered to the hypermyelinated mouse models through intravenous injections in the tail vein. The fluorescence signals were recorded at 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110 and 120 min postinjection. The images recorded at 2, 4, 6, 8, 10, 15, 20, 25, 30 min postinjection are shown in Figure 5. To exclude the influence of autofluorescence from the mouse tissues and impact from the solution, the vehicle (0.05% DMSO/PBS (V/V)) was also injected and fluorescence was recorded at 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110 and 120 min to compare with the DBT solution (images not shown). The fluorescent radiance of Plp-Akt-DD mice is much stronger than that observed in the wild-type mice, which is consistent with the fact that the myelin content in the Plp-Akt-DD mice is much higher than that in the wild-type mice.
Figure 5.

A, B, Near-infrared fluorescence in vivo imaging in Plp-Akt-DD mice (A) and wild-type mice (B), recorded at 0, 2, 4, 6, 8, 10, 15, 20, 25, and 30 min after the intravenous injection of 0.1 mg/kg DBT.
Quantitative analysis of these images was then conducted as shown in Figure 6. Over a period of 30 min, the average fluorescent intensity of DBT in Plp-Akt-DD mice is fourfold higher than that observed in wild-type mice. This is consistent with the enhanced myelin content in Plp-Akt-DD mice. The significant difference between the hypermyelinated and wild-type models appeared as early as 2 min postinjection with a much stronger fluorescent signal being detected in the hypermyelinated mouse brain, indicating facile brain entry of DBT in proportion to the myelin content. Compared with the fast clearance of DBT in the WT control, relatively slower clearance was observed in the hypermyelinated mice. These results show the potential of DBT used as a probe for in vivo near-infrared fluorescence imaging.
Figure 6.
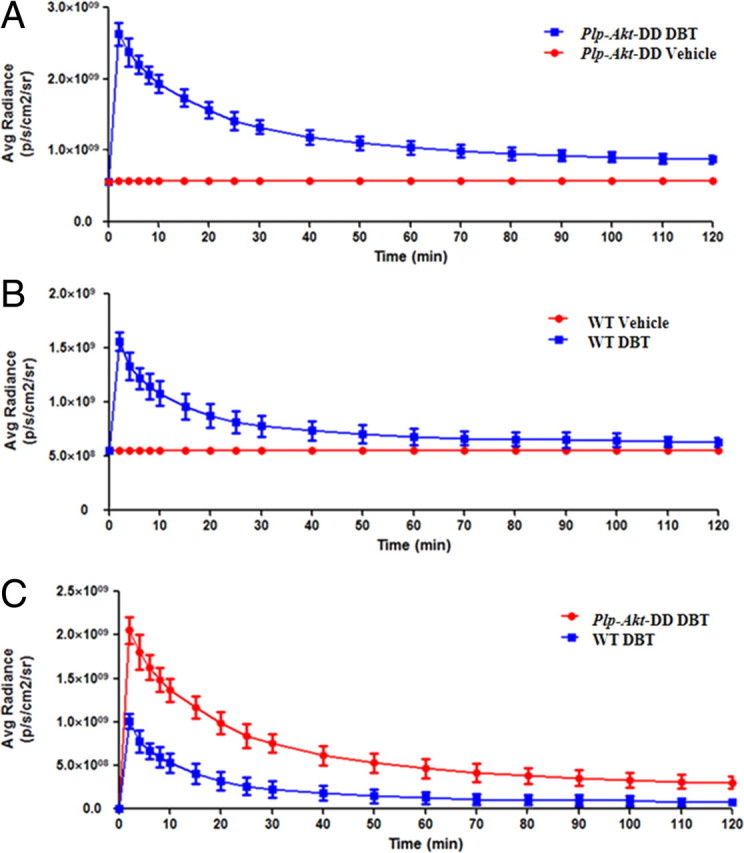
Quantification of in vivo imaging of the myelin sheath in living mice. A, The average radiance of Plp-Akt-DD mice after the injection of DBT (blue) and vehicle (red) at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, and 120 min [p < 0.0001, two-tailed t test, confidence interval (CI) 99%]. B, The average radiance of wild-type mice after injection of DBT (blue) and vehicle (red) at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, and 120 min (p < 0.0001, two-tailed t test, CI 99%). C, Comparison of the average radiance between the Plp-Akt-DD mice (red) and wild-type mice (blue) after deducting the vehicle signals (p = 0.0012, two-tailed t test, CI 99%). Values are given as mean ± SD.
In vivo NIRF imaging in shiverer mouse model
We then evaluated the pharmacokinetic profiles of DBT in 3-month old female shiverer mice (n = 3) and age-matched wild-type mice (n = 2). In vivo NIRF imaging was conducted using the same dosage as in the hypermyelinated mouse models. The fluorescent signals were recorded at 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110 and 120 min after intravenous injection of DBT and the vehicle (0.05% DMSO/PBS (V/V)). As shown in Figure 7, the fluorescent intensities in the brain between the two shiverer and wild-type models were significantly different.
Figure 7.

A, B, Near-infrared fluorescence in vivo imaging in shiverer mice (A) and wild-type mice (B) recorded at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30 min after intravenous injection of 0.1 mg/kg DBT.
Based on these images, quantitative analyses were conducted to determine the time course of fluorescent radiance. As shown in Figure 8, the average radiance at 30 min in wild-type mice is eight fold greater than that observed in shiverer mice. Since shiverer mice are deficient in CNS myelin, the fluorescent signal originating from shiverer mice is very weak, even at its peak at 2 min. These results further demonstrated that accumulation of DBT in the brain is proportional to the myelin content and can be used as a surrogate marker of myelination.
Figure 8.
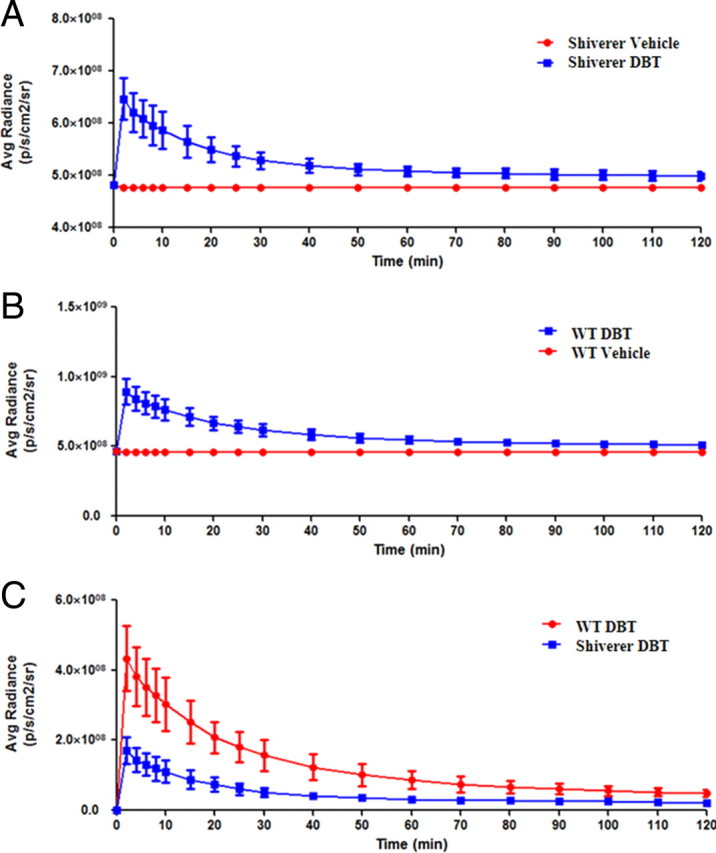
The quantification of in vivo imaging of the myelin sheath in the living mice. A, The average radiance of shiverer mice after the injection of DBT (blue) and vehicle (red) at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, and 120 min (p < 0.0001, two-tailed t test, CI 99%). B, The average radiance of wild-type mice after the injection of DBT (blue) and vehicle (red) at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120 min(p < 0.0001, two-tailed t test, CI 99%). C, The comparison of the average radiance between the shiverer mice (blue) and wild-type mice(red) after deducting the vehicle signals (p = 0.0019, two-tailed t test, CI 99%). Values are given as mean ± SD.
Longitudinal NIRF imaging of myelin changes in a cuprizone-induced mouse model of demyelination and remyelination
We then conducted longitudinal NIRF imaging studies in a cuprizone-treated mouse model of demyelination and remyelination at the sixth week of demyelination and the fourth and eighth week of remyelination. The longitudinal NIRF imaging was conducted using the same dosage as previously used in other mouse models. Fluorescent signals were recorded at 5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80 and 90 min after intravenous injection of DBT (Fig. 9). Quantitative image analyses were then conducted to determine the time course of fluorescent radiance detected using the whole brain as region of interest (Fig. 10A). Compared with wild-type control brains, the fluorescent intensity was significantly reduced in the same mouse brains following 6 weeks of cuprizone treatments. The mice were then allowed to recover with normal diets for up to 8 weeks. At the fourth week of recovery, the fluorescent intensity was increased, which was slightly above the level detected after 6-weeks of demyelination. At the eighth week of recovery, the fluorescent intensity was further increased, yet still below the level detected in the normal control brains. After imaging, mice were killed by perfusion and immunochemical staining was conducted on the frozen sections to examine the lesion load in the corpus callosum at different time points. As shown in Figure 10B–E, immunochemical staining suggested that the lesion loads at each time point were consistent with the optical imaging results. Compared with the peak of demyelination, the myelin content was gradually increased at the fourth week and eighth week of remyelination. Yet, the myelin content was still significantly below the level present in the normal control brain as previously demonstrated by others. These results demonstrated that DBT is a sensitive marker of myelin that is capable of longitudinal monitoring of myelin content in vivo.
Figure 9.

A–D, Representative in vivo near-infrared fluorescence images in a normal control mouse brain (A) mouse brain treated with cuprizone for 6 weeks (B), cuprizone-treated mouse brain after remyelination for 4 weeks (C), and cuprizone-treated mouse brain after remyelination for 8 weeks (D). NIRF images of each mouse brain were recorded at 5, 10, 15, and 20 min after the intravenous injection of 0.1 mg/kg DBT.
Figure 10.

A, Quantification and comparison of fluorescent intensity detected in mouse brains after NIRF imaging in different mouse models. The time courses of the average radiance of DBT detected in normal control mouse brain (red), mouse brain treated with cuprizone for 6 weeks (green), cuprizone-treated mouse brain after remyelination for 4 weeks (blue) and cuprizone-treated mouse brain after remyelination for 8 weeks (purple) (p = 0.0006, one-way ANOVA test, CI 95%). Value are given as mean ± SD. B–E, After imaging, corresponding immunochemical staining was conducted at different stages including wild-type (B), cuprizone-induced demyelination (C), remyelination of 4 weeks (D), and remyelination of 8 weeks (E). Scale bar, 300 μm.
Discussion
Near-infrared-based fluorescence imaging represents a relatively new imaging modality of potential value in in vivo studies. To enhance its potential in biomedical imaging, efforts have been focused on the development of small molecular probes (SMPs) possessing appropriate characteristics. Ideal near-infrared SMPs should exhibit emission/excitation wavelengths in the range between 650 and 900 nm. To reach the desired level of sensitivity, the near-infrared SMPs should also have a high quantum yield, high molar absorptivity, large Stokes shifts, large two-photon excitation cross-sections (Albota et al., 1998) as well as short lifetimes of ∼1 ns. In addition to the above physical properties, the SMPs should readily penetrate the blood–brain barrier and bind to myelin fibers with high sensitivity and specificity. The SMPs should also be relatively stable so that no metabolites interfere with the specific signals.
DBT was found to meet most of the preceding requirements. With an excitation peak at 782 nm and an emission peak at 797 nm, the fluorescence of DBT falls in the middle of the near-infrared range with a relatively large Stokes shift of 15 nm. The extinction coefficients at 780 nm is 43,000 (m cm)−1, the quantum yield is 0.034, and the lifetime is 1.18 ns. DBT is a charged, highly conjugated molecule with large changes of quadrupole moment. According to a previous study (Albota et al., 1998), such molecules normally possess large two-photon absorption cross sections. Although DBT is a charged molecule, the overall lipophilicity in terms of logP is estimated at 3.27 (Log P was calculated by ChemAxon's Marvin and Calculator Plugins on-line), which falls in the optimal range for brain permeability.
Taking advantage of the inherent fluorescence, the binding specificity of DBT for myelin was first evaluated through tissue staining of mouse brain sections. As shown in Figure 1, myelinated regions such as corpus callosum and caudate putamen were selectively labeled by DBT. To determine which forms of myelin fibers can be stained by DBT, we further examined tissue staining of additional brain regions in both white matter and gray matter including the cerebellum and frontal cortex. As shown in Figure 2, in addition to the binding of compact myelin fibers present in the caudate putamen, DBT stains granule layers present in the cerebellum. More interestingly, DBT also stains small myelin fibers present in the frontal cortex. Following DBT staining, subsequent immunohistochemical staining of MBP was significantly reduced or even completely blocked, which suggests that DBT binds to MBP present in the myelin sheaths and stains both compact myelin fibers and small myelin fibers. The binding specificity of DBT for myelin were further validated by tissue staining in relevant areas of the hyper- and hypomyelinated mouse mutants as well as immunohistochemical staining of neurons (Fig. 3). These studies indicated that DBT is a small molecular probe that is sensitive and specific for all the forms of myelin present in the CNS. This is the first time that a small molecular probe has been developed with a myelin-binding property similar to MBP antibody.
To examine the sensitivity and specificity in vivo, we conducted in vivo DBT-NIRF imaging in two animal models. The Plp-Akt-DD model was used to represent a myelin-rich state while the shiverer mouse model was used to represent a myelin-deficient state relative to wild-type controls. At 2 min after tail vein injection, strong fluorescent signals were detected in the hypermyelinated Plp-Akt-DD mouse model relative to the wild-type under the same attenuation power (Fig. 5). To confirm that the fluorescent signals were from inside the brain, the fluorescent images were colocalized with the corresponding x-ray images, which were used to define the brain region (Fig. 4). Quantitative analysis of the fluorescent intensity showed that DBT retention in the Plp-Akt-DD mouse brain is significantly higher than that in the wild-type control (Fig. 6). In contrast, opposite results were observed in myelin-deficient shiverer mice. The fluorescent signals were relatively weak in the shiverer mice after tail vein injection compared with the wild-type controls under the same attenuation power (Fig. 7). Quantitative fluorescent intensity showed that DBT retention in the shiverer mouse brain is significantly lower than that in the wild-type mouse brain (Fig. 8). In addition, a dose of DBT as low as 0.1 mg/kg was used in the imaging studies. No animals experienced any adverse pharmacologic effects during these studies. The pharmacokinetic profiles of DBT correlate with the well defined myelin levels in both mouse models suggesting that DBT-NIRF is a sensitive and specific imaging marker of myelination.
To broaden the application of DBT to multiple sclerosis studies, we then conducted longitudinal imaging studies to monitor demyelination and remyelination in a cuprizone-induced mouse model. In this model, young adult mice are fed with the copper chelator cuprizone (bis-cyclohexanone oxaldihydrazone). This leads to consistent demyelination followed by spontaneous remyelination after the neurotoxin is replaced with normal diets. Based on NIRF imaging and quantitative analysis (Fig. 9), the fluorescent intensity of DBT can be used to monitor the level of demyelination and subsequent remyelination in the cuprizone model, which correlate well with histochemical staining (Fig. 10). Thus, DBT-NIRF can be used as an imaging marker of myelination for longitudinal studies of myelin-related diseases in animal models.
Each DBT-NIRF study takes only several hours. After disappearance of the fluorescent signal, the same study can be repeated in the same animals, which can be used for longitudinal studies and fast throughput screening of drugs targeted at myelination. In addition, DBT showed no adverse pharmacological and behavioral effects in vivo when up to 50 mg/kg DBT were administered through tail vein injection. Thus, DBT-NIRF provides a very economical and practical tool for myelin imaging that complements current imaging modalities.
In summary, we demonstrated that DBT is a promising probe for near-infrared fluorescence imaging of myelination in vivo. It readily enters the brain and selectively binds to myelin sheaths. In vivo NIRF studies in animal models of both hypermyelination and hypomyelination showed that DBT-NIRF is a sensitive and specific imaging marker of myelination.
Footnotes
This work is supported in part by the National Multiple Sclerosis Society, National Institutes of Health–National Institute of Neurological Disorders and Stroke (R01 NS061837), and Department of Defense (MS090082).
References
- Albota M, Beljonne D, Brédas JL, Ehrlich JE, Fu JY, Heikal AA, Hess SE, Kogej T, Levin MD, Marder SR, McCord-Maughon D, Perry JW, Röckel H, Rumi M, Subramaniam G, Webb WW, Wu XL, Xu C. Design of organic molecules with large two-photon absorption cross sections. Science. 1998;281:1653–1656. doi: 10.1126/science.281.5383.1653. [DOI] [PubMed] [Google Scholar]
- Bottrill M, Kwok L, Long NJ. Lanthanides in magnetic resonance imaging. Chem Soc Rev. 2006;35:557–571. doi: 10.1039/b516376p. [DOI] [PubMed] [Google Scholar]
- Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) Chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- Flores AI, Narayanan SP, Morse EN, Shick HE, Yin X, Kidd G, Avila RL, Kirschner DA, Macklin WB. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JS, Wolf AP. Working against time: rapid radiotracer synthesis and imaging the human brain. Acc Chem Res. 1997;30:181–188. [Google Scholar]
- Frullano L, Meade TJ. Multimodal MRI contrast agents. J Biol Inorg Chem. 2007;12:939–949. doi: 10.1007/s00775-007-0265-3. [DOI] [PubMed] [Google Scholar]
- Guy C, Ffytche D. An introduction to the principles of medical imaging. London: Imperial College, London; 2005. [Google Scholar]
- Hildebrand C, Remahl S, Persson H, Bjartmar C. Myelinated nerve fibres in the CNS. Prog Neurobiol. 1993;40:319–384. doi: 10.1016/0301-0082(93)90015-k. [DOI] [PubMed] [Google Scholar]
- Jennings LE, Long NJ. ‘Two is better than one’—probes for dual-modality molecular imaging. Chem Commun. 2009;24:3511–3524. doi: 10.1039/b821903f. [DOI] [PubMed] [Google Scholar]
- Lee S, Chen X. Dual-modality probes for in vivo molecular imaging. Mol Imaging. 2009;8:87–100. [PubMed] [Google Scholar]
- Lowe MP. MRI contrast agents: the next generation. Aust J Chem. 2002;55:551–556. [Google Scholar]
- Martin A, Aguirre J, Sarasa-Renedo A, Tsoukatou D, Garofalakis A, Meyer H, Mamalaki C, Ripoll J, Planas AM. Imaging changes in lymphoid organs in vivo after brain ischemia with three-dimensional fluorescence molecular tomography in transgenic mice expressing green fluorescent protein in T lymphocytes. Mol Imaging. 2008;7:157–167. [PubMed] [Google Scholar]
- Merbach AE, Toth E. The chemistry of contrast agents in medical magnetic resonance imaging. Hoboken, NJ: Wiley; 2001. [Google Scholar]
- Morell P, Quarles RH. Basic neurochemistry: molecular, cellular, and medical aspects. In: Siegel GJ, editor. Myelin formation, structure, and biochemistry. Philadelphia: Lippincott-Raven; 1999. pp. 79–93. [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A. 2000;97:9226–9233. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin M. Molecular imaging—basic principles and applications in biomedical research. London: Imperial College, London; 2005. [Google Scholar]
- Sevick-Muraca EM, Lopez G, Reynolds JS, Troy TL, Hutchinson CL. Fluorescence and absorption contrast mechanisms for biomedical optical imaging using frequency-domain techniques. Photochem Photobiol. 1997;66:55–64. doi: 10.1111/j.1751-1097.1997.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Stankoff B, Wang Y, Bottlaender M, Aigrot M-S, Dolle F, Wu C, Feinstein D, Huang GF, Semah F, Mathis CA, Klunk W, Gould RM, Lubetzki C, Zalc B. Imaging of CNS myelin by positron emission tomography. Proc Natl Acad Sci U S A. 2006;103:9304–9309. doi: 10.1073/pnas.0600769103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Jiang H. DOT guided fluorescence molecular tomography of arbitrarily shaped objects. Med Phys. 2008;35:5703–5707. doi: 10.1118/1.3020594. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu C, Caprariello AV, Somoza E, Zhu W, Wang C, Miller RH. In vivo quantification of myelin changes in the vertebrate nervous system. J Neurosci. 2009;29:14663–14669. doi: 10.1523/JNEUROSCI.4082-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580–589. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight PA, Duchala CS, Readhead C, Macklin WB. A myelin proteolipid protein-LacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J Cell Biol. 1993;123:443–454. doi: 10.1083/jcb.123.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Tian D, Feng Y, Polak P, Wei J, Sharp A, Stankoff B, Lubetzki C, Zalc B, Mufson EJ, Gould RM, Feinstein DL, Wang Y. A novel fluorescent probe that is brain permeable and selectively binds to myelin. J Histochem Cytochem. 2006;54:997–1004. doi: 10.1369/jhc.5A6901.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wei J, Tian D, Feng Y, Miller RH, Wang Y. Molecular probes for imaging myelinated white matter in CNS. J Med Chem. 2008;51:6682–6688. doi: 10.1021/jm8003637. [DOI] [PMC free article] [PubMed] [Google Scholar]