

Abstract
The modification of DNA by methylation is an important epigenetic mechanism that affects the spatial and temporal regulation of gene expression. Methylation patterns have been described in many contexts within and across a range of species. However, the extent to which changes in methylation might underlie inter-species differences in gene regulation, in particular between humans and other primates, has not yet been studied. To this end, we studied DNA methylation patterns in livers, hearts, and kidneys from multiple humans and chimpanzees, using tissue samples for which genome-wide gene expression data were also available. Using the multi-species gene expression and methylation data for 7,723 genes, we were able to study the role of promoter DNA methylation in the evolution of gene regulation across tissues and species. We found that inter-tissue methylation patterns are often conserved between humans and chimpanzees. However, we also found a large number of gene expression differences between species that might be explained, at least in part, by corresponding differences in methylation levels. In particular, we estimate that, in the tissues we studied, inter-species differences in promoter methylation might underlie as much as 12%–18% of differences in gene expression levels between humans and chimpanzees.
Author Summary
It has long been hypothesized that changes in gene regulation have played an important role in primate evolution. However, despite the wealth of comparative gene expression data, there are still only few studies that focus on the mechanisms underlying inter-primate differences in gene regulation. In particular, we know relatively little about the degree to which changes in epigenetic profiles might explain differences in gene expression levels between primates. To this end, we studied DNA methylation and gene expression levels in livers, hearts, and kidneys from multiple humans and chimpanzees. Using these comparative data, we were able to study the evolution of gene regulation in the context of conservation of or changes in DNA methylation profiles across tissues and species. We found that inter-tissue methylation patterns are often conserved between humans and chimpanzees. In addition, we also found a large number of gene expression differences between species, which might be explained, at least in part, by corresponding differences in methylation levels. We estimate that, in the tissues we studied, inter-species differences in methylation levels might underlie as much as 12%–18% of differences in gene expression levels between humans and chimpanzees.
Introduction
Changes in the regulation of gene expression levels have long been hypothesized to play an important role in primate evolution [1], [2]. To begin to address this hypothesis, a large number of studies have characterized gene expression differences across primates, in particular between humans and chimpanzees [3]–[9]. These studies have pointed to several classes of biological processes (such as transcriptional regulation, oxidative stress response, and a number of metabolic pathways), which might have evolved under natural selection in primates. In addition, in a few cases, comparative studies in primates have been able to draw strong connections between regulatory adaptations and ultimate physiological or anatomical phenotypes [10]–[15].
Despite the wealth of comparative gene expression data, there are many fewer studies of the mechanisms that underlie inter-primate differences in gene regulation (e.g., [12], [13], [16]–[18]). In particular, we know relatively little about the degree to which changes in epigenetic profiles might explain differences in gene expression levels between primates.
One of the most extensively studied epigenetic mechanisms is DNA methylation – an epigenetic modification that facilitates fine-tuned regulation of transcription rates [19], [20]. Spatial and temporal regulation of transcription by DNA methylation has been shown to play an important role in many contexts, including in female X-chromosome inactivation [21], [22], genomic imprinting [23], [24], and susceptibility to complex diseases in humans, especially cancers [25], [26]. Methylation is also essential for proper differentiation and development of mammalian tissues [27], [28]. For instance, the knockout of genes encoding for the DNA-methyl-transferase (DNMT) enzymes, which are responsible for de-novo methylation of DNA, results in embryonic lethality in mice [29], [30].
The causal relationship between changes in promoter DNA methylation and differences in gene regulation has been well established [28], [31]. It has been shown that hyper-methylation at promoter CpG islands typically results in decreased transcription of downstream genes [32]. When methylation is experimentally removed from promoter regions, transcription levels rise [33]. The specific mechanisms by which DNA methylation affects gene regulation are less clear, though DNA methylation is thought to interact with proteins (such as methyl-DNA binding proteins) that associate with histone modifications or the nucleosome in order to maintain a silenced chromatin state [28], [31], [34], [35]. Additionally, it has been proposed that the binding of the transcriptional machinery and enhancer-related transcription factors to methylated genomic regions is less frequent, resulting in decreased transcription levels or absolute gene silencing [28], [36].
Previous studies have typically described patterns of DNA methylation in a single or few tissues across species [26], [37]–[41] or in multiple tissues or developmental stages within a single organism [26], [27], [34], [42]–[45]. Comparative studies of DNA methylation across mammals have suggested that the role of DNA methylation in tissue-specific gene regulation is generally conserved. For example, after identifying Tissue-specific Differentially Methylated Regions (T-DMRs [42]), in heart, colon, kidney, testis, spleen, and muscle tissues in mice, Kitamura and colleagues were able to use the methylation status in orthologous human regions to distinguish between the corresponding human tissues [44]. Irizarry and colleagues [26], who studied genome-wide DNA methylation patterns in spleen, liver, and brain tissues from human and mouse, reported that 51% of T-DMRs are shared across both species. However, there also are a large number of potentially functional differences in methylation levels across species. In particular, in primates, Gama-Sosa and colleagues [39] found that relative methylation levels within tissues generally differ between species, with the exception of hyper-methylation in the brain and thymus, which were observed regardless of species. In addition, Enard and colleagues [38], who compared methylation profiles of 36 genes in livers, brains, and lymphocytes from humans and chimpanzees, reported significant inter-species methylation level differences in 22 of the 36 genes, in at least one tissue.
With few exceptions, however (e.g., [46]), comparative studies in primates have not explored the extent to which methylation differences between species might contribute to the genome-wide regulation of inter-species differences in gene expression levels. Towards this goal, we compared genome-wide gene expression levels and DNA methylation data in tissue samples from humans and chimpanzees.
Results
We characterized DNA promoter methylation across the genome in samples from heart, liver, and kidney tissues from both humans and chimpanzees, using two technical replicates from six individual samples of each tissue from each species (see Figure S1 for an illustration of the study design). Since genome-wide gene expression data were previously collected from the same tissue samples [8], we were able to study the relationships between DNA methylation and gene expression levels across tissues and species. The gene-specific expression level estimates and methylation profiles, for all samples, are provided in Table S1.
DNA methylation varies more across tissues than between humans and chimpanzees
We obtained methylation profiles from each sample (using two independent DNA extraction replicates) by using the Illumina HumanMethylation27 DNA Analysis BeadChip assay, which provides reproducible (Figure S2) quantitative estimates of methylation levels at 27,578 CpG-loci near transcription start sites. Since the 50 bp probes on the Illumina array were designed to interrogate human samples, we limited our analysis to probes that were a perfect sequence match to the chimpanzee genome. In addition, we only used probes that were associated with genes for which we had expression measurements across the three tissues [8]. Following these exclusion criteria, we retained 10,575 CpG site probes in the putative promoter regions of 7,723 genes (see Methods for more details). At each probe, DNA methylation levels were estimated using the Illumina-recommended β values, which are essentially estimates of the proportion of methylated DNA at each CpG site (see Methods).
We note that limiting our analysis to identical methylation probes in humans and chimpanzees resulted in a slight (0.5%) but significant decrease of the median sequence divergence estimates within 500 bp windows around the retained probes (Figure S3). As a result, it is possible that, in what follows, we slightly underestimate the proportion of inter-species differences in methylation levels. However, we confirmed that limiting our analysis to identical methylation probes in the two species did not result in a noticeable shift in the distribution of expression levels of the associated genes, nor in the proportion of observed differences in gene expression levels between the two species.
As a first step of our analysis, we examined patterns of promoter methylation across tissues and species. As expected [28], [31], we found a negative correlation between methylation and gene expression levels in each individual, whereby, regardless of tissue and species, the promoters of highly expressed genes tended to be lowly methylated while the promoters of lowly expressed genes were usually highly methylated (Figure 1A; Figure S4). We also confirmed that methylation patterns on the X-chromosome account for variation due to sex, regardless of species, as expected due to X-inactivation in mammalian females [21] (the first component of variance, corresponding to sex, accounts for 67% of the overall variation in the X-chromosome data; Figure 1B). Finally, we found that genes known to be imprinted in humans tend to show a similar hemi-methylation pattern in chimpanzees (permutation tests P<0.001; Figure 1C), suggesting that the imprinted status of this set of genes is conserved in the two species.
Figure 1. Genome-wide methylation patterns across all samples.

(A) Probability density functions (y-axis) of estimated promoter methylation levels (plotted as β-values on the x-axis) for genes whose expression levels are in the lowest (black) and highest (red) quartiles. (B) Principal components analysis of the methylation data from the 365 X-chromosome probes from all samples (PC1 is plotted on the y-axis, with sample indices on the x-axis). (C) Density (y-axes) histograms of β-values in humans (left) and chimpanzees (right) for 90 CpG-sites associated with 27 genes previously identified as imprinted in humans. The red lines indicate the β-value distributions in the genes with evidence for imprinting, while the grey lines are β-value distributions in ten sets of 90 randomly chosen probes.
For the remainder of the analyses, we considered only the methylation data from autosomal probes. We observed that methylation patterns across different tissues and species were quite distinct (Figure 2; similar patterns for the expression data in Figure S5). The first component of variance for the autosomal probes, accounting for 69.3% of the overall variation in methylation, distinguished samples based on tissue, while the second principal component (accounting for 12.7% of the overall variation), separated the species. Overall, an average of 14.5% (range of 8.2–26.1%, depending on the pairwise comparison) of the assayed promoter CpG sites were differentially methylated between tissues within a species, while an average of 8.6% of the CpG sites (range of 3.4–13.5%, depending on the tissue) were differentially methylated between humans and chimpanzees (at FDR<0.001). Reassuringly, these patterns recapitulate previous observations in human and mouse [26], [44].
Figure 2. Principal components analysis of the methylation profiles.

This analysis only considers the 9,911 autosomal CpG sites from all samples (PC1 and PC2 are plotted on the x-axis and y-axis, respectively).
Methylation patterns in T-DMRs are often conserved
We identified regions with tissue-specific patterns of methylation (T-DMRs [26], [42]) by analyzing the data from each species separately (Figure 3). Specifically, we modeled the methylation data (namely, the β values) from each autosomal CpG site independently, using a linear mixed-effects model with a fixed effect for the tissue and a random effect to account for variation between individuals. We tested for differences in methylation levels between tissues by using likelihood ratio tests within the framework of the linear model (see Methods). Using this approach, we identified 1,578 and 1,401 T-DMRs in humans and chimpanzees, respectively (at an FDR<0.001; Figure 3A; Table S1).
Figure 3. Conservation of tissue-specific differentially methylated regions.

(A) Venn diagrams of the number of T-DMRs classified in each species per tissue. (B) Probability density functions (y-axis) of distributions of Pearson correlations (x-axis) between methylation and gene expression levels across tissues, in human (solid lines) and chimpanzee (broken lines), for all genes expressed in at least one tissue (black), genes associated with a T-DMR in only one species (blue), and genes associated with a conserved T-DMRs (red). See Figure S4 for plots of the tissue-specific data (C) A representative example of a heart-specific T-DMR associated with the CASQ1 gene. Plotted in the left panels are the methylation β-values (y-axis), and in the right panels are the normalized gene expression levels (y-axis) in liver, kidney, and heart samples from human (top) and chimpanzee (bottom).
Tissue-specific methylation profiles are of interest because they may underlie tissue-specific patterns of gene expression levels. To test this hypothesis, we calculated, separately for each species, Pearson correlation values between promoter methylation profiles and the corresponding gene expression levels, across the three tissues. If methylation was consistently used to silence tissue-specific gene expression across the genome, we would expect to observe an abundance of negative correlations between the estimates of methylation and gene expression levels. However, when we considered the data for all genes that were expressed in at least one tissue, we found no evidence for an enrichment of negative correlations between methylation and gene expression levels (Figure 3B, Figure S6; 48% and 49% of the correlation values were negative in human and chimpanzee, respectively). In contrast, when we restricted the analysis to species-specific T-DMRs, we found an enrichment of negative correlations between methylation and gene expression levels (Figure 3B; 64% and 67% of correlation values were negative in human and chimpanzee, respectively; Fisher's exact P<10−16). This result suggests that T-DMRs underlie a subset of gene expression differences across tissues, a notion that is consistent with the important role played by DNA methylation in tissue differentiation in a wide range of species [42].
We then focused on the subset of T-DMRs with the same methylation pattern in both species. We found that 18–26% (depending on the tissue) of loci classified as T-DMRs in either human or chimpanzee are shared between the two species (Figure 3A, Table S2), a highly significant overlap compared to that expected by chance alone (hypergeometric distribution P values across all pairwise tissue comparisons <10−16). Importantly, the observation of a significant overlap in T-DMRs across species is robust with respect to the statistical cutoff used to classify T-DMRs (0.001≤FDR≤0.05; Table S2). Interestingly, when we considered correlations of methylation and gene expression levels only at conserved T-DMRs, we found an even more pronounced enrichment of negative correlations (Figure 3B and 3C; 72% of the correlation values were negative, regardless of species; Fisher's exact for an enrichment of negative correlations: P<10−23), suggesting that conservation of T-DMRs often relates to functionally important tissue-specific patterns of gene regulation.
It is perhaps interesting to note that we did not find a difference in the correlation of methylation and expression levels between T-DMR CpG sites that are located within or outside an annotated CpG island (as defined by [47]; Figure S7).
When we examined the functional annotations of genes associated with species-specific T-DMRs as well as conserved T-DMRs (using gene ontology annotations), we found an expected enrichment of genes annotated as important in ‘developmental’ processes, regardless of tissue (P<5×10−3; FDR<0.3; Table S3), congruent with the importance of epigenetic modification in tissue differentiation. We also found enrichments of tissue-specific biological processes, such as genes associated with cardiac muscle cell differentiation processes among heart T-DMRs (P<5×10−3; FDR<0.3), genes associated with embryonic organ morphogenesis and embryonic organ development processes among kidney T-DMRs (P<5×10−4; FDR<0.05), and genes associated with blood coagulation and with the regulation of body fluid levels (putatively involved in homeostatic functions) among liver T-DMRs (P<10−5; FDR<6×10−3 and P<10−4; FDR<0.007, respectively). The enrichment of genes associated with both developmental and tissue-specific processes among genes associated with T-DMRs is consistent with previous observations [27], [42]. Furthermore, when we considered only conserved T-DMRs, we observed a significant under-representation of genes associated with nucleic-acid and primary metabolic processes in all three tissues studied (all P<5×10−3; FDR<0.01; Table S4). This result suggests that the epigenetically-mediated tissue-specific regulation of these core processes tends to be conserved between humans and chimpanzees.
Inter-species differences in methylation
We next focused on the relationships between inter-species differences in methylation profiles and differences in gene expression levels between humans and chimpanzees. To estimate the relative contribution of changes in DNA methylation to inter-species differences in gene expression levels, we used linear regression analysis to account for promoter methylation effects (per autosomal CpG site) before analyzing the gene expression data from both species. We analyzed methylation and gene expression data in each tissue using a linear model framework similar to the one described in Blekhman et al. 2008 [8]. We then compared the evidence supporting an inter-species difference in gene expression levels before and after correcting for methylation profiles (see Methods for more details).
For the majority of genes (78%, 82%, and 77% in liver, kidney, and heart, respectively; Figure 4A), the evidence for a difference in expression level between the species was similar, regardless of whether or not methylation status was taken into account. For a small subset of genes (1%, 3%, and 2% in liver, kidney, and heart, respectively), we did not find compelling evidence for a difference in expression level between the species using the uncorrected expression level data, but after correcting for methylation levels using regression analysis, we rejected the null hypothesis of no inter-species differences in gene expression level (at an FDR<0.01). This observation, however, is unlikely to be biologically meaningful, since it is expected by chance alone (by permutation analysis; P>0.434 for all tissues; Figure S8).
Figure 4. Inter-species methylation and gene expression differences.

(A) Scatter-plots of the p-values obtained by testing the null hypothesis of no differences in gene expression levels between human and chimpanzee before (x-axis) and after (y-axis) regressing out methylation levels. The solid purple lines correspond to a 1% FDR threshold. (B) Boxplots of the distributions, based on 1000 permutations, of the percentage of genes for which the evidence for inter-species differences in gene expression levels is expected to be reduced following the correction for methylation levels, by chance alone. Yellow points indicate the percentages seen in the actual data. (C) A representative example of the ZBTB80S gene, which is associated with inter-species promoter methylation differences in the kidney. In the left and middle panels are the human and chimpanzee methylation β-values and normalized gene expression levels, respectively. In the right panel are the normalized gene expression levels for ZBTB80S, after correcting for the methylation β-values.
In contrast, in all three tissues, we found a significant enrichment of genes for which the evidence for inter-species differences in expression level was compelling (FDR<0.01) before, but not after we corrected for the methylation levels (21%, 15%, and 21% in liver, kidney, and heart, respectively, permutation analysis yields P<0.001 for all tissues; Figure 4B and 4C). Based on the expectation of such a pattern by chance alone (by permutations – see Methods for details), we estimated that, in the three tissues we studied, inter-species differences in promoter DNA methylation might underlie as much as 12–18% of differences in gene expression levels between humans and chimpanzees.
When we analyzed the data considering only the sets of genes that have negative correlations between methylation and gene expression levels (as expected if methylation is used to silence gene expression), we found that 8.1%, 7.6%, and 8.8% of interspecies differences in gene expression levels in liver, kidney, and heart, respectively, might be explained by corresponding methylation differences. The extent to which inter-species gene expression differences might be explained by methylation differences between the species was similar regardless of whether the methylated site was within or outside an annotated CpG islands (Figure S9).
Discussion
We explored the extent to which putatively functional DNA methylation differences between tissues are conserved in humans and chimpanzees, and estimated the relative contribution of inter-species changes in methylation levels to gene expression differences between the two species. To do so, we collected DNA methylation and gene expression data from frozen human and chimpanzee primary tissue samples. While we chose to work with tissues that are relatively homogenous with respect to their cellular composition, we could not measure the precise composition or choose to work with particular cell types, because the samples were frozen. Similarly, we could not stage the tissues or control the environment of the donor individuals because the samples were collected post mortem. These are limitations shared by nearly all comparative molecular studies of primary tissues from humans and other apes (see [8], [9] for more detailed discussions of the limitations associated with studying gene regulation in primate tissues).
The challenge is therefore to focus on patterns in the data that should be robust with respect to the aspects of the study design that could not be controlled. For example, it is reasonable to expect that differences in environment, staging, and cellular composition across samples will tend to increase variation of measurements within, and especially between species. For that reason, our analysis of conserved inter-tissue gene expression differences and tissue-specific methylation patterns is likely to be conservative. Indeed, because of our inability to minimize environmental differences across the donor individuals, it is likely that we are underestimating the proportion of conserved inter-tissue gene expression differences and conserved T-DMRs.
In turn, when we focus on inter-species differences in DNA methylation and gene expression levels, it is important to note that our study design does not allow us to distinguish between regulatory differences due to either heritable or environmental effects. Studies in model organisms typically do so by controlling the environments of all subjects, a restriction we cannot apply when studying primate tissues. However, we have previously shown that estimates of differences in gene regulation between humans and chimpanzees based on six randomly sampled individuals are stable [8], [9]. Regardless of the underlying mechanism, it is likely that the analysis of the data uncovered mostly steady-state inter-species regulatory differences. Thus, even if differences in environments underlie a subset of the observed regulatory differences between humans and chimpanzees, our previous work suggests that it is likely that, in most cases, we capture the effects of general environmental differences between the species, not just between the samples used.
DNA methylation and differences in gene expression across tissues
We found a substantial degree of conservation of tissue-specific methylated regions in human and chimpanzee. This observation is not surprising given that previous studies found a marked conservation of T-DMRs between human and mouse, which are much more distantly related [26], [41], [43], [44]. On the other hand, 7.0%, 21.6%, and 23.8% of the kidney, heart, and liver T-DMRs, respectively (identified in either species), were differentially methylated (in the relevant tissue) between humans and chimpanzees, while only 3.3%, 8.0%, and 11.8% of non-TDMRs in these three tissues were differentially methylated between the two species (P<10−10 for all pairwise comparisons).
The conservation of T-DMR profiles yet the generally faster rate of inter-species change in promoter methylation at T-DMRs compared to non-T-DMRs are intriguing. These observations are difficult to explain by technical or uncontrolled aspects of the study design, because it is unlikely that those confounding factors would affect methylation at T-DMRs differently than at non-T-DMRs. Instead, it is likely that the different patterns truly reflect a functional difference between methylation at T-DMRs and at non-T-DMR CpG sites (in the studied tissues).
Though there is substantial evidence that DNA methylation levels upstream of genes are often inversely correlated with gene expression levels [24], [28], [31], recent studies proposed that methylation of promoters may play only a relatively minor role in the regulation of tissue-specific gene expression [34]. In particular, Maunakea et al. [48] posited that methylation of gene body regions (in regions that putatively serve as alternative promoters) might have a greater influence on regulatory differences across tissues. While we cannot use our data to ask about the relative importance of different types and locations of epigenetic marks to tissue-specific gene regulation, our observations strongly imply that any such debate would benefit from further investigation into the evolution of epigenetic profiles. Indeed, in addition to a faster rate of evolutionary change of the methylation profiles in T-DMRs, we found evidence for an enrichment of inverse correlations between inter-tissue gene expression patterns and promoter methylation profiles at genes associated with T-DMRs, but not when we considered all genes (the latter observation is consistent with the findings of Weber et al. [34] and Maunakea et al. [48]). Our results, therefore, imply that tissue-specific promoter methylation patterns may play especially important roles in regulating gene expression. The data also suggest that altered methylation levels, primarily at these sites, may underlie regulatory differences between species.
DNA methylation and inter-species differences in gene regulation
We estimated that as much as 12–18% (depending on the tissue) of inter-species differences in gene expression levels might be explained, at least in part, by changes in DNA methylation patterns. It is important to note that this statement is based on the proposed mechanism by which DNA methylation affects the rate of transcription and overall levels of gene expression [28], [31]. Though we did not perform experiments from which causality can be directly deduced, a causal relationship between changes in DNA methylation and gene regulation is strongly supported by previous studies (e.g., [24], [28], [31]). When we only consider negative correlations between methylation and gene expression levels to be indicative of a putative causal relationship, 8–9% of inter-species differences in gene expression levels might be explained by corresponding changes in DNA methylation.
However, other mechanisms are also likely [34], [43]. While DNA methylation is typically considered a silencing mechanism, high levels of methylation may be causally linked to increased gene expression levels. For example, the methylation of a repressor site could prevent the binding of repressor transcription factors, or enhancer transcription factors could favor binding to a methylated site rather than to the unmethylated site [49]–[51]. The observation of a small enrichment of positive correlations between methylation and expression when only T-DMRs are considered provides additional support for these types of mechanisms. Thus, perhaps as much as 12–18% of differences in gene expression levels between humans and chimpanzees might be explained by inter-species changes in DNA methylation.
Either way, our results suggest that DNA methylation differences in promoter regions might account for, at most, a modest proportion of inter-primate differences in gene expression levels (we confirmed that our estimates do not rely on arbitrary choices of specific statistical cutoffs; Tables S2 and S5). Many inter-species differences in promoter methylation are not associated with gene expression differences between the species. One explanation for that observation may simply be that these methylation patterns are not regulatory or functional. An alternative, more interesting possibility to consider, is that a subset of genes whose regulation differed between species later acquired modifications in nearby DNA methylation patterns to accommodate (or even partially counteract) the original expression level changes.
Since we assayed methylation using a pre-designed microarray, changes in DNA methylation in un-assayed genomic regions might explain additional regulatory differences between the species. In particular, while our assay focused on methylation at promoter regions, it has been recently shown that as a class, gene-body methylation profiles might explain a larger proportion of variation in gene expression levels than methylation profiles at currently annotated promoters [26], [48]. With the advent of new sequencing technologies, it will soon be feasible to extend our comparative approach to characterize genome-wide patterns of methylation.
In summary, we have taken some of the first steps towards characterizing variation in one mechanism that affects gene expression differences between closely related primate species [16], [17]. In a broader context, DNA methylation is just one of many mechanisms that have been posited to regulate gene expression levels [28], [31], [52]. In that sense, our study is a step towards the ultimate goal of understanding the relative importance of changes in different regulatory mechanisms to human evolution. Our observations indicate that at least 82% of gene expression differences between humans and chimpanzees (in the three studied tissues and specific promoter CpG sites examined) are not likely to be explained by differences in promoter DNA methylation.
Methods
DNA methylation data
We collected methylation data from the same human and chimpanzee liver, kidney, and heart tissue samples used in Blekhman et al. 2008 [8] (Figure S1; see Table S6 for details on the samples). DNA was extracted from each sample (6 human and 6 chimpanzee samples from each of the three tissues) in two independent technical replicates using the QIAamp DNA Mini Kit (Qiagen) (with the exception of chimpanzee sample CK2, for which DNA was only available for one replicate – see Table S4). The methylation profile of each sample was assayed using the Illumina HumanMethylation27 DNA Analysis BeadChip, which assays methylation at 27,578 CpG sites. Methylation array data are deposited to the NCBI GEO database under the accession number GSE26033 (http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE26033).
To facilitate an unbiased comparison of methylation and gene expression levels in the human and chimpanzee samples, we first mapped the 27,578 50-bp Illumina probes to the human genome sequence (hg18) using BLAT [53] and MAQ [54]. We retained only the 26,690 probes that unambiguously mapped to a single location in the human genome with a maximum of two mismatches. These probes were then associated with the nearest gene using Ensembl gene annotation, and we retained only the subset of probes associated with genes that were represented on the multi-species gene expression microarray used by Blekhman et al. 2008 [8]. This resulted in the retention of 19,849 probes, associated with 11,059 genes. Finally, since the Illumina array was designed based on human genomic sequence, we limited our analysis to probes that were a perfect sequence match to a single location in the chimpanzee genome, by mapping the remaining 19,849 probes to the chimpanzee genome (panTro2) using BLAT [53] and MAQ [54]. We retained 10,575 probes that mapped uniquely to the chimpanzee genome with no sequence mismatches. This step ensures that our relative methylation measurements are not biased due to the effect of sequence mismatches on hybridization intensities. The resulting set of 10,575 probes is associated with 7,723 genes, which are present on every chromosome in the genome except for the Y-chromosome (Figure S10). The majority (97%) of retained probes are located within 2 kb of an annotated transcription start site of the associated gene (Figure S11). We note that a similar screen for probes that were a perfect match to the genomes of human, chimpanzee, and rhesus macaque resulted in the retention of only 1,944 probes (associated with 1,715 genes). For that reason, we limited our current study to a comparison between human and chimpanzee samples.
All samples were hybridized to the Illumina HumanMethylation27 DNA Analysis BeadChip at the Southern California Genotyping Consortium facility following standard manufacturer's instructions. Basic quality checks were performed using Illumina's BeadStudio software. Of the 10,575 probes we considered as the final dataset, 299 had missing data for one or more individuals and were discarded in all subsequent analyses. This resulted in 9,911 autosomal probes (corresponding to 7,291 genes) and 365 probes on the X-chromosome (corresponding to 266 genes). Since the probes map to distinct CpG island regions, which can affect downstream gene expression independently, we treated methylation levels from each CpG probe as distinct data points in all subsequent analyses. We further classified each probe as being located confidently within a CpG island region or outside of a strict CpG island region using the CpG Islands track information downloaded from UCSC [47].
For each sample, the methylation status at a probed location was summarized as:
where M and U denote the signal emitted from the beads assaying the methylated and unmethylated versions at each site, respectively. Due to the number of samples being interrogated, it was necessary to hybridize the samples in two balanced batches. We observed a small difference in the mean β-value between batches, and corrected for this difference by standardizing the means across batches. After this correction, there was no further evidence for a batch effect.
To further assess the quality of the data, we calculated pairwise correlations between the β-values for all hybridized samples (Figure S2). As expected, technical replicates (which were independent DNA extractions) were the most highly correlated (36 comparisons; median r = 0.99), followed by samples from the same tissue and species (396 comparisons; median r = 0.98), samples from the same tissue across species (432 comparisons; median r = 0.97), samples from different tissues from the same species (864 comparisons; median r = 0.95), and samples from different tissues and different species (864 comparisons; median r = 0.93).
To look for evidence of imprinting in both humans and chimpanzees, we focused on a set of 27 genes (associated with 90 methylation probes) known to be imprinted based on the Imprinted Gene Catalog (IGC) at http://igc.otago.ac.nz/. To assess whether the patterns of DNA methylation at these imprinted genes were likely to occur by chance, we compared the observed proportion of hemi-methylated sites (defined as 0.3<β<0.7) to the distribution obtained by analyzing methylation patterns in 1000 randomly chosen sets of 90 methylation probes, associated with an average of 27 genes (range 26–28).
Gene expression data
Measurements of gene expression levels for all samples in our study were previously described by Blekhman et al. (2008) [8]. These data are available at the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under series accession number GSE11560. In that study, a multi-species microarray was used to estimate gene expression levels in cDNA samples from humans, chimpanzees, and rhesus macaques. The multi-species array includes orthologous probes for 18,109 genes, thus facilitating comparisons of gene expression levels between species without the confounding effects of sequence mismatches on hybridization intensities [8]. Since our current study focused only on the human and chimpanzee gene expression data, we re-normalized the expression data using only the human and chimpanzee probes on the array, using the same modified quantile normalization approach described in Blekhman et al. (2008) [8]. All further analyses used these re-normalized gene expression estimates. When examining the relationships between gene expression and methylation levels, we limited our analyses to genes that were either expressed in at least one tissue (for inter-tissue comparisons within a species) or expressed in at least one species (for the inter-species comparisons within a tissue), using a conservative threshold for defining expression, based on the entire distribution of expression values (normalized expression value of 8; see Figure S14 in Blekhman et al. (2008) [8]).
Statistical analysis
All statistical analyses were performed using the R statistical framework (http://www.r-project.org).
Identifying tissue-differentially methylated regions (T-DMRs)
To identify T-DMRs, we modeled the methylation level of each CpG site separately within both humans and chimpanzees using a linear mixed-effects model. Specifically, for each of the 9,913 probes (associated with 7,291 genes) located on the autosomal chromosomes, if yijk represents the β value for technical replicate k (k = 1 or 2), for individual j (j = 1,…,6), from tissue i (i = heart, liver, or kidney), we assume that:
| (1) |
where:
Here, αi represents the mean methylation value at a given site in tissue i. To account for correlation between samples of the same tissue from different individuals, a random effect, ρij, which follows a N(0,σ2rand) distribution, is also included in the model.
To determine whether a CpG site was likely to fall within a T-DMR, we assessed how well the model (1) fitted the data under various parameterizations of μijk. The three types of parameterizations considered are:
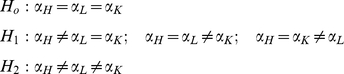 |
In the simplest model (H0), the region's methylation value is assumed to be constant across all three tissues, while in the second alternative (H2) the methylation value is allowed to differ between all three tissues. The first alternative (H1) models the situation where the methylation level at the site of interest is constant in the two non-target tissues but differs in the target tissue. All models are fitted using a restricted maximum likelihood (REML) framework, and the maximum likelihoods were calculated.
In this study, we are interested in identifying sites whose methylation levels are best modeled by H1. To find such sites, we first used a likelihood-ratio test statistic (with one degree of freedom) to exclude sites where H2 provides a better fit to the data than H1 (specifically, if the likelihood-ratio p-value was less than 0.05, we removed these sites from the analysis). H2 provides a better fit for 1220 and 886 (in humans and chimpanzees, respectively) of the total 9911 autosomal CpG sites. For the remaining positions, we examined whether there was significant evidence to reject H0 in favor of H1 using a likelihood-ratio test statistic (which we compared to a χ2 distribution with 1 degree of freedom). We corrected for multiple testing using the FDR approach of Storey and Tibshirani [55].
Gene ontology analysis for T-DMRs
We used GeneTrail (http://genetrail.bioinf.uni-sb.de) [56] to test for enrichments of functional annotations among different classes of T-DMRs. In all tests, we used a background set of genes that were present in our study and classified as expressed in at least one tissue (conditional on a normalized expression value of 8). The tests were performed using all GO categories and KEGG pathways. We calculated p-values using a Hyper-geometric distribution and report false discovery rates for each p-value.
A joint analysis of methylation and gene expression levels
To examine whether changes in gene expression levels between humans and chimpanzees (within each tissue) can be explained by inter-species differences in methylation levels, we extended the linear mixed-effects model framework described in Blekhman et al. (2008) [8] to include methylation as a covariate. However, since we have to correct the multi-species array data for probe-effects [8], it is difficult to interpret the methylation coefficient when it is added directly to the model, since it is confounded with the probe effects. Consequently, we used an alternative approach in which we used regression to correct for the methylation effect. Specifically, for each gene-tissue combination, we tested for differences in expression level between human and chimpanzee after regressing out the following effects:
Expression microarray probe effects only
Expression microarray probe effects and CpG-specific methylation levels
To do this, we used a fully parameterized model where gene expression probe effects, CpG-probe methylation values, and species effects were explanatory variables. Additionally, a random effect was used to account for variability between biological replicates. Specifically, if ysroi denotes the normalized log2 intensity expression value for individual i (i = 1,…,6), from species s (s = human or chimpanzee) measure at probe r (r = 1,…,7), which is derived from species o, we assume that:
where:
Here, μs denotes the species effect, πro is a fixed-effect representing the probe effect for each individual probe within a probe-set and the composition effect of species-specific orthologous probes, and κsro is a fixed-effect representing the attenuation of hybridization intensities due to sequence mismatches between species of RNA and a species-specific derived probe, which are different for each individual probe within a probe set (see [8] for more details). Additionally, γsi is a random effect (following a N(0,σ2rand) distribution) and βsi denotes the β value for the methylation probe of interest for individual i from species s. Upon fitting this model, using the lmer package within the R statistical framework, estimates of the parameters and the residuals were obtained. To obtain corrected measures of expression for each individual from each species, 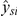 when probe and methylation effects are regressed out (scenario 2), we defined
when probe and methylation effects are regressed out (scenario 2), we defined 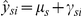 . When we only regressed out probe effects (scenario 1), the corrected values are defined as
. When we only regressed out probe effects (scenario 1), the corrected values are defined as 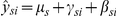 . In both of these scenarios, once the corrected data were obtained, we tested for differences in gene expression levels as follows. If, for each tissue-gene combination, xsik denotes the (corrected) level of expression for replicate k of individual i from species s, we modeled these data as follows:
. In both of these scenarios, once the corrected data were obtained, we tested for differences in gene expression levels as follows. If, for each tissue-gene combination, xsik denotes the (corrected) level of expression for replicate k of individual i from species s, we modeled these data as follows:
where:
Here, αs is a species effect, and ρsi is a random individual effect. Subsequently, to test for inter-species differences in expression levels, we compare the following hypotheses:
Here, the null model assumes equal expression level between the two species, and the alternative assumes different expression levels. Evidence against the null model was determined using a likelihood-ratio test statistic (compared against a chi-squared distribution with one degree of freedom). By performing this analysis independently for each CpG-gene combination in all tissues, we obtained a p-value indicating the strength of the evidence against the null hypothesis, before (under scenario 1 above) and after (under scenario 2 above) accounting for the region's DNA methylation status. By comparing these p-values, we were able to identify genes within each tissue where the difference in expression level between human and chimpanzee was likely explained by inter-species differences in DNA methylation.
To assess the statistical significance of our observations, we permuted the methylation values for a given gene across all individuals (maintaining replicate correlations, but allowing labels to permute across species classifications). Subsequently, we repeated the analysis described above to obtain an expected distribution of discrepancies between the methylation-corrected and uncorrected data. We performed 1000 permutations and p-values were calculated based on the number of times we observed as many or more discrepancies in the permuted compared to the real data.
In order to estimate the proportion of genes for which methylation differences might underlie gene expression differences, we treated the medians of the permutation distributions from each tissue as background levels. For each tissue, we then subtracted the background level from the observed proportion of genes with reduced evidence for inter-species differences in gene expression levels, once methylation was taken into account.
Supporting Information
An illustration of the methylation profiling study design.
(1.48 MB TIF)
Boxplots of distributions of pairwise Pearson correlations between methylation array data, by category.
(0.82 MB TIF)
Boxplots of the distributions of the proportion of divergent nucleotides between human and chimpanzee in 500bp windows around each 50bp probe. The probes are divided into those that are a perfect and unique match to both the human and chimpanzee genomes (left) and those that are a perfect and unique match to the human genome but not the chimpanzee genome (right).
(0.15 MB TIF)
Distributions of methylation values (x-axis) within each species-tissue type, for CpG islands associated with genes whose expression levels rank at different quartiles (colored lines).
(0.89 MB TIF)
Principal components analysis of the expression profiles. Considering the 7,291 autosomal genes from all samples (PC1 and PC2 are plotted on the x-axis and y-axis, respectively).
(0.11 MB TIF)
Density (y-axis) histograms of distributions of Pearson correlations (x-axis) between methylation and gene expression levels across tissues in human (solid lines) and chimpanzee (broken lines) for all genes expressed in at least one tissue (black; plots are identical in the three panels), genes associated with a T-DMR in only one species (blue; data from T-DMRs in liver, kidney, or heart are plotted separately in each panel), and genes associated with a conserved T-DMRs (red; separated by tissue).
(0.49 MB TIF)
Boxplots of distributions of Pearson correlations (y-axis) between methylation and gene expression levels across tissues in human (bold lines) and chimpanzee (non-bold lines) for all genes expressed in at least one tissue (black), genes associated with a T-DMR in only one species (blue), and genes associated with conserved T-DMRs (red). Each category is further divided into CpG sites that are located within CpG islands (“In”) or outside of a CpG island (“Out”). The P-values for a significant difference between the “inside” and “outside” distributions for each category are located below the x-axis.
(0.18 MB TIF)
Boxplots of the distributions, based on 1,000 permutations, of the percentage of genes for which the evidence for inter-species differences in gene expression levels is expected to increase following the correction for methylation levels, by chance alone. Yellow points indicate the percentages seen in the actual data.
(0.07 MB TIF)
Barplots of the percentages of genes in each tissue for which the evidence for inter-species differences in gene expression levels is reduced following the correction for methylation levels. Each tissue is further divided into CpG sites that are located within CpG islands (“In”) or outside of a CpG island (“Out”). The P-values for a significant difference (compared to expectations from a chi-squared distribution) between the “inside” and “outside” percentages for each category are located above the barplots.
(0.16 MB TIF)
Histogram of the number of CpG site probes (y-axis) on each chromosome, except the Y-chromosome (x-axis).
(0.23 MB TIF)
Frequency (y-axis) histogram of the distances from CpG sites assayed in our study to the transcription start sites (TSS) of the nearest annotated genes. The inset is an expanded view of frequency (y-axis) histogram near the 2kb around the TSS, where 97% of the probes are located.
(0.22 MB TIF)
Summary of the data for all CpG probes.
(6.64 MB XLS)
Percentages of negative correlation between methylation and expression levels in T-DMRs across a range of statistical cutoffs used to classify T-DMRs.
(0.03 MB XLS)
Gene ontology categories that are the most significantly enriched or depleted GO annotations among genes that are classified as T-DMRs in either species (P<0.01). For this analysis, we used a background of all genes assayed in our study that are expressed in at least one tissue. GO tables for T-DMRs specific to each tissue are located on individual sheets.
(0.06 MB XLS)
Gene ontology categories that are the most significantly enriched or depleted GO annotations among genes that are classified as T-DMRs in both species (shared T-DMRs) (P<0.001). For this analysis, we used a background of all genes assayed in our study that are expressed in at least one tissue. GO tables for T-DMRs specific to each tissue are located on individual sheets.
(0.03 MB XLS)
Estimates of the percentage of genes for which methylation differences could underlie inter-species expression differences, across a range of significance thresholds.
(0.03 MB XLS)
Information on the 36 samples used in this study.
(0.09 MB DOC)
Acknowledgments
We thank the Yerkes primate center, the Southwest Foundation for Biomedical Research, MD Anderson Cancer Center, and Anne Stone for providing primate tissue samples. We thank Ran Blekhman for scripts and help with re-normalizing the gene expression data and Jenny Tung for help estimating divergence rates, as well as members of the Gilad and Pritchard labs for discussions and/or for comments on the manuscript.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by the Howard Hughes Medical Institute and NIGMS grants GM084996 and GM077959 to YG and by MH084703 to JKP. AAP is supported by an American Heart Association pre-doctoral fellowship. JTB is supported by a Sir Henry Wellcome postdoctoral fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.King M-C, Wilson AC. Evolution at Two Levels in Humans and Chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 2.Britten RJ, Davidson EH. Gene Regulation for Higher Cells: A Theory. Science. 1969:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 3.Enard W, Khaitovich P, Klose J, Zöllner S, Heissig F, et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296:340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- 4.Khaitovich P, Muetzel B, She X, Lachmann M, Hellmann I, et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Research. 2004;14:1462–1473. doi: 10.1101/gr.2538704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- 6.Cáceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, et al. Elevated gene expression levels distinguish human from non-human primate brains. Proc Natl Acad Sci USA. 2003;100:13030–13035. doi: 10.1073/pnas.2135499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karaman MW, Houck ML, Chemnick LG, Nagpal S, Chawannakul D, et al. Comparative analysis of gene-expression patterns in human and African great ape cultured fibroblasts. Genome Research. 2003;13:1619–1630. doi: 10.1101/gr.1289803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blekhman R, Oshlack A, Chabot AE, Smyth GK, Gilad Y. Gene regulation in primates evolves under tissue-specific selection pressures. PLoS Genet. 2008;4:e1000271. doi: 10.1371/journal.pgen.1000271. doi: 10.1371/journal.pgen.1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. Sex-specific and lineage-specific alternative splicing in primates. Genome Research. 2010;20:180–189. doi: 10.1101/gr.099226.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, et al. Human-specific gain of function in a developmental enhancer. Science. 2008;321:1346–1350. doi: 10.1126/science.1159974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babbitt CC, Silverman JS, Haygood R, Reininga JM, Rockman MV, et al. Multiple Functional Variants in cis Modulate PDYN Expression. Molecular Biology and Evolution. 2010;27:465–479. doi: 10.1093/molbev/msp276. [DOI] [PubMed] [Google Scholar]
- 12.Warner LR, Babbitt CC, Primus AE, Severson TF, Haygood R, et al. Functional consequences of genetic variation in primates on tyrosine hydroxylase (TH) expression in vitro. Brain Research. 2009;1288:1–8. doi: 10.1016/j.brainres.2009.06.086. [DOI] [PubMed] [Google Scholar]
- 13.Loisel DA, Rockman MV, Wray GA, Altmann J, Alberts SC. Ancient polymorphism and functional variation in the primate MHC-DQA1 5′ cis-regulatory region. Proc Natl Acad Sci USA. 2006;103:16331–16336. doi: 10.1073/pnas.0607662103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rockman MV, Hahn MW, Soranzo N, Zimprich F, Goldstein DB, et al. Ancient and recent positive selection transformed opioid cis-regulation in humans. Plos Biol. 2005;3:e387. doi: 10.1371/journal.pbio.0030387. doi: 10.1371/journal.pbio.0030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 16.Blekhman R, Oshlack A, Gilad Y. Segmental Duplications Contribute to Gene Expression Differences Between Humans and Chimpanzees. Genetics. 2009;182:627–630. doi: 10.1534/genetics.108.099960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chabot A, Shrit RA, Blekhman R, Gilad Y. Using reporter gene assays to identify cis regulatory differences between humans and chimpanzees. Genetics. 2007;176:2069–2076. doi: 10.1534/genetics.107.073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babbitt CC, Fedrigo O, Pfefferle AD, Boyle AP, Horvath JE, et al. Both Noncoding and Protein-Coding RNAs Contribute to Gene Expression Evolution in the Primate Brain. Genome Biology and Evolution. 2010;2:67–79. doi: 10.1093/gbe/evq002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goren A, Simchen G, Fibach E, Szabo PE, Tanimoto K, et al. Fine Tuning of Globin Gene Expression by DNA Methylation. PLoS ONE. 2006;1:e46. doi: 10.1371/journal.pone.0000046. doi: 10.1371/journal.pone.0000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes & Development. 2006;20:1848–1867. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- 21.Heard E, Clerc P, Avner P. X-Chromosome Inactivation in Mammals. Annu Rev Genet. 1997;31:571–610. doi: 10.1146/annurev.genet.31.1.571. [DOI] [PubMed] [Google Scholar]
- 22.Sado T, Fenner MH, Tan SS, Tam P, Shioda T, et al. X inactivation in the mouse embryo deficient for Dnmt1: distinct effect of hypomethylation on imprinted and random X inactivation. Dev Biol. 2000;225:294–303. doi: 10.1006/dbio.2000.9823. [DOI] [PubMed] [Google Scholar]
- 23.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 24.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 25.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 26.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature Genetics. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illingworth R, Kerr A, DeSousa D, Jorgensen H, Ellis P, et al. A Novel CpG Island Set Identifies Tissue-Specific Methylation at Developmental Gene Loci. Plos Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome intergrates intrinsic and environmental signals. Nature Genetics. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 29.Li E, Bestor TH, Jaenisch R. Targeted Mutation of the DNA Methyltransferase Gene Results in Embryonic Lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 30.Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 31.Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum Mol Genet 14 Spec No. 2005;1:R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- 32.Stein R, Razin A, Cedar H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci USA. 1982;79:3418–3422. doi: 10.1073/pnas.79.11.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen RS, Gartler SM. 5-Azacytidine-induced reactivation of the human X chromosome- linked PGK1 gene is associated with a large region of cytosine demethylation in the 5′ CpG island. Proc Natl Acad Sci USA. 1990;87:4174–4178. doi: 10.1073/pnas.87.11.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nature Genetics. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 35.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes & Development. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- 37.Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, et al. Conservation and divergence of methylation patterning in plants and animals. Proceedings of the National Academy of Sciences. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enard W, Fassbender A, Model F, Adorjan P, Paabo S, et al. Differences in DNA methylation patterns between humans and chimpanzees. Current Biology. 2004;14:R148–R149. [PubMed] [Google Scholar]
- 39.Gama-Sosa MA, Midgett RM, Slagel VA, Githens S, Kuo KC, et al. Tissue-specific differences in DNA methylation in various mammals. Biochimica et Biophysica Acta. 1983;740:212–219. doi: 10.1016/0167-4781(83)90079-9. [DOI] [PubMed] [Google Scholar]
- 40.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science. 2010:1–7. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 41.Igarashi J, Muroi S, Kawashima H, Wang X, Shinojima Y, et al. Quantitative analysis of human tissue-specific differences in methylation. Biochemical and Biophysical Research Communications. 2009;376:658–664. doi: 10.1016/j.bbrc.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rakyan VK, Down TA, Thorne NP, Flicek P, Kulesha E, et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Research. 2008;18:1518–1529. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nature Genetics. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitamura E, Igarashi J, Morohashi A, Hida N, Oinuma T, et al. Analysis of tissue-specific differentially methylated regions (TDMs) in humans. Genomics. 2007;89:326–337. doi: 10.1016/j.ygeno.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, et al. Abundant quantitative trait Loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farcas R, Schneider E, Frauenknecht K, Kondova I, Bontrop R, et al. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol Biol Evol. 2009;26:1379–1389. doi: 10.1093/molbev/msp046. [DOI] [PubMed] [Google Scholar]
- 47.Gardiner-Garden M, Frommer M. CpG Islands in Vertebrate Genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 48.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'souza C, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rishi V, Bhattacharya P, Chatterjee R, Rozenberg J, Zhao J, et al. CpG methylation of half-CRE sequences creates C/EBP{alpha} binding sites that activate some tissue-specific genes. Proc Natl Acad Sci USA. 2010;107:20311–20316. doi: 10.1073/pnas.1008688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun L, Huang L, Nguyen P, Bisht KS, Bar-Sela G, et al. DNA methyltransferase 1 and 3B activate BAG-1 expression via recruitment of CTCFL/BORIS and modulation of promoter histone methylation. Cancer Res. 2008;68:2726–2735. doi: 10.1158/0008-5472.CAN-07-6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gius D, Cui H, Bradbury CM, Cook J, Smart DK, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361–371. doi: 10.1016/j.ccr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 52.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes & Development. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kent WJ. BLAT--The BLAST-Like Alignment Tool. Genome Research. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Backes C, Keller A, Kuentzer J, Kneissl B, Comtesse N, et al. GeneTrail--advanced gene set enrichment analysis. Nucleic Acids Res. 2007;35:W186–192. doi: 10.1093/nar/gkm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An illustration of the methylation profiling study design.
(1.48 MB TIF)
Boxplots of distributions of pairwise Pearson correlations between methylation array data, by category.
(0.82 MB TIF)
Boxplots of the distributions of the proportion of divergent nucleotides between human and chimpanzee in 500bp windows around each 50bp probe. The probes are divided into those that are a perfect and unique match to both the human and chimpanzee genomes (left) and those that are a perfect and unique match to the human genome but not the chimpanzee genome (right).
(0.15 MB TIF)
Distributions of methylation values (x-axis) within each species-tissue type, for CpG islands associated with genes whose expression levels rank at different quartiles (colored lines).
(0.89 MB TIF)
Principal components analysis of the expression profiles. Considering the 7,291 autosomal genes from all samples (PC1 and PC2 are plotted on the x-axis and y-axis, respectively).
(0.11 MB TIF)
Density (y-axis) histograms of distributions of Pearson correlations (x-axis) between methylation and gene expression levels across tissues in human (solid lines) and chimpanzee (broken lines) for all genes expressed in at least one tissue (black; plots are identical in the three panels), genes associated with a T-DMR in only one species (blue; data from T-DMRs in liver, kidney, or heart are plotted separately in each panel), and genes associated with a conserved T-DMRs (red; separated by tissue).
(0.49 MB TIF)
Boxplots of distributions of Pearson correlations (y-axis) between methylation and gene expression levels across tissues in human (bold lines) and chimpanzee (non-bold lines) for all genes expressed in at least one tissue (black), genes associated with a T-DMR in only one species (blue), and genes associated with conserved T-DMRs (red). Each category is further divided into CpG sites that are located within CpG islands (“In”) or outside of a CpG island (“Out”). The P-values for a significant difference between the “inside” and “outside” distributions for each category are located below the x-axis.
(0.18 MB TIF)
Boxplots of the distributions, based on 1,000 permutations, of the percentage of genes for which the evidence for inter-species differences in gene expression levels is expected to increase following the correction for methylation levels, by chance alone. Yellow points indicate the percentages seen in the actual data.
(0.07 MB TIF)
Barplots of the percentages of genes in each tissue for which the evidence for inter-species differences in gene expression levels is reduced following the correction for methylation levels. Each tissue is further divided into CpG sites that are located within CpG islands (“In”) or outside of a CpG island (“Out”). The P-values for a significant difference (compared to expectations from a chi-squared distribution) between the “inside” and “outside” percentages for each category are located above the barplots.
(0.16 MB TIF)
Histogram of the number of CpG site probes (y-axis) on each chromosome, except the Y-chromosome (x-axis).
(0.23 MB TIF)
Frequency (y-axis) histogram of the distances from CpG sites assayed in our study to the transcription start sites (TSS) of the nearest annotated genes. The inset is an expanded view of frequency (y-axis) histogram near the 2kb around the TSS, where 97% of the probes are located.
(0.22 MB TIF)
Summary of the data for all CpG probes.
(6.64 MB XLS)
Percentages of negative correlation between methylation and expression levels in T-DMRs across a range of statistical cutoffs used to classify T-DMRs.
(0.03 MB XLS)
Gene ontology categories that are the most significantly enriched or depleted GO annotations among genes that are classified as T-DMRs in either species (P<0.01). For this analysis, we used a background of all genes assayed in our study that are expressed in at least one tissue. GO tables for T-DMRs specific to each tissue are located on individual sheets.
(0.06 MB XLS)
Gene ontology categories that are the most significantly enriched or depleted GO annotations among genes that are classified as T-DMRs in both species (shared T-DMRs) (P<0.001). For this analysis, we used a background of all genes assayed in our study that are expressed in at least one tissue. GO tables for T-DMRs specific to each tissue are located on individual sheets.
(0.03 MB XLS)
Estimates of the percentage of genes for which methylation differences could underlie inter-species expression differences, across a range of significance thresholds.
(0.03 MB XLS)
Information on the 36 samples used in this study.
(0.09 MB DOC)