

Abstract
Objective
Impaired endothelium-independent vasodilation is a known consequence of type-1 and 2 diabetes, and the mechanism of impaired vasodilation is not well understood. The following study investigated the effects of type-1 and 2 diabetes in endothelial-independent vasodilation associated with coronary vascular smooth muscle (VSM) relaxation and contractile signaling mechanisms.
Methods
Type-1 diabetes was induced in Yucatan mini-swine via alloxan injection and treated with or without insulin(DM and IDM). Non-diabetic swine served as controls(ND). Expression and/or phosphorylation of determinants of VSM relaxation and contraction signaling were examined in coronary arteries and microvessels. Coronary microvessel relaxation was assessed using sodium nitroprusside(SNP). In addition, SNP-induced vasodilation and myosin light chain (MLC) phosphorylation was determined in coronary microvessels isolated from ND and type-2 diabetic human atrial appendage.
Results
Diabetic impairment in SNP-induced relaxation was completely normalized by insulin. Soluble guanylate cyclase (sGC) VSM expression decreased in both DM and IDM groups and did not correlate with vasorelaxation. Phosphorylation of MLC and myosin phosphatase increased in the DM group and MLC phosphorylation strongly correlated with impaired VSM relaxation(r=.670, p<0.01). Coronary microvessels from type-2 diabetic human patients exhibited similarly impaired vasodilation and enhanced VSM MLC phosphorylation.
Conclusions
Impaired vasodilation in type-1 diabetes correlates with enhanced VSM MLC phosphorylation. In addition, enhanced VSM MLC phosphorylation is associated with impaired vasodilation in type 2 diabetes in humans.
Introduction
Diabetes Mellitus (DM) Type 1 and 2 are both characterized by impaired vasomotor regulation. Impaired vasodilation is a contributing factor to diabetes-induced peripheral and coronary vascular abnormalities including diminished angiogenesis, altered tissue perfusion, coronary artery disease (CAD), and peripheral vasculopathy. Endothelium-dependent vasodilation in diabetes is markedly impaired and is thought to be regulated by decreases in bioavailability of nitric oxide (NO) 1,2. However, there is also a clear role for diabetes-induced impairments in endothelium-independent vasodilation suggesting an underlying dysfunction in smooth muscle vasomotor regulation regardless of endothelial dysfunction. Diabetes limits coronary vascular responses to NO donors or the largely endothelium-independent vasodilator adenosine 3,4,5,6. Similar impairments in smooth muscle dependent vasodilation have been observed in both type 1 and 2 diabetic patients and animal models. Impaired endothelium-independent vasodilation is also a prognostic indicator of CAD 3,4,5,6,7. Therefore, we hypothesized that in addition to endothelial alterations, diabetes induces impaired vascular relaxation through alterations in smooth muscle relaxation or contractile signaling mechanisms,
The control of vascular smooth muscle (VSM) tone depends on pro-contractile and relaxation signaling mechanisms. The major end-effecter of VSM contraction is the molecular motor myosin which is regulated through phosphorylation on ser19 and thr18 of myosin light chain (MLC)8. MLC is phosphorylated via activation of specific signaling cascade including PKC/CPI-17, Rho/Rho-kinase, and Ca++/MLCK. 8,9,10 In addition, Insulin signals are implicated in antagonistic effects on MLC phosphorylation cascades 11. Endothelial derived NO activates soluble Guanylate Cyclase (sGC) to produce cGMP and activation of cGMP dependent protein kinase (PKG) 12,13 The NO/sGC/PKG signaling cascade subsequently exerts antagonistic effects on smooth muscle MLC phosphorylation to induce vasorelaxation 13.
Alterations in both the NO/PKG and Rho/RhoK signaling have been implicated in impaired NO-dependent vasodilation in both type 1 and 2 models of diabetes14,15,16. In addition, the majority of studies evaluating these phenomena have been performed in VSM cells in culture or small animal models of type 1 and 2 diabetes. We have recently examined both NO signaling constituents and MLC phosphorylation as related to endothelium independent coronary vasodilation in a large animal model of type I diabetes. These experiments demonstrate that enhanced contractile VSM signals (and not impaired NO activated signaling) greatly reduce NO-dependent vessel dilation. In addition, these findings were extended by demonstrating similar enhanced contractile signaling in coronary smooth muscle of type 2 diabetic patients.
Materials and Methods
Animal model and tissue collection
Eighteen male Yucatan mini-swine [Non-diabetic (ND) n=6, Diabetic (DM) n=6, and Diabetic + Insulin (IDM) n=6] were used for the studies. Diabetes was induced in 8 month old animals using a single intravenous injection of alloxan (150 mg/kg) as previously described 17. Only animals that achieved and maintained blood glucose levels greater than 200 mg/dL were included in the diabetes group. Animals with blood glucose levels exceeding 400 mg/dL were treated with daily insulin (70% amorphous, 30% crystalline) given intramuscularly to maintain blood glucose levels between 250 and 400 mg/dL. In the diabetic + Insulin group glycemic control was achieved (fasting blood sugar < 150 mg/dL) through daily intramuscular insulin administration. Age matched miniswine served as controls. Fasting blood glucose was monitored every two days early after diabetes induction and weekly thereafter. Diabetes was maintained for a total of 15 weeks.
Following euthanasia, sections of the left anterior descending coronary artery (LAD) were excised and frozen in liquid N2 for western blot analysis. Sections of the LAD dependent myocardial territory were immediately harvested in Krebs and/or 10 % formalin for microvessel and confocal microscopy studies respectively. Animals received humane care in compliance with the Harvard Medical Area Institutional Animal Care and Use Committee and the National Research Council’s Guide for the Care and Use of Laboratory Animals,
Human tissue collection
Samples were obtained from patients undergoing cardiac surgery as described previously 18. Briefly, samples of right atrium were obtained during placement of the venous cannula and immediately placed in 10% formalin for subsequent miscroscopy studies. For human microvessel studies, samples of collected right atrial appendage were immediately placed in ice cold Krebs-Heinslet. Tissue was used for microvessel studies within 1 hour of tissue harvest. Patients were determined to be diabetic based on a previous diagnosis of type 2 diabetes and Hemoglobin A1c (HbA1c) > 6.0%. Control vessels were from patients with no previous documentation of diabetes and a history of normal blood glucose levels. Experiments with patient tissues were approved under the CCI/IRB of the Beth Israel Deaconness Medical Center.
In-Vitro Assessment of Coronary Microvessel Reactivity
After cardiac harvest, epicardial coronary arterioles (100 to 200 μm in diameter and 1 to 2 mm in length) originating from branches of the LAD were dissected from the surrounding tissue and examined in isolated organ chambers with video based dimension measurements, as described previously 19 Vessels were cannulated with dual glass micropipettes and maintained in a no-flow pressurized (40 mmHg) state. Vessels were bathed in Krebs solution (37 C) for the length of the experiment. The responses to sodium nitroprusside (SNP) (1 nM to 100 μM), an endothelium-independent cGMP-mediated vasodilator were examined following pre-constriction with the thromboxane A2 analog U46619 (1 μM). In both the pig and human studies, % relaxation to SNP was expressed as the SNP dependent change in the diameter of the vessel as a percentage of the U46619-induced change from the baseline diameter. Vessels were not used for data analysis if there was no response to U46619. To generate EC50 values of the SNP dose response, a best-fit variable slope sigmoidal dose response curve was generated for each vessel using Graph Pad Prism software. EC50 values are expressed as the mean of the 50% effective relaxation concentration of SNP for each vessel
SDS-PAGE and Immunoblot
SDS-PAGE and immunoblot were performed as previously described 18. Briefly, LAD vessles (~1–2 cm in length) were homegenized in RIPA buffer with 50 mM NaF, protease inhibitors (Complete, Boehringer Mannheim, Germany), and phosphatase inhibitor cocktail (I + 2, 1:100, Sigma). Following homogenization, lysates were centrifuged at 10,000 g for 10 minutes, and bicinchroninic protein assay performed to allow equal gel loading. Forty μg of lysate was loaded on Tris-Glycine 8–16% gradient gels and electrophoresed for approximately 1 hour at 150 volts. Gels were transferred to PVDF membranes for 1 hour at 100V. Gels were blocked in 3% non-fat dry milk in Tris-buffered saline (TBS) for 1 hour followed by incubation in primary antibodies in either 3% TBS- milk or 3% Bovine Serum Albumin (BSA) according to the manufacturers recommendation. Blots were washed 3 times in TBS and incubated with the appropriate HRP-conjugated secondary antibody for 1 hour, washed 3x in TBS and detected using chemilluminescent detection (Pierce). Antibodies for immunoblot were as follows: PKG (Stressgen, Vancouver, BC), soluble Guanylate Cyclase (abcam, Cambridge, UK), anti-pthr38-CPI-17 (Upstate Biotechnology, NY), total CPI-17 (Santa Cruz Biotech, CA), smooth Muscle Actin (Sigma), Akt and pser473-Akt (Cell Signaling, Beverly, MA) and anti-ser19/thr18-MLC (gift of P.A. Vincent, Albany Medical College, NY)20.
Confocal Immunofluorescent Microscopy
Harvested tissue was immediately placed in 10 % formalin in PBS, fixed overnight, embedded in paraffin blocks, and cut into 4 um sections. Deparafinized slides were boiled in 10 mM Na-citrate pH 6.0 for 10 minutes. Slides were blocked with 2% BSA in TBS and incubated in 2% BSA-TBS with primary antibodies overnight at 4C. Slides were washed 3x with TBS and incubated with the appropriate Alexa-fluor secondary antibodies, and topro-3 (Invitrogen, San Diego, CA). Slides were washed 4x with TBS and mounted with fluorescent mounting medium (Vector Labs: Barlingame, CA). Tissue was visualized using a Zeiss 510meta confocal microscope system (Germany). Antibodies used were as follows: rabbit anti thr696-MYPT (Upstate Biotechnology, NY) and rabbit anti-ser19/thr18-MLC. Mouse anti-smooth muscle actin was used to label and identify vascular smooth muscle. Labeling of the tissues with either secondary alone or primary incubation with normal rabbit IgG or rabbit serum were used as negative controls.
Statistical Analysis
Data are presented as the mean ± S.E.M. Student’s T-test or One Way ANOVA with Student-Newman-Keuls post hoc analysis was used to determine statistical significance (P < .05) as appropriate. For determination of significance in SNP dose response curves, a Two Way Repeated Measures ANOVA was employed. Correlation anlaysis was performed using Pearson Correlation (P < .05)Statistical tests were performed using Sigmastat (SYSTAT: San Jose, CA). The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Injection of 32 week old pigs with alloxan did not result in significant changes in body weight between the three experimental groups (Figure 1A). Although there was a trend for decreased weight in the Diabetic swine group (DM, p=.06 vs. ND), little difference between the IDM and ND group body weight argues against weight differences due to the toxic effects of Alloxan independent of Diabetes induction. In addition, BUN levels were initially elevated following injection in the majority of pigs but returned to normal in approximately two weeks (data not shown). Alloxan injection resulted in significantly increased blood glucose levels in the DM group which was partially rescued by insulin treatment (Figure 1B).
Figure 1. Swine treated with Alloxan did not lose weight and exhibited increased blood glucose that was reduced with insulin.

A) Weight of 8 month old swine treated with Alloxan (DM and IDM) and weight gain over the subsequent 15 weeks. 40 week old control swine (ND) were acquired prior to the second surgery. B) Blood glucose levels at time of sacrifice for ND, DM and IDM groups. * indicates p<.05 vs. ND, One Way Anova on Ranks, Dunn’s method.
Diabetic swine (DM) exhibited significantly impaired coronary microvessel relaxation to the NO donor sodium nitroprusside (SNP) in comparison to both Non-diabetic (ND) and insulin treated diabetic swine (IDM) (Figure 2A). The concentration of SNP required to achieve 50% relaxation (EC50 SNP) was approximately 5 fold greater in the DM group compared to both control and IDM groups (Figure 2B). Impaired relaxation was not due to enhanced vessel preconstriction as the % contraction with U46619 and the vessel baseline diameter were similar between groups (Figures 2C and D). In contrast, there was a nonsignificant trend for decreased U46619-induced preconstriction and increased baseline diameter in the DM group.
Figure 2. Type I Diabetic swine have impaired coronary microvessel dilation to the NO donor SNP.

A) SNP dose response curves of coronary microvessels following preconstriction with U46619 from ND, DM and IDM swine. B) Mean EC50 values of microvessel responses to SNP determined from the individual curves used to generate A. C) The % contraction (relative to baseline diameter) of ND, DM, and IDM coronary microvessels to 1 uM U46619. D) Baseline diameter of coronary microvessels before preconstriction with U46619. * indicates p<.05 vs ND, One Way ANOVA, and # indicates p<.05 for dose response curves versus ND, Two Way RM ANOVA.
Western analysis of constituents of smooth muscle relaxation pathways, cyclic GMP dependent protein kinase (PKG) and soluble guanylate cyclase (sGC), in the LAD conduit vessels, showed that diabetes depressed expression of sGC (Figure 3A and B). In contrast to vascular relaxation responses, sGC expression was significantly diminished in both the diabetic groups of animals, with no effects of insulin on diminished sGC expression (Figure 3A and B). There were no significant changes in the levels of PKG.
Figure 3. Type 1 DM swine Coronary vessels exhibit reduced expression of soluble Guanylate Cyclase with or without insulin treatment.

A) Representative immunoblots sGC (top panel) and PKG (bottom panel) in LAD lysates from ND, DM, and IDM swine. B) Quantitation of the data shown in A expressed as fold difference from ND. * indicates p<.05 versus control, One Way ANOVA, student Neuman-Keuls post hoc analysis.
Western analysis of constituents of smooth muscle contractile signaling, phosphorylated myosin light chain (ser19/thr18 MLC), protein kinase C inhibitor -17 (CPI-17) and phosphorylated Akt (pser473-Akt), in the LAD displayed significant increases in the phosphorylation of MLC on ser19 and thr18 that were normalized with insulin (Figure 4A and B). There were no significant changes in CPI-17 or Akt phosphorylation in either the DM or IDM groups.
Figure 4. Diabetic swine exhibit enhanced phosphorylation of MLC on ser19 and thr18 independent of enhanced CPI17 phosphorylation and reduced Akt activation.
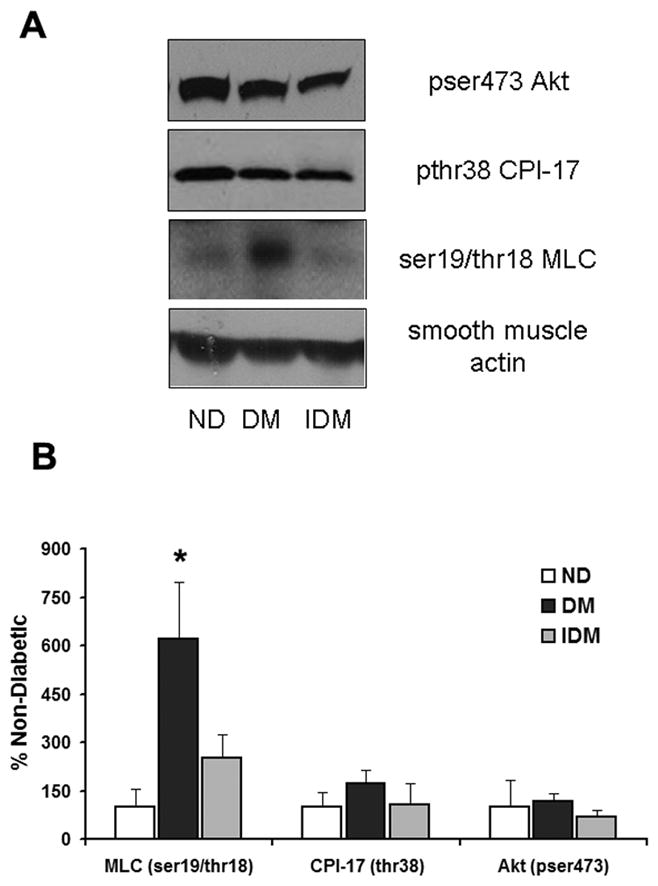
A) Representative immunoblots of pser473-Akt (top panel) pthr38-CPI17 (upper middle panel), and ser19/thr18 phosphorylated MLC (lower middle panel) in LAD lysates from ND, DM, and IDM swine. B) Quantitation of the data shown in A expressed as fold difference from ND for each protein examined. *indicates p<.05 versus ND, One Way ANOVA, student Neuman-Keuls post hoc analysis.
DM-induced increases in smooth muscle MLC phosphorylation were confirmed in resistance coronary microvessels (~50 – 100 um) from the LAD dependent territory (Figure 5A, red). In addition, larger diameter vessels displayed DM-induced increases in MLC phosphorylation in both the smooth muscle and endothelium (Figure 5A, arrows), that was normalized by insulin. DM-induced increases in microvessel MLC phosphorylation were associated with increases in inhibitory phosphorylation of thr696 of myosin phosphatase (pthr696-MYPT) (Figure 5B). However, there were no apparent changes in pthr696-MYPT in larger diameter vessels (Figure 5B last column).
Figure 5. Diabetic coronary microvessels display enhanced phosphorylation of MYPT on thr696 and MLC on ser19 and thr18.

A) Confocal micrographs of coronary microvessels from the LAD dependent ventricular myocardium stained for smooth muscle actin (green), pser19/thr18 MLC (red), and topro3 (nuclei, blue). B) Coronary microvessels as in A stained for smooth muscle actin (green), pthr696-MYPT (red), and topro3 (nuclei, blue). Bar = 30μm,
Correlation analysis demonstrated a significant direct correlation between the EC50 SNP relaxation response and blood glucose (Table 1). In addition, there was a highly significant direct correlation between the level of phosphorylated MLC and both the EC50 SNP and the blood glucose level (Table 1). There were no significant correlations between the other relaxation and contractile signaling proteins (Table 1).
Table 1.
Pearson correlation between fold change densitometry values and the EC50 SNP or fasting blood glucose levels
| SNP EC50 | p= | Glucose | p= | |
|---|---|---|---|---|
| PKG | −0.182 | 0.50 | −0.328 | 0.22 |
| sGC | −0.065 | 0.81 | −0.300 | 0.26 |
| pser19/thr18-MLC | 0.670 | <0.01 | 0.613 | 0.01 |
| pthr38-CPI-17 | 0.044 | 0.88 | 0.070 | 0.80 |
| pser473-Akt | 0.387 | 0.14 | 0.262 | 0.33 |
| SNP EC50 | N.D. | N.D. | 0.533 | 0.03 |
Depressed SNP-induced vasodilation and coronary smooth muscle MLC phosphorylation was examined in non-diabetic and type 2 diabetes patients using right atrial appendage tissue obtained during cardiac surgery. The characteristics of the patients used in this study are presented in Table 2. Type 2 diabetes patients exhibited depressed coronary microvessel vasodilation to SNP as compared to non diabetic patients (Figure 6A). The concentration of SNP required to achieve 50% relaxation (EC50 SNP) was approximately 3 fold greater in the DM group compared to ND patients (Figure 6B). Impaired relaxation was not due to enhanced vessel pre-constriction as the vessel baseline diameter was similar between groups (Figures 6C) and there was significantly decreased preconstriction to U46619 (Figure 6D). Similar to DM swine, DM patients exhibited enhanced phosphorylation of MLC in coronary resistance vessels (Figure 6E, red).
Table 2.
Characteristics of patients used in diabetes microvessel study.
| Patient Characteristics | ||
|---|---|---|
| Control | Diabetes | |
| Patients | 7 | 7 |
| Sex (M, F) | (5, 2) | (4, 3) |
| Age (range) | 71.3 (43, 84) | 70 (64, 84) |
| Hypertension | 3 | 6 |
| Hypercholesterolemia | 5 | 7 |
| Diabetes (NIDDM) | 0 | 7 |
| HbA1c | 5.5 ± 0.3 | 6.8 ± 0.3 |
| Tobacco Hx | 3 | 5 |
| CAD | 7 | 7 |
| Vessels | 3.6 | 3.5 |
| Medications: | ||
| Ca++ channel blocker | 2 | 3 |
| beta-blocker | 5 | 4 |
| Aspirin | 5 | 4 |
| plavix/wafarin/coumadin | 2 | 5 |
| ACE inhibitor | 4 | 4 |
| statin | 4 | 6 |
| sulfonylurea | 0 | 2 |
| metformin | 0 | 1 |
| diuretic | 3 | 4 |
Figure 6. Enhanced coronary MLC phosphorylation and impaired SNP-induced vasodilation are common to Type 2 diabetes in humans.

A) SNP dose response curves of coronary microvessels following preconstriction with U46619 from ND and DM CABG patients. B) Mean EC50 values of microvessel responses to SNP determined from the individual curves used to generate A. C) Baseline diameter of coronary microvessels before preconstriction with U46619. D) The % contraction (relative to baseline diameter) of ND and DM-2, coronary microvessels to 1 uM U46619. E) Confocal micrographs of coronary microvessels stained for smooth muscle actin (green), pser19/thr18 MLC (red), and Topro3 (nuclei, blue), Bar = 30 μm. * indicates p<.05 vs ND, One Way ANOVA, and # indicates p=.05 for dose response curves versus ND, Two Way RM ANOVA.
Discussion
The majority of studies examining diabetes-induced impaired vasodilation have focused on endothelial dependent alterations in NO-mediated vasorelaxation 1,21,17. In Diabetes, reduced NO production and subsequent vasodilation in response to endothelial acting vasodilators is well established22. In this study, we chose to examine Diabetes-induced impaired vasodilation independent of diabetes-induced reductions in endothelial derived NO. In the presence of equal concentrations of the NO donor SNP, Type 1 and 2 diabetic coronary vessels exhibited impaired vasodilation compared to non-diabetic vessels. These results are in agreement with earlier studies demonstrating diabetes-impaired myocardial blood flow in response to the largely endothelium independent vasodilators adenosine and/or NO donors4,6,23,3. Collectively, these results suggest an underlying diabetes-induced maladaption of coronary vascular smooth muscle reactivity in vivo, regardless of diabetes-induced perturbations in NO production and consumption. Although smooth muscle-mediated impaired responsiveness may appear minimal to described pathological decreases in endothelial-dependent responses, estimates of local vessel wall concentration of NO are in the 1–5 × 10−7 molar range 24, and agonist stimulated production of NO results in relatively discrete increases in NO concentration 25. These concentrations compare well to the doses of SNP used in this study, and diabetes in both pigs and humans results in significant reductions in SNP-induced dilation at these concentrations (Figure 2B and 6B). Therefore, diabetes-induced impaired endothelial dependent and independent coronary vasodilation likely reflects overall VSM insensitivity to multiple vasodilators.
The current study is in contrast to numerous studies in the literature. In both human26,27,28,29 and animal models30,31,32,33,34, of type 1 and 2 diabetes, SNP has been demonstrated to have minimal effect on the vasodilatory response to SNP. There are several possible reasons for these discrepancies. Differences in diabetes-dependent vascular responses may be attributable to many factors including disease progression35, gender 34, and type of diabetes 36,29. In addition, differences in species and/or specific vascular beds may play a role, with altered responses to SNP demonstrated within specific circulations 34. Finally, differences in data analysis and experimental design may also affect interpretation and results, including differences in specific contractile agonists (U46619, endothelin, KCl) or whether relaxation measured is passive or following agonist-induced contraction. One large similar study examining diabetes differences in atrial arterioles found no difference in SNP-induced vasodilation 27. However, these differences may be due to data analysis, as in the above study SNP responses were normalized to the 100% maximal SNP response. This method may have the effect of minimizing any absolute differences in SNP responses between the two populations. In contrast the SNP results (in both pigs and humans) of the current study were normalized to the baseline diameter (100% relaxation) and the luminal diameter following 1 μM U46619 pre-constriction (0%). In contrast to the above mentioned studies, numerous experimental diabetic models in multiple species and circulatory beds support a role for reduced endothelium independent vasorelaxation in type 1 and 2 diabetes 37,36,38,39,40,41.
The diabetes-induced decreases in vasodilation associated with both decreased sGC expression and enhanced phosphorylation of MYPT and MLC. Importantly, insulin completely reversed impaired vasodilation to NO, but had no effect on sGC expression, indicating that the reduced sGC expression is insufficient to account for diabetes-induced impaired vasodilation. In addition, the reduced levels of sGC were sufficient to produce normal vasodilation in the IDM group. However, it should be noted that there was no direct measurement of sGC activity. The possibility remains that insulin may alter responsiveness of this enzyme to NO. In addition, sGC has additional pro-relaxant effects other than modulation of myosin phosphatase (see below) and these effects may play a role in the current study11 In contrast to the effects on sGC, the effect of diabetes to promote VSM contractile signaling appeared more important than the observed alterations in VSM relaxation signal pathways. The greatly enhanced MLC phosphorylation in both type 1 diabetic swine and type 2 diabetic patients correlated with depressed vasodilation in response to SNP, and both were normalized by administration of insulin in the type 1 diabetes swine model. In addition, MLC phosphorylation was the only contractile or relaxation signaling parameter that correlated with both the level of blood glucose and depressed Diabetes-induced vasodilation (Table 1). Overall, these results suggest that impaired vasodilation is the result of a greatly enhanced contractile phenotype due to a loss of inhibitory effects of insulin and/or direct effects of hyperglycemia to induce MLC phosphorylation cascades. Indeed, a role for enhanced vessel reactivity has been previously implicated in diabetes, with demonstrated agonist induced increased vessel contraction to endothelin-1 and thromboxane 34,42. Surprisingly, in our study, despite clear increases in MLC phosphorylation, there were no corresponding increases in U46619-induced contraction in both type 1 diabetic swine and type 2 diabetic patients. In contrast, there was a trend for decreased contraction in type 1 diabetic pigs and significantly decreased contraction in human type 2 diabetic vessels. Furthermore, a trend for increased baseline diameter of diabetic vessels argues against an overall precontracted state. It is currently unclear why enhanced MLC phosphorylation doesn’t result in increased agonist-induced contraction. A similar dissociation between enhanced MLC phosphorylation and contraction in diabetes has been previously reported. Urinary bladder smooth muscle in diabetic rabbits exhibit enhanced MLC phosphorylation, but have a considerably reduced force production when compared to non-diabetic animals 43. Further investigation and understanding of this apparent paradoxical effect of diabetes may yield valuable mechanistic insight into smooth muscle contraction. Nevertheless, the present results are consistent with the view that impaired vasodilation in diabetes is the result of enhanced MLC phosphorylation that limits responsiveness to NO-induced relaxation signaling. Therefore, pharmacologic treatments that inhibit enhanced smooth muscle MLC phosphorylation may be a more relevant therapeutic target (vs. VSMC relaxation signaling: i.e. NO, eNOS, sGC, PKG) to alleviate impaired vasodilation in diabetes.
Diabetes-induced impaired vasodilation in both type 1 diabetic pigs and type 2 human coronary microvessels are likely mediated via the reduced effects of insulin. In type 1 diabetes, the reduced levels of insulin, and in type 2 diabetes, the reduced sensitivity to insulin, signaling appear to result in the common consequence of enhanced VSMC MLC phosphorylation. However, the precise mechanism of enhanced MLC phosphorylation is unclear. Studies in VSMC in culture and small animal models of diabetes 1 and 2 demonstrate numerous insulin dependent effects on signaling cascades capable of altering MLC phosphorylation. These include the NO-sGC-PKG, RhoA-Rho-kinase-MYPT, and the PI3K/Akt signaling cascades 8,11.
Insulin is known to promote eNOS dependent production of NO and subsequently increase activation of sGC and PKG resulting in stimulatory phosphorylation of MYPT on ser69544. In addition, PKG dependent phosphorylation of RhoA and subsequent reductions in activity are implicated in NO-dependent reductions in VSMC MLC phosphorylation. The actions of insulin on the NO/sGC/PKG VSMC relaxation pathways are mediated through insulin dependent activation of the PI3K/Akt signaling cascade45,11. Furthermore, the expression levels of sGC and PKG are under tight feedback control and are also believed to be linked to effects of insulin and/or beneficial effects of insulin on endothelial NO production 46,47,11. It is unlikely that the impaired vessel relaxation and enhanced MLC phosphorylation observed in the diabetes pig models is due to modulation of the sGC/PKG pathway for the following reasons. First, as discussed above, insulin which rescued MLC phosphorylation and SNP-induced vasodilation did not change the diabetes-induced impairment in expression of relaxation signaling enzymes. Second, there were no changes in Akt phosphorylation associated with diabetes or insulin treated diabetic animals. Third, we were unable to detect any phosphorylation of MYPT on ser695 in vivo. Finally, insulin dependent enhanced NO generation and direct effects to activate sGC would not be expected to alter the vasodilatory response in these experiments as NO was delivered via SNP at equal concentrations to all groups.
A more likely explanation for the observed effects in this model is enhanced signaling via the Rho-kinase/MYPT signaling mechanism. Upon GTP binding, RhoA binds and activates Rho-kinase which can inhibit MYPT via phosphorylation on thr696 or directly phosphorylate MLC on ser19/thr1848. In addition both Rho-kinase and PKC can phosphorylate CPI-17 which subsequently binds and inhibits MYPT. In cultured VSMC and small animal models insulin reduces activation of both RhoA and Rho Kinase, thereby relieving the inhibition of MYPT and reducing MLC phosphorylation. Similar to insulin dependent effects on relaxation signaling pathways, many of these effects are attributed to Akt dependent effects of insulin 11, the lack of change in Akt phosphorylation indicates that the effects of insulin observed in this chronic in vivo model were independent of Akt. Another possibility is that hyperglycemia directly activates the Rho-kinase cascade. Hyperglycemia in cultured VSMC increases Rho-Kinase expression, CPI-17 phosphorylation, and subsequent MLC phosphorylation. In the type 1 diabetes pig microvessels we demonstrate increased phosphorylation of MYPT at the Rho-Kinase site (thr696). However, no diabetes-induced changes in CPI-17 phosphorylation were detected in the coronary LAD’s (Figure 4) or microvessels (data not shown). A final possibility may be the diabetes-induced upregulation of specific mediators that induce activation of the RhoA/Rho-kinase signaling cascade34. Determining the major upstream molecular mechanisms of enhanced MLC phosphorylation will require further investigation. However, the main determinants of depressed vasodilation in diabetes appear to be mediated by enhanced VSMC MLC phosphorylation and inhibitory phosphorylation of MYPT associated with reduced negative regulation by insulin (Figure 7).
Figure 7. Schematic of coronary smooth muscle contraction and relaxation signaling pathways perturbed in DM in vivo.

Black arrows and bars indicate normal positive and negative regulation respectively. Thick grey arrows indicate Diabetes-induced alterations in coronary smooth muscle signaling. Diabetes-induced effects on MLC phosphorylation may be due to the absence of insulin and/or hyperglycemia, whereas the effects on sGC are independent of insulin.
In conclusion, we have demonstrated, in both patients and a large animal model, impaired coronary smooth muscle vasodilation to NO is common to type 1 and 2 diabetic disease. Furthermore, diabetes mediated increases in MLC phosphorylation correlate with impaired coronary vasodilation, and may contribute to the multitude of vascular disorders associated with diabetes.
Acknowledgments
This study was supported by grants R01-HL69024 and R01-HL46716 from the National Institutes of Health (Dr Sellke). Drs. Clements and Sodha were supported by a postdoctoral training grant from the National Institutes of Health (5T32-HL076130-02) and the Irving Bard Memorial Fellowship. We thank Peter A. Vincent of Albany Medical College for providing the anti-ser19/thr18 MLC antibody.
References
- 1.Britten MB, Zeiher AM, Schachinger V. Clinical importance of coronary endothelial vasodilator dysfunction and therapeutic options. J Intern Med. 1999;245:315–327. doi: 10.1046/j.1365-2796.1999.00449.x. [DOI] [PubMed] [Google Scholar]
- 2.Milsom AB, Jones CJ, Goodfellow J, Frenneaux MP, Peters JR, James PE. Abnormal metabolic fate of nitric oxide in Type I diabetes mellitus. Diabetologia. 2002;45:1515–1522. doi: 10.1007/s00125-002-0956-9. [DOI] [PubMed] [Google Scholar]
- 3.Nahser PJ, Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal Coronary Flow Reserve and Metabolic Coronary Vasodilation in Patients With Diabetes Mellitus. Circulation. 1995;91:635–640. doi: 10.1161/01.cir.91.3.635. [DOI] [PubMed] [Google Scholar]
- 4.Nitenberg A, Valensi P, Sachs R, Dali M, Aptecar E, Attali JR. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42:1017–1025. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- 5.Natali A, Toschi E, Baldeweg S, Casolaro A, Baldi S, Sironi AM, Yudkin JS, Ferrannini E. Haematocrit, type 2 diabetes, and endothelium-dependent vasodilatation of resistance vessels. Eur Heart J. 2005;26:464–471. doi: 10.1093/eurheartj/ehi113. [DOI] [PubMed] [Google Scholar]
- 6.Di Carli MF, Janisse J, Grunberger G, Ager J. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol. 2003;41:1387–1393. doi: 10.1016/s0735-1097(03)00166-9. [DOI] [PubMed] [Google Scholar]
- 7.Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- 8.Pfitzer G. Invited Review: Regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:497–503. doi: 10.1152/jappl.2001.91.1.497. [DOI] [PubMed] [Google Scholar]
- 9.Noda M, Yasuda-Fukazawa C, Moriishi K, Kato T, Okuda T, Kurokawa K, Takuwa Y. Involvement of rho in GTPgammaS-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS Letter. 1995;367:246–250. doi: 10.1016/0014-5793(95)00573-r. [DOI] [PubMed] [Google Scholar]
- 10.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho- kinase) [see comments] Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 11.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular Actions of Insulin. Endocr Rev er- 2007 doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura M, Ichikawa K, Ito M, Yamamori B, Okinaka T, Isaka N, Yoshida Y, Fujita S, Nakano T. Effects of the Phosphorylation of Myosin Phosphatase by Cyclic GMP-dependent Protein Kinase. Cellular Signalling. 1999;11:671–676. doi: 10.1016/s0898-6568(99)00036-4. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann F. The Biology of Cyclic GMP-dependent Protein Kinases. J Biol Chem. 2005;280:1–4. doi: 10.1074/jbc.R400035200. [DOI] [PubMed] [Google Scholar]
- 14.Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular Consequences of Endothelial Nitric Oxide Synthase Uncoupling for the Activity and Expression of the Soluble Guanylyl Cyclase and the cGMP-Dependent Protein Kinase. Arterioscler Thromb Vasc Biol. 2005;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- 15.Chang S, Hypolite JA, DiSanto ME, Changolkar A, Wein AJ, Chacko S. Increased basal phosphorylation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase beta and CPI-17. Am J Physiol Renal Physiol. 2006;290:F650–F656. doi: 10.1152/ajprenal.00235.2005. [DOI] [PubMed] [Google Scholar]
- 16.Xie Z, Su W, Guo Z, Pang H, Post SR, Gong MC. Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovascular Research. 2006;69:491–501. doi: 10.1016/j.cardiores.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Boodhwani M, Sodha NR, Mieno S, Xu SH, Feng J, Ramlawi B, Clements RT, Sellke FW. Functional, cellular, and molecular characterization of the angiogenic response to chronic myocardial ischemia in diabetes. Circulation. 2007;116:I31–I37. doi: 10.1161/CIRCULATIONAHA.106.680157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clements RT, Sodha NR, Feng J, Mieno S, Boodhwani M, Ramlawi B, Bianchi C, Sellke FW. Phosphorylation and translocation of heat shock protein 27 and alphaB-crystallin in human myocardium after cardioplegia and cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2007;134:1461–1470. doi: 10.1016/j.jtcvs.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 19.Tofukuji M, Metais C, Li J, Hariawala MD, Franklin A, Vassileva C, Li J, Simons M, Sellke FW. Effects of ischemic preconditioning on myocardial perfusion, function, and microvascular regulation. Circulation. 1998;98:II197–II204. [PubMed] [Google Scholar]
- 20.Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA. RhoA and Rho-kinase dependent and independent signals mediate TGF-beta-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol. 2005;288:L294–L306. doi: 10.1152/ajplung.00213.2004. [DOI] [PubMed] [Google Scholar]
- 21.Hsueh WA, Quinones MJ. Role of endothelial dysfunction in insulin resistance. The American Journal of Cardiology. 2003;92:10–17. doi: 10.1016/s0002-9149(03)00611-8. [DOI] [PubMed] [Google Scholar]
- 22.Goodfellow J. Microvascular heart disease in diabetes mellitus. Diabetologia. 1997;40(Suppl 2):S130–S133. doi: 10.1007/s001250051428. [DOI] [PubMed] [Google Scholar]
- 23.Sundell Sundell J, Laine Laine H, Nuutila Nuutila P, Rönnemaa R+T, Luotolahti Luotolahti M, Raitakari Raitakari O, Knuuti Knuuti J. The effects of insulin and short-term hyperglycaemia on myocardial blood flow in young men with uncomplicated Type I diabetes. Diabetologia. 2002;45:775–782. doi: 10.1007/s00125-002-0819-4. [DOI] [PubMed] [Google Scholar]
- 24.Clough GF, Bennett AR, Church MK. Measurement of nitric oxide concentration in human skin in vivo using dermal microdialysis. Exp Physiol. 1998;83:431–434. doi: 10.1113/expphysiol.1998.sp004126. [DOI] [PubMed] [Google Scholar]
- 25.Simonsen U, Wadsworth RM, Buus NH, Mulvany MJ. In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. J Physiol. 1999;516:271–282. doi: 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szerafin T, Erdei N, Fulop T, Pasztor ET, Edes I, Koller A, Bagi Z. Increased Cyclooxygenase-2 Expression and Prostaglandin-Mediated Dilation in Coronary Arterioles of Patients With Diabetes Mellitus. Circ Res. 2006;99:e12–317. doi: 10.1161/01.RES.0000241051.83067.62. [DOI] [PubMed] [Google Scholar]
- 27.Miura H, Wachtel RE, Loberiza FR, Jr, Saito T, Miura M, Nicolosi AC, Gutterman DD. Diabetes mellitus impairs vasodilation to hypoxia in human coronary arterioles: reduced activity of ATP-sensitive potassium channels. Circ Res. 2003;92:151–158. doi: 10.1161/01.res.0000052671.53256.49. [DOI] [PubMed] [Google Scholar]
- 28.Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia. 2000;43:1435–1438. doi: 10.1007/s001250051551. [DOI] [PubMed] [Google Scholar]
- 29.Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA. Impaired endothelium-dependent vasodilation in patients with insulin- dependent diabetes mellitus. Circulation. 1993;88:2510–2516. doi: 10.1161/01.cir.88.6.2510. [DOI] [PubMed] [Google Scholar]
- 30.Zhou W, Wang XL, Lamping KG, Lee HC. Inhibition of protein kinase Cbeta protects against diabetes-induced impairment in arachidonic acid dilation of small coronary arteries. J Pharmacol Exp Ther. 2006;319:199–207. doi: 10.1124/jpet.106.106666. [DOI] [PubMed] [Google Scholar]
- 31.Chakraphan D, Sridulyakul P, Thipakorn B, Bunnag S, Huxley VH, Patumraj S. Attenuation of endothelial dysfunction by exercise training in STZ-induced diabetic rats. Clin Hemorheol Microcirc. 2005;32:217–226. [PubMed] [Google Scholar]
- 32.Ammar RF, Jr, Gutterman DD, Brooks LA, Dellsperger KC. Impaired dilation of coronary arterioles during increases in myocardial O2 consumption with hyperglycemia. Am J Physiol Endocrinol Metab. 2000;279:E868–E874. doi: 10.1152/ajpendo.2000.279.4.E868. [DOI] [PubMed] [Google Scholar]
- 33.Koltai MZ, Hadhazy P, Posa I, Kocsis E, Winkler G, Rosen P, Pogatsa G. Characteristics of coronary endothelial dysfunction in experimental diabetes. Cardiovasc Res. 1997;34:157–163. doi: 10.1016/s0008-6363(97)00050-3. [DOI] [PubMed] [Google Scholar]
- 34.Sanz E, Fernandez N, Monge L, Martinez MA, Climent B, Dieguez G, Garcia-Villalon AL. Effects of diabetes on the vascular response to nitric oxide and constrictor prostanoids: gender and regional differences. Life Sciences. 2003;72:1537–1547. doi: 10.1016/s0024-3205(02)02444-x. [DOI] [PubMed] [Google Scholar]
- 35.Prior JO, Quinones MJ, Hernandez-Pampaloni M, Facta AD, Schindler TH, Sayre JW, Hsueh WA, Schelbert HR. Coronary Circulatory Dysfunction in Insulin Resistance, Impaired Glucose Tolerance, and Type 2 Diabetes Mellitus. Circulation. 2005;111:2291–2298. doi: 10.1161/01.CIR.0000164232.62768.51. [DOI] [PubMed] [Google Scholar]
- 36.Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- 37.Brooks BA, Franjic B, Ban CR, Swaraj K, Yue DK, Celermajer DS, Twigg SM. Diastolic dysfunction and abnormalities of the microcirculation in type 2 diabetes. Diabetes Obes Metab. 2007 doi: 10.1111/j.1463-1326.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- 38.Brooks BA, McLennan SV, Twigg SM, Yue DK. Detection and characterisation of microcirculatory abnormalities in the skin of diabetic patients with microvascular complications. Diab Vasc Dis Res. 2008;5:30–35. doi: 10.3132/dvdr.2008.006. [DOI] [PubMed] [Google Scholar]
- 39.Gatenby PA. The role of complement in the aetiopathogenesis of systemic lupus erythematosus. Autoimmunity. 1991;11:61–66. doi: 10.3109/08916939108994709. [DOI] [PubMed] [Google Scholar]
- 40.McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1992;35:771–776. doi: 10.1007/BF00429099. [DOI] [PubMed] [Google Scholar]
- 41.Watts GF, O’Brien SF, Silvester W, Millar JA. Impaired endothelium-dependent and independent dilatation of forearm resistance arteries in men with diet-treated non-insulin-dependent diabetes: role of dyslipidaemia. Clin Sci(Lond) 1996;91:567–573. doi: 10.1042/cs0910567. [DOI] [PubMed] [Google Scholar]
- 42.Bender SB, Klabunde RE. Altered role of smooth muscle endothelin receptors in coronary endothelin-1 and {alpha}1-adrenoceptor-mediated vasoconstriction in Type 2 diabetes. Am J Physiol Heart Circ Physiol. 2007;293:H2281–H2288. doi: 10.1152/ajpheart.00566.2007. [DOI] [PubMed] [Google Scholar]
- 43.Su X, Changolkar A, Chacko S, Moreland RS. Diabetes decreases rabbit bladder smooth muscle contraction while increasing levels of myosin light chain phosphorylation. Am J Physiol Renal Physiol. 2004;287:F690–F699. doi: 10.1152/ajprenal.00027.2004. [DOI] [PubMed] [Google Scholar]
- 44.Wooldridge AA, MacDonald JA, Erdodi F, Ma C, Borman MA, Hartshorne DJ, Haystead TA. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J Biol Chem. 2004;279:34496–34504. doi: 10.1074/jbc.M405957200. [DOI] [PubMed] [Google Scholar]
- 45.Lee JH, Ragolia L. AKT phosphorylation is essential for insulin-induced relaxation of rat vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;291:C1355–C1365. doi: 10.1152/ajpcell.00125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Browner NC, Dey NB, Bloch KD, Lincoln TM. Regulation of cGMP-dependent Protein Kinase Expression by Soluble Guanylyl Cyclase in Vascular Smooth Muscle Cells. J Biol Chem. 2004;279:46631–46636. doi: 10.1074/jbc.M408518200. [DOI] [PubMed] [Google Scholar]
- 47.Liu S, Ma X, Gong M, Shi L, Lincoln T, Wang S. Glucose down-regulation of cGMP-dependent protein kinase I expression in vascular smooth muscle cells involves NAD(P)H oxidase-derived reactive oxygen species. Free Radical Biology and Medicine. 2007;42:852–863. doi: 10.1016/j.freeradbiomed.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 48.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho- kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]