

Abstract
Family studies of individual tissues have shown that gene expression traits are genetically heritable. Here, we investigate cis and trans components of heritability both within and across tissues by applying variance-components methods to 722 Icelanders from family cohorts, using identity-by-descent (IBD) estimates from long-range phased genome-wide SNP data and gene expression measurements for ∼19,000 genes in blood and adipose tissue. We estimate the proportion of gene expression heritability attributable to cis regulation as 37% in blood and 24% in adipose tissue. Our results indicate that the correlation in gene expression measurements across these tissues is primarily due to heritability at cis loci, whereas there is little sharing of trans regulation across tissues. One implication of this finding is that heritability in tissues composed of heterogeneous cell types is expected to be more dominated by cis regulation than in tissues composed of more homogeneous cell types, consistent with our blood versus adipose results as well as results of previous studies in lymphoblastoid cell lines. Finally, we obtained similar estimates of the cis components of heritability using IBD between unrelated individuals, indicating that transgenerational epigenetic inheritance does not contribute substantially to the “missing heritability” of gene expression in these tissue types.
Author Summary
An important goal in biology is to understand how genotype affects gene expression. Because gene expression varies across tissues, the relationship between genotype and gene expression may be tissue-specific. In this study, we used heritability approaches to study the regulation of gene expression in two tissue types, blood and adipose tissue, as well as the regulation of gene expression that is shared across these tissues. Heritability can be partitioned into cis and trans effects by assessing identity-by-descent (IBD) at the genomic location close to the expressed gene or genome-wide, respectively, and applying variance-components methods to partition the heritability of each gene. We estimated the proportion of gene expression heritability explained by cis regulation as 37% in blood and 24% in adipose tissue. Notably, the heritability shared across tissue types was primarily due to cis regulation. Thus, the relative contribution of cis versus trans regulation is expected to increase with the number of cell types present in the tissue being assayed, just as observed in our study and in a comparison to previous work on lymphoblastoid cell lines (LCL). We specifically ruled out a substantial contribution of transgenerational epigenetic inheritance to heritability of gene expression in these cohorts by repeating our heritability analyses using segments shared IBD in distantly related Icelanders.
Introduction
The genome contains a complex set of instructions for the assembly and maintenance of an organism. A fundamental goal in biology is to understand the relationship between genotype and phenotype. This goal can be achieved in part by studying the genetic basis of gene expression, as many genotype-phenotype correlations are a consequence of genetically driven variation in gene expression [1]. A number of studies have mapped individual cis and trans regulatory variants in humans, and recent work has suggested that the majority of regulators act in trans [2]-[5]; regulation of gene expression has also been widely studied in animal models [6]-[9]. However, the bulk of variability in gene expression remains unexplained. Heritability analyses can shed light on the genetic basis of gene expression. Several previous studies have demonstrated substantial overall heritability of gene expression in family data sets, and heritability approaches have also been broadly applied to other phenotypes [10]-[14].
In this study, we used gene expression measurements [11] and genome-wide single nucleotide polymorphism (SNP) data [15] from 722 Icelanders from family cohorts to examine the heritability of gene expression in blood and adipose tissue. By studying more than one tissue type, we were able to analyze the regulation of gene expression both within and across tissues. Our goal was to answer three key questions about gene expression heritability. First, can heritability be partitioned into cis and trans components using local and genome-wide IBD between pairs of individuals? Second, to what extent are heritable components of variance shared across tissues? Third, to what extent does heritability extend to distantly related individuals inheriting IBD segments from distant ancestors?
We sought to partition the heritability of gene expression into cis versus trans components by comparing the effects of IBD at the genome-wide level (trans) to those of IBD at the local level (cis), defined as the number of chromosomes (0, 1 or 2) shared IBD at the genomic location containing the expressed gene. Our results show a substantially higher proportion of heritability due to cis regulation, 37% in blood and 24% in adipose tissue, than the 12% reported in a previous ancestry-based study of lymphoblastoid cell lines (LCL) in African Americans [16]. One possible explanation for this discrepancy is transgenerational epigenetic inheritance, which is one of the explanations proposed to account for the “missing heritability” in genetic studies of human traits [17]-[23]. Epigenetic inheritance would regulate gene expression at the cis locus, and would be expected to contribute to cis heritability in family-based analyses but not in ancestry-based analyses, given that this mode of inheritance persists over a relatively short time scale. However, by using IBD in distantly related individuals to produce similar estimates of cis heritability, we were able to rule out this hypothesis. Instead, our analyses indicate that the proportion of heritability attributable to cis regulation is tissue-specific, and that similarities in gene expression across tissues are primarily due to heritable cis effects. Thus, the proportion of gene expression heritability attributable to cis regulation is expected to increase as a function of the number of different cell types present in the tissue being assayed, consistent with results obtained from blood, adipose tissue and LCL.
Methods
Ethics statement
This research was approved by the Data Protection Commission of Iceland and the National Bioethics Committee of Iceland. The appropriate informed consent was obtained for all sample donors.
Icelandic Family Blood cohort
Relative abundances of 23,720 transcripts were obtained for blood samples from each of 1,001 individuals from the IFB cohort, as described previously [11] (see Web Resources). Values were adjusted for sex and age. We removed 4,985 transcripts that either had >5% missing data, did not map to an autosomal chromosome, or mapped to more than one genomic location. We removed 16 individuals with >5% missing data and 269 individuals for which long-range phased SNP data were not available. This left 18,735 transcripts and 716 individuals. Most of out analyses focused on 2,233 related pairs (individuals from the same family pedigree with genome-wide IBD >0.05) spanning a subset of 687 individuals.
Icelandic Family Adipose cohort
Relative abundances of 23,720 transcripts were obtained for adipose tissue samples from each of 673 individuals from the IFA cohort, as described previously [11] (see Web Resources). Values were adjusted for sex, age and body mass index (BMI), restricting to 638 individuals with BMI data. We removed 4,621 transcripts that either had >5% missing data, did not map to an autosomal chromosome, or mapped to more than one genomic location. We removed 2 individuals with >5% missing data and 67 individuals for which long-range phased SNP data were not available. This left 19,099 transcripts and 569 individuals. Most of our analyses focused on 1,700 related pairs (individuals from the same family pedigree with genome-wide IBD >0.05) spanning a subset of 531 individuals.
Local IBD estimates
Individuals were genotyped using the Illumina 300K chip. Owing to the sensitive nature of genotype data, access to these data can only be granted at the headquarters of deCODE Genetics in Iceland. Given long-range phased Illumina 300K data [15] for a pair of individuals, we partitioned the genome into 2cM blocks and for each block performed 2×2 = 4 comparisons between haplotypes from the two individuals. We declared two haplotypes to be IBD if they matched at >95% of alleles in the block, non-IBD if they matched at <85% of alleles, and unknown-IBD otherwise. We excluded SNPs with missing data in one or both individuals, so that lack of a match implies a mismatch, and set IBD status to unknown for pairs of haplotypes with >5% of SNPs excluded. We defined local IBD as the total number of comparisons producing a match. We verified that this approach infers 0∶1∶2 copies IBD between parent-child pairs with probabilities 0.2%∶99.3%∶0.4% and 0∶1∶2: copies IBD between sibling pairs with probabilities 24.9%∶50.1%∶24.9%, excluding from this computation the 7% of pairs and blocks for which inferred IBD was unknown. These numbers are a function of the thresholds we used to define IBD and non-IBD; the thresholds were largely chosen for specificity rather than sensitivity since for our application it does not matter that inferred IBD is sometimes unknown. The numbers are very close to the expected theoretical probabilities (for parent-child pairs, 2 copies IBD is expected to occasionally occur due to IBD in “unrelated” parents). This validates our use of long-range phased SNP genotypes to compute local IBD estimates. We computed genome-wide IBD estimates as the average of local IBD estimates across all 2cM blocks.
Heritability estimates using genome-wide IBD only
We applied variance-components methods to estimate narrow-sense heritability [14], [24]. The source code used in our heritability analyses is available for download (see Web Resources). Let egs denote the gene expression for gene g and individual s, normalized to have mean 0 and variance 1 across individuals. Let θst denote the genome-wide IBD between individuals s and t (0≤θst≤1) and 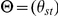 be the N×N matrix of genome-wide IBD, where N is the number of individuals. Let Vg denote the covariance matrix of normalized gene expression for gene g. We consider the model
be the N×N matrix of genome-wide IBD, where N is the number of individuals. Let Vg denote the covariance matrix of normalized gene expression for gene g. We consider the model 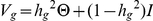 and fit hg2, the heritability of gene g, to the observed normalized gene expression values egs by maximizing the likelihood
and fit hg2, the heritability of gene g, to the observed normalized gene expression values egs by maximizing the likelihood 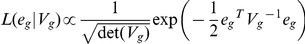 , where
, where 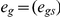 . Values of egs, hg and Vg vary with tissue type, but we view tissue type as an implicit index rather than an explicit index for simplicity of notation. For both blood and adipose tissue, the estimated values of hg2 were ∼80% correlated to values that were computed previously using similar methods [11], despite the fact that the current analysis was restricted to a subset of individuals for which long-range phased SNP data was available for local IBD inference. We declare hg2>0 to be nominally significant (P<0.05) if hg2 is larger than each value analogously estimated from 19 data sets with sample labels randomly permuted (thus, 5% of genes will be nominally significant even in the absence of a true effect). We estimated average h2 as the average of hg2 across genes g. We computed standard errors on h2 by performing independent runs with sample labels randomly permuted; we obtained identical standard errors using either five permutations or 20 permutations. Similar procedures were used in the cis vs. trans and cross-tissue analyses described below.
. Values of egs, hg and Vg vary with tissue type, but we view tissue type as an implicit index rather than an explicit index for simplicity of notation. For both blood and adipose tissue, the estimated values of hg2 were ∼80% correlated to values that were computed previously using similar methods [11], despite the fact that the current analysis was restricted to a subset of individuals for which long-range phased SNP data was available for local IBD inference. We declare hg2>0 to be nominally significant (P<0.05) if hg2 is larger than each value analogously estimated from 19 data sets with sample labels randomly permuted (thus, 5% of genes will be nominally significant even in the absence of a true effect). We estimated average h2 as the average of hg2 across genes g. We computed standard errors on h2 by performing independent runs with sample labels randomly permuted; we obtained identical standard errors using either five permutations or 20 permutations. Similar procedures were used in the cis vs. trans and cross-tissue analyses described below.
Heritability estimates using both cis and trans IBD
We extended the variance-components approach to cis and trans heritability via the model 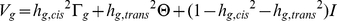 , where
, where 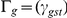 is the N×N matrix of local (cis) IBD between individuals s and t at the genomic location proximal to gene g. We used the midpoint of the gene expression probe to define genomic location, but the value of γgst is not sensitive to this choice as local IBD segments between related individuals span many megabases. We scale γgst to have value 0.0, 0.5, or 1.0 (for 0, 1, or 2 copies shared). We fit the cis heritability hg,cis2 and trans heritability hg,trans2 by maximizing the usual likelihood, fitting hg2 = hg,cis2+hg,trans2 and hg,cis2 in turn. We average across genes g to estimate hcis2 and htrans2. We define the proportion of heritable gene expression variation that is due to cis regulation as πcis = hcis2/(hcis2+htrans2). As above, all values vary with tissue type, which we view as an implicit index.
is the N×N matrix of local (cis) IBD between individuals s and t at the genomic location proximal to gene g. We used the midpoint of the gene expression probe to define genomic location, but the value of γgst is not sensitive to this choice as local IBD segments between related individuals span many megabases. We scale γgst to have value 0.0, 0.5, or 1.0 (for 0, 1, or 2 copies shared). We fit the cis heritability hg,cis2 and trans heritability hg,trans2 by maximizing the usual likelihood, fitting hg2 = hg,cis2+hg,trans2 and hg,cis2 in turn. We average across genes g to estimate hcis2 and htrans2. We define the proportion of heritable gene expression variation that is due to cis regulation as πcis = hcis2/(hcis2+htrans2). As above, all values vary with tissue type, which we view as an implicit index.
Cross-tissue analysis
The cross-tissue correlation ρ was computed as the correlation between normalized expression levels in blood and adipose tissue across genes and individuals. Due to the normalization, this is equal to the average of gene-specific correlations ρg. We computed standard errors of both gene-specific and average cross-tissue correlations via jackknife, repeating the computation with each individual removed in turn and estimating the standard error as 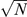 times the standard deviation of the N estimates. We now describe our estimation of cross-tissue heritability. Let
times the standard deviation of the N estimates. We now describe our estimation of cross-tissue heritability. Let 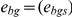 and
and 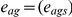 denote normalized expression levels for gene g and individual s in blood and adipose tissue, respectively. Let Wg denote the covariance matrix of the vector (ebg,eag) of length 2N. Here the relevant equations are
denote normalized expression levels for gene g and individual s in blood and adipose tissue, respectively. Let Wg denote the covariance matrix of the vector (ebg,eag) of length 2N. Here the relevant equations are
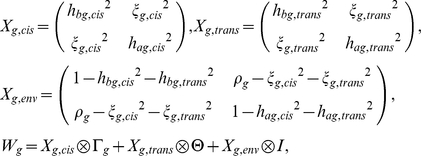 |
where ξ2 denotes cross-tissue heritability, ρ denotes cross-tissue correlation, and  denotes the tensor product of a 2×2 matrix with an N×N matrix to form a 2N×2N matrix. For example, the first term of Wg has entries hbg,cis2γgst in the upper left N×N block, ξg,cis2γgst in the upper right N×N block, and so on. This generalization of the variance-components approach to cross-phenotype analyses has been previously described (for the case of genome-wide IBD) in an analysis of two height phenotypes, self-reported height and clinically measured height [25]. The likelihood is defined in the usual way, replacing Vg with Wg and eg with (ebg,eag). We fit hbg2 = hbg,cis2+hbg,trans2, hbg,cis2, hag2 = hag,cis2+hag,trans2, hag,cis2, ρg, ξg2 = ξg,cis2+ξg,trans2 and ξg,cis2 in turn. For each of the parameters estimated, we compute average values by averaging across genes g.
denotes the tensor product of a 2×2 matrix with an N×N matrix to form a 2N×2N matrix. For example, the first term of Wg has entries hbg,cis2γgst in the upper left N×N block, ξg,cis2γgst in the upper right N×N block, and so on. This generalization of the variance-components approach to cross-phenotype analyses has been previously described (for the case of genome-wide IBD) in an analysis of two height phenotypes, self-reported height and clinically measured height [25]. The likelihood is defined in the usual way, replacing Vg with Wg and eg with (ebg,eag). We fit hbg2 = hbg,cis2+hbg,trans2, hbg,cis2, hag2 = hag,cis2+hag,trans2, hag,cis2, ρg, ξg2 = ξg,cis2+ξg,trans2 and ξg,cis2 in turn. For each of the parameters estimated, we compute average values by averaging across genes g.
Web Resources
http://www.ncbi.nlm.nih.gov/geo/ (Gene Expression Omnibus). Gene expression data sets have been deposited into the GEO database under accession numbers GSE7965 and GPL3991, as described previously [11].
http://www.hsph.harvard.edu/faculty/alkes-price/software/. The source code used in our heritability analyses is available for download, along with the results presented in Table S1.
Results
Overall heritability of gene expression
For the analysis of gene expression in blood, we analyzed normalized intensity values for 18,735 mRNA transcripts. Analysis was restricted to 687 individuals from the IFB cohort for whom long-range phased SNP data were available (see Methods). For each pair of individuals, we used the long-range phased SNP data to compute the number of chromosomes shared IBD at each location in the genome, and computed the genome-wide IBD as an average of these values (Figure 1; see Methods). Our initial analyses focused on 2,233 related pairs with genome-wide IBD >0.05. For the analysis of gene expression in adipose tissue, we similarly analyzed 19,099 mRNA transcripts of 531 individuals from the IFA cohort, focusing on 1,700 related pairs with genome-wide IBD >0.05 (see Methods). The IFA cohort largely overlaps the IFB cohort, with 496 of the 722 individuals analyzed appearing in both cohorts.
Figure 1. Local and genome-wide IBD.

We plot the local relatedness (0, 1 or 2 copies IBD) between two siblings from the IFB cohort at each 2cM block on chromosome 1. The dotted line represents their genome-wide relatedness of 0.568, which is within the expected range for siblings [46].
We estimated the overall heritability hg2 for each gene g using variance-component methods [14] (see Methods). Although estimates for each gene g are statistically noisy at these sample sizes, histograms show a clear positive bias for both IFB and IFA cohorts (Figure S1 and Table S1), and hg2>0 was nominally significant (P = 0.05; see Methods) for an excess of genes: 42% for IFB and 63% for IFA. We computed the average h2 as the average of hg2 across genes g. A relevant question is whether or not to allow negative values of hg2 when computing this average [26]. Such values have no biological interpretation (except in the case of negative correlation among siblings in traits that depend on birth order). However, because values close to zero may be either increased or decreased by statistical noise—leading to negative estimates of hg2 for 3,031 of 18,735 genes for IFB and 1,038 of 19,099 genes for IFA—we elected to allow negative values in our main computations so as to produce an unbiased estimate of average h2. We obtained estimates of h2 = 0.150 for blood and h2 = 0.234 for adipose tissue. We obtained similar results when using a regression-based approach to estimate average h2 (Text S1), which more readily lends itself to visualization (Figure 2A and 2B). (When clipping negative hg2 values to zero, we obtained h2 = 0.159 for blood and h2 = 0.237 for adipose tissue.) Our results are consistent with previous analyses reporting that expression levels of a substantial fraction of genes are significantly heritable at the level of h2 = 0.3 or higher [10]-[13], [26].
Figure 2. Family heritability in the IFB and IFA cohorts.

(A) Gene expression covariance (average value of product of normalized gene expression measurements) between related individuals in the IFB cohort varies with genome-wide IBD. Each point represents one pair of related individuals. The slope of this plot corresponds to the regression-based estimate of h2. (B) Same as (A), for IFA cohort. (C) Gene expression covariance between siblings for genes with 0, 1 or 2 copies IBD at the cis locus, minus total covariance as displayed above. The slope of this plot corresponds to the regression-based estimate of hcis2. The signal to noise ratio is higher in this plot due to reduced effects of systematic noise covariance. (D) Same as (C), for IFA cohort.
Cis versus trans heritability of gene expression
While estimates of overall heritability are based on genome-wide IBD, it is possible to estimate cis versus trans heritability by extending variance components to consider both local (cis) IBD at the genomic location close to the expressed gene, and genome-wide (trans) IBD (see Methods). As before, analyses were restricted to 2,233 and 1,700 related pairs from the IFB and IFA cohorts, respectively. Histograms of hg,cis2 and hg,trans2 estimates for each gene g show a clear positive bias for both IFB and IFA cohorts (Figure S2 and Table S1), with an excess of nominally significant (P<0.05) genes for IFB (hg,cis2>0: 16%; hg,trans2>0: 19%) and IFA (hg,cis2>0: 16%; hg,trans2>0: 30%). For IFB, we obtained average cis and trans heritability estimates of hcis2 = 0.055 and htrans2 = 0.095, respectively, which sum to h2 = 0.150. This leads to the conclusion that the proportion of heritability of expression due to cis variants in blood is πcis = 37%. For IFA, we obtained estimates of hcis2 = 0.057 and htrans2 = 0.177, which sum to h2 = 0.234. This yields an estimate of πcis = 24% in adipose tissue. The values of h2 and htrans2 in adipose tissue are significantly higher than for blood, but hcis2 is similar, leading to a lower value of πcis. We obtained similar results when using a regression-based approach to estimate average hcis2 and htrans2 (Text S1; Figure 2C and 2D). We note that there is considerably less statistical uncertainty in estimates of hcis2 (Figure 2C and 2D) than in estimates of h2 (Figure 2A and 2B). Indeed, we obtained standard errors of h2 = 0.150±0.011, hcis2 = 0.055±0.001 and htrans2 = 0.095±0.010 for blood and h2 = 0.234±0.011, hcis2 = 0.057±0.002 and htrans2 = 0.177±0.010 for adipose tissue (see Methods). These standard errors are 7-100 times lower than standard errors for single-gene heritability estimates, which are inadequate for estimating πcis (see Text S1). The much lower standard errors for hcis2 are a consequence of variation in cis IBD across the genome that decouples the estimation of this parameter from the systematic noise covariance structure across all pairs of individuals (see Text S1). Based on these standard errors for hcis2 and htrans2, πcis has little statistical uncertainty, although results may be affected by modeling uncertainty.
Our heritability model does not account for the possibility of phenotypic similarity in related individuals due to shared environment, which can confound estimates of heritability [14]. We note that such effects would inflate estimates of h2 and htrans2, but have a negligible impact on hcis2, since the extent of shared environment would be related to genome-wide (trans) rather than local (cis) IBD. To investigate the possibility of confounding due to shared environment, we computed the average correlation in gene expression between spouses, who are genetically unrelated but have a shared environment. We observed average correlations of 0.074±0.042 in 33 IFB spouse pairs and 0.076±0.035 in 28 IFA spouse pairs, which are similar in magnitude to correlations between sib-sib or parent-child pairs that correspond to the average heritabilities reported above (see Text S1 and Table S2). Thus, there is strong evidence that shared environment can lead to similarity in gene expression phenotypes. We further investigated whether the gene by gene signature of correlations in spouse pairs matches the signature of correlations in sib-sib or parent-child pairs or estimates of hg2, but found that it does not (see Text S1 and Table S3). Thus, we hypothesize that the correlations in spouse pairs are due to very recent shared environment (e.g. diet) arising from sharing the same household, whereas the correlations in sib-sib and parent-child pairs in this study (who are unlikely to share the same household, since only adult individuals were sampled) are due to genetic heritability. However, we cannot rule out a small amount of inflation in h2 and htrans2 estimates due to shared environment in related individuals.
Assessing the impact of epigenetic inheritance on cis heritability
Our family-based estimates of πcis in blood and adipose tissue are considerably greater than a previous estimate of 12±3% obtained using lymphoblastoid cell lines (LCL) from African-Americans, in which local versus genome-wide European ancestry was used to infer the relative contribution of cis versus trans heritability [16]. An analogous ancestry-based analysis of LCL gene expression data [27] from admixed HapMap 3 Mexican-Americans [28] has produced a similarly low value of πcis = 13±9%. One possible explanation for the lower values as compared to family-based estimates could be the epigenetic inheritance of cis-acting factors other than DNA sequence that are transmitted from parent to offspring. Given the relatively short time scale of epigenetic inheritance, this would be expected to have a much greater impact on family-based estimates of πcis than those based on ancestry [22]-[23].
To further explore the epigenetic hypothesis, we repeated the cis versus trans analysis using subsets of unrelated or distantly related individuals (genome-wide IBD <0.01) from the IFB and IFA cohorts. The mean genome-wide IBD for all such pairs of individuals was 0.0044, with a standard deviation of 0.0018, consistent with the known properties of distant relatedness between “unrelated” individuals from Iceland as well as other world populations [29]-[31]. We independently generated five random subsets of IFB individuals (85, 87, 92, 93, 91 individuals) and five random subsets of IFA individuals (127, 85, 92, 95, 89 individuals) with genome-wide IBD <0.01 between each pair of individuals in each subset, such that each subset was maximal subject to this constraint. The resulting estimates of hcis2 were 0.057±0.008 for blood and 0.067±0.005 for adipose tissue (mean ± standard deviation across five subsets). These estimates of hcis2 were close to our previous estimates based on closely related pairs, thereby ruling out a substantial contribution of epigenetic inheritance to cis heritability (see Discussion). However, we did not obtain meaningful estimates of htrans2 using distantly related individuals, due to the systematic noise covariance structure (see Text S1), and therefore πcis could not be estimated. We note that similar results for distantly related pairs were obtained using different IBD estimation algorithms (see Text S1).
Cross-tissue analysis
We conducted a cross-tissue analysis of expression heritability in blood and adipose tissue in 496 individuals who overlapped between the IFB and IFA cohorts. We determined that an individual’s blood expression for a particular gene is slightly but significantly correlated to the same individual’s adipose expression for the same gene, with an average correlation of ρ = 0.041±0.005 (mean ± standard error) (see Methods). Although estimates for each gene g are statistically noisy at these sample sizes, histograms show a clear positive bias in ρg (Figure S3), and ρg>0 was nominally significant (P = 0.05) for 20% of genes, a significant excess.
We next investigated the relationship between an individual’s blood expression and a related individual’s adipose expression, using variance-components methods (see Methods). This revealed that cross-tissue similarity varies with the level of family relatedness, with an average cross-tissue heritability estimate of ξ2 = 0.030±0.006. Analogous to the analyses for single tissues, we partitioned the cross-tissue heritability into cis and trans components, yielding values of ξcis2 = 0.031±0.001 and ξtrans2 = -0.001±0.006. We obtained similar results using regression-based approaches (Text S1; Figure 3A and 3B). Histograms of cross-heritability estimates for each gene g show a positive bias for ξg2 and ξg,cis2, but not ξg,trans2, for which the histogram is symmetric about zero (Figure S4). While our estimate of ξtrans2 is not significantly different from zero, ξcis2 is highly significant and explains the bulk of our estimate of ρ. This implies that the extent to which gene expression in blood and adipose tissue is similar across genes and individuals is dominated by heritable effects at the cis locus.
Figure 3. Cross-tissue heritability in the IFB and IFA cohorts.

Plots are analogous to those in Figure 2, except that we analyze the covariance between related individuals in different tissues, instead of between related individuals in the same tissue. (A) Cross-tissue covariance between related individuals in the intersection of IFB and IFA varies with genome-wide IBD. The slope of this plot corresponds to the regression-based estimate of ξ2. (B) Cross-tissue covariance between siblings in the intersection of IFB and IFA with 0, 1 or 2 copies IBD at the cis locus, minus total covariance as displayed in (A). The slope of this plot corresponds to ξcis2.
Averaging across cell types with shared cis effects increases the value of πcis
Our finding that cross-tissue similarities are dominated by heritable cis effects leads to the mathematical result that πcis is expected to increase with tissue heterogeneity: as the number of cell types represented in a tissue increases, the strongly correlated cis effects will add linearly but the uncorrelated trans effects will be diluted. In detail, let x and y denote cells types and suppose that Cov(exgs,exgt) = Cov(eygs,eygt) = hcis2γgst+htrans2θst for all genes g and individuals s≠t, and that all cis effects (but no trans or non-genetic effects) are shared across cell types. Thus, Cov(exgs,eygt) = hcis2γgst. Now consider a tissue z containing cell types x and y. Up to a normalization constant, Cov(ezgs,ezgt) = Cov(0.5(exgs+eygs),0.5(exgt+eygt)) = hcis2γgst+0.5htrans2θst, so that πcis,z = hcis2/(hcis2+0.5htrans2) is larger than πcis,x = πcis,y = hcis2/(hcis2+htrans2).
We verified this theoretical result empirically by defining ezgs = ebgs + eags as the average of normalized gene expression in blood and adipose tissue, normalized to mean 0 and variance 1. For synthetic tissue z, we obtained the value πcis = 0.41, which is larger than the value of πcis for either blood or adipose tissue, and similar to the predicted value of 0.055/(0.055+0.25(0.095+0.177)) = 0.45 based on hcis2 and htrans2 (πcis < 0.45 is actually expected since not all cis effects are shared). Thus, the variability in πcis across tissue types (0.12 for LCL, 0.24 for adipose, 0.37 for blood) is consistent with the fact that LCL represent a single cell type, whereas adipose tissue and blood contain many cell types: adipose tissue contains smooth muscle cells, fibroblasts, adipocytes, mast-cells and endothelial cells, while blood contains erythrocytes, thrombocytes, neutrophils, lymphocytes, monocytes, eosinophils and basophils in proportions that vary across individuals [32]-[34]. This also explains why studies of individual cell types have been more successful in identifying trans eQTLs than studies of whole tissues, and why most replications across tissue types occur at cis eQTLs [11], [34]-[37].
Discussion
In this study, we observed a greater contribution of cis regulation in blood and adipose tissue than in a previous ancestry-based analysis of LCL in African-Americans [16]. This result is not sensitive to sample size, because although estimates for individual genes are statistically noisy, we considered averages across genes. We also observed that cross-tissue similarity between blood and adipose expression is genetically heritable and dominated by cis effects. These two results are highly concordant. Due to the dilution of trans effects that are not shared across cell types, cis regulation is expected to explain a greater proportion of heritability in tissue types that are heterogeneous in their cell composition, such as blood and adipose tissue—particularly blood, in which cell type proportions may vary among individuals. This highlights the importance of considering different tissue types [16]. However, other explanations for the higher contribution of cis regulation in this study than in the ancestry-based analysis are also possible. For example, epistasis between two neighboring cis variants would be included in cis heritabilities estimated via IBD, but not in the ancestry-based analysis in which ancestry is a partial proxy for SNP genotype but a very poor proxy for both genotypes of two interacting SNPs. In addition, epistatic interactions involving multiple loci may potentially be important, and may confound estimates of narrow-sense heritability, but are outside the scope of this study. A further possibility is that trans effects in LCL could be overstated due to genetically heritable variation of in vitro factors such as the response to EBV virus, which would mimic trans regulation in heritability analyses but does not reflect true biological trans regulation [38]. Distinguishing between these possibilities is an important direction of future work.
Efforts to understand cis regulation are likely to benefit from combining information from many cell or tissue types, since underlying mechanisms can be either shared or cell-type specific. Indeed, our finding that on average roughly half single-tissue cis heritability (hcis2) is shared across tissues (ξcis2) is consistent with a recent study focusing on cis eQTLs, which reported that 54%, 50% and 54% of cis eQTLs in fibroblasts, LCLs and T cells, respectively, are cell-type specific [36]. Those percentages would be expected to be higher when considering only two cell types, but lower at larger sample sizes. On the other hand, studies of trans regulation should focus on a single cell type to avoid diluting trans effects that are not shared across cell types. New technologies to assay cell type-specific gene expression in complex tissues may also prove valuable [39]. Future experiments will shed light on whether similarity between tissues other than blood and adipose is also predominantly explained by heritable cis effects. Results may vary by organism as well as tissue type. Recent studies of fat, kidney, adrenal and heart tissues in rat recombinant inbred strains also observed reduced trans effects in more heterogeneous tissues, but reported some evidence of cross-tissue regulation in trans as well as in cis [8]-[9].
The similarity of cis heritability results using IBD in closely related versus distantly related individuals has significant implications. It has been suggested that epigenetic inheritance, defined as the transmission across generations of epigenetic changes not due to variation in DNA sequence, is a potential source of the “missing heritability” in genetic association studies [17]-[21]. Epigenetic inheritance would be expected to influence expression at the cis locus, and would be expected to contribute to cis heritability between closely related individuals but not between distantly related individuals, given that this mode of inheritance persists over a relatively short time scale [22]-[23]. Our failure to observe any such discordance suggests that transgenerational epigenetic inheritance is unlikely to play a major role in the missing heritability of gene expression and other traits, although it does not rule out a very small aggregate effect across all genes or large effects at certain metastable epialleles [40]-[41], nor does it shed light on the importance of mitotically conserved epigenetic effects that are not transmitted from parent to offspring.
Our results highlight the utility of using IBD in distantly related individuals to make inferences about heritability. This approach will be particularly valuable as sample sizes increase, since the number of pairs of individuals increases quadratically with sample size. Indeed, IBD in distantly related individuals has already proven useful for mapping specific loci [42], and heritability-related analyses using identity-by-state (IBS) instead of IBD have also yielded important insights [43]-[45]. By using IBD segments shorter than those analyzed here to consider IBD sharing at different distances from genes, it may even be possible draw conclusions about the distribution of genomic distances at which cis regulation contributes to heritability.
Supporting Information
Histograms of heritability estimates for each gene. We plot histograms of (a) hg2 estimates for IFB and (b) hg2 estimates for IFA, across genes g.
(0.23 MB TIF)
Histograms of cis and trans heritability estimates for each gene. We plot histograms of (a) hg,cis2 estimates for IFB, (b) hg,trans2 estimates for IFB, (c) hg,cis2 estimates for IFA and (d) hg,trans2 estimates for IFA, across genes g.
(0.19 MB TIF)
Histograms of cross-tissue correlations for each gene. We plot a histogram of observed gene-specific cross-tissue correlations ρg.
(0.14 MB TIF)
Histograms of cross-tissue heritability estimates for each gene. We plot histograms of (a) ξg2 estimates, (b) ξg,cis2 estimates and (c) ξg,trans2 estimates, across genes.
(0.20 MB TIF)
Heritability results for each gene.
(1.82 MB TXT)
Average correlations between spouse-spouse, sib-sib, and parent-child pairs. We list the average correlation for each pair type and cohort, averaging across correlations for each gene g. We also list standard errors, computed via jackknife.
(0.03 MB DOC)
Concordance of gene-by-gene signatures of correlations in each pair type. We list values of Rsib-sib,parent-child, Rspouse,sib-sib and Rspouse,parent-child for each cohort (see text), along with the number of pairs of each type used to compute those values. For comparison purposes, we also list (in italics) values of Rsib-sib,parent-child computed using smaller subsets of pairs to match the number of pairs used to compute Rspouse,sib-sib or Rspouse,parent-child, as a smaller number of pairs leads to lower values of R.
(0.03 MB DOC)
Supplementary Note.
(0.04 MB DOC)
Acknowledgments
We are grateful to David Reich and Aditi Hazra for valuable discussions. We thank Barbara Stranger and Manolis Dermitzakis for sharing LCL gene expression data from HapMap 3 samples; the International HapMap 3 Consortium for providing genotype data; and Benjamin Raby, Liming Liang, and Noah Zaitlen for helpful comments. Finally, we thank the Icelanders who participated in this study.
Footnotes
Authors whose affiliations are listed as deCODE Genetics are shareholders and/or employees of deCODE Genetics, a biotechnology company.
This work was supported by NIH grant R03HG005732 (ALP) and by HSPH incubator funding (ALP). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- 3.Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung VG, Nayak RR, Wang IX, Elwyn S, Cousins SM, Morley M, Spielman RS. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 8:e1000480. doi: 10.1371/journal.pbio.1000480. doi: 10.1371/journal.pbio.1000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chesler EJ, Lu L, Shou S, Qu Y, Gu J, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. 2005;37:233–242. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- 7.Williams RB, Cotsapas CJ, Cowley MJ, Chan E, Nott DJ, et al. Normalization procedures and detection of linkage signal in genetical-genomics experiments. Nat Genet. 2006;38:855–856; author reply 856-859. doi: 10.1038/ng0806-855. [DOI] [PubMed] [Google Scholar]
- 8.Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, et al. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2006;2:e172. doi: 10.1371/journal.pgen.0020172. doi: 10.1371/journal.pgen.0020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petretto E, Bottolo L, Langley SR, Heinig M, McDermott-Roe C, et al. New insights into the genetic control of gene expression using a Bayesian multi-tissue approach. PLoS Comput Biol. 2010;6:e1000737. doi: 10.1371/journal.pcbi.1000737. doi: 10.1371/journal.pcbi.1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, et al. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet. 2004;75:1094–1105. doi: 10.1086/426461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 12.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 14.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 15.Kong A, Masson G, Frigge ML, Gylfason A, Zusmanovich P, et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price AL, Patterson N, Hancks DC, Myers S, Reich D, et al. Effects of cis and trans genetic ancestry on gene expression in African Americans. PLoS Genet. 2008;4:e1000294. doi: 10.1371/journal.pgen.1000294. doi: 10.1371/journal.pgen.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008;17:R156–165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 19.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 22.Youngson NA, Whitelaw E. Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet. 2008;9:233–257. doi: 10.1146/annurev.genom.9.081307.164445. [DOI] [PubMed] [Google Scholar]
- 23.Jablonka E, Raz G. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol. 2009;84:131–176. doi: 10.1086/598822. [DOI] [PubMed] [Google Scholar]
- 24.Amos CI. Robust variance-components approach for assessing genetic linkage in pedigrees. Am J Hum Genet. 1994;54:535–543. [PMC free article] [PubMed] [Google Scholar]
- 25.Macgregor S, Cornes BK, Martin NG, Visscher PM. Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum Genet. 2006;120:571–580. doi: 10.1007/s00439-006-0240-z. [DOI] [PubMed] [Google Scholar]
- 26.McRae AF, Matigian NA, Vadlamudi L, Mulley JC, Mowry B, et al. Replicated effects of sex and genotype on gene expression in human lymphoblastoid cell lines. Hum Mol Genet. 2007;16:364–373. doi: 10.1093/hmg/ddl456. [DOI] [PubMed] [Google Scholar]
- 27.Stranger BE, Montgomery SB, Dimas A, Parts L, Ingle CE, et al. Patterns of cis regulatory variation in diverse human populations. doi: 10.1371/journal.pgen.1002639. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The International HapMap 3 Consortium. An integrated haplotype map of rare and common genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helgason A, Palsson S, Gudbjartsson DF, Kristjansson T, Stefansson K. An association between the kinship and fertility of human couples. Science. 2008;319:813–816. doi: 10.1126/science.1150232. [DOI] [PubMed] [Google Scholar]
- 30.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, et al. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Astori G, Vignati F, Bardelli S, Tubio M, Gola M, et al. "In vitro" and multicolor phenotypic characterization of cell subpopulations identified in fresh human adipose tissue stromal vascular fraction and in the derived mesenchymal stem cells. J Transl Med. 2007;5:55. doi: 10.1186/1479-5876-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josephson B, Dahlberg G. Variations in the cell-content and chemical composition of the human blood due to age, sex and season. Scand J Clin Lab Invest. 1952;4:216–236. doi: 10.3109/00365515209060661. [DOI] [PubMed] [Google Scholar]
- 34.Cheung VG, Spielman RS. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schadt EE, Molony C, Chudin E, Hao K, Yang X, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Idaghdour Y, Czika W, Shianna KV, Lee SH, Visscher PM, et al. Geographical genomics of human leukocyte gene expression variation in southern Morocco. Nat Genet. 2010;42:62–67. doi: 10.1038/ng.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choy E, Yelensky R, Bonakdar S, Plenge RM, Saxena R, et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genet. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, et al. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7:287–289. doi: 10.1038/nmeth.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 41.Rakyan VK, Chong S, Champ ME, Cuthbert PC, Morgan HD, et al. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci U S A. 2003;100:2538–2543. doi: 10.1073/pnas.0436776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenny EE, Gusev A, Riegel K, Lutjohann D, Lowe JK, et al. Systematic haplotype analysis resolves a complex plasma plant sterol locus on the Micronesian Island of Kosrae. Proc Natl Acad Sci U S A. 2009;106:13886–13891. doi: 10.1073/pnas.0907336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powell JE, Visscher PM, Goddard ME. Reconciling the analysis of IBD and IBS in complex trait studies. Nat Rev Genet. 2010 doi: 10.1038/nrg2865. [DOI] [PubMed] [Google Scholar]
- 46.Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006;2:e41. doi: 10.1371/journal.pgen.0020041. doi: 10.1371/journal.pgen.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histograms of heritability estimates for each gene. We plot histograms of (a) hg2 estimates for IFB and (b) hg2 estimates for IFA, across genes g.
(0.23 MB TIF)
Histograms of cis and trans heritability estimates for each gene. We plot histograms of (a) hg,cis2 estimates for IFB, (b) hg,trans2 estimates for IFB, (c) hg,cis2 estimates for IFA and (d) hg,trans2 estimates for IFA, across genes g.
(0.19 MB TIF)
Histograms of cross-tissue correlations for each gene. We plot a histogram of observed gene-specific cross-tissue correlations ρg.
(0.14 MB TIF)
Histograms of cross-tissue heritability estimates for each gene. We plot histograms of (a) ξg2 estimates, (b) ξg,cis2 estimates and (c) ξg,trans2 estimates, across genes.
(0.20 MB TIF)
Heritability results for each gene.
(1.82 MB TXT)
Average correlations between spouse-spouse, sib-sib, and parent-child pairs. We list the average correlation for each pair type and cohort, averaging across correlations for each gene g. We also list standard errors, computed via jackknife.
(0.03 MB DOC)
Concordance of gene-by-gene signatures of correlations in each pair type. We list values of Rsib-sib,parent-child, Rspouse,sib-sib and Rspouse,parent-child for each cohort (see text), along with the number of pairs of each type used to compute those values. For comparison purposes, we also list (in italics) values of Rsib-sib,parent-child computed using smaller subsets of pairs to match the number of pairs used to compute Rspouse,sib-sib or Rspouse,parent-child, as a smaller number of pairs leads to lower values of R.
(0.03 MB DOC)
Supplementary Note.
(0.04 MB DOC)