

Abstract
Oculopharyngeal muscular dystrophy (OPMD) is an adult-onset syndrome characterized by progressive degeneration of specific muscles. OPMD is caused by extension of a polyalanine tract in poly(A) binding protein nuclear 1 (PABPN1). Insoluble nuclear inclusions form in diseased muscles. We have generated a Drosophila model of OPMD that recapitulates the features of the disorder. Here, we show that the antiprion drugs 6-aminophenanthridine (6AP) and guanabenz acetate (GA), which prevent formation of amyloid fibers by prion proteins in cell models, alleviate OPMD phenotypes in Drosophila, including muscle degeneration and nuclear inclusion formation. The large ribosomal RNA and its activity in protein folding were recently identified as a specific cellular target of 6AP and GA. We show that deletions of the ribosomal DNA locus reduce OPMD phenotypes and act synergistically with sub-effective doses of 6AP. In a complementary approach, we demonstrate that ribosomal RNA accelerates in vitro fibril formation of PABPN1 N-terminal domain. These results reveal the conserved role of ribosomal RNA in different protein aggregation disorders and identify 6AP and GA as general anti-aggregation molecules.
Keywords: anti-aggregation drug, Drosophila model, muscular dystrophy, PABPN1, ribosomal RNA
INTRODUCTION
Protein aggregation diseases form a set of disorders that are characterized by intracellular aggregation and deposition of pathogenic proteins in the form of insoluble inclusions or plaques (Taylor et al, 2002). Most of these diseases are neurodegenerative, including prion-based diseases, Alzheimer's, Parkinson's, as well as Huntington's and other polyglutamine expansion-based diseases. Polyglutamine diseases result from triplet expansion leading to an increased length of a polyglutamine tract. The protein with an expanded polyglutamine tract aggregates as inclusions in affected cells (Cooper et al, 1998; Davies et al, 1997; DiFiglia et al, 1997). However, whether the mature insoluble fibril or an oligomeric intermediate play a role in the pathology remains unknown. A protective role of inclusions has even been described (Arrasate et al, 2004). Anti-aggregation drugs, such as trehalose, that can reduce the number of inclusions in a cell model of Huntington's disease, have also a beneficial effect in a mouse model (Tanaka et al, 2004), indicating that either inclusions per se or the process of inclusion formation is an important aspect in pathogenesis.
Oculopharyngeal muscular dystrophy (OPMD) is another triplet expansion disease in which the mutation expands a polyalanine tract. The affected protein, poly(A) binding protein nuclear 1 (PABPN1), has a 10-alanine tract following the initiation codon in its normal form, and this tract is expanded to 12–17 in the mutant forms found in OPMD patients (Brais et al, 1998). PABPN1 is an ubiquitous protein with general and essential functions in messenger RNA (mRNA) polyadenylation (Benoit et al, 2005; Wahle, 1991, 1995). During the process of nuclear polyadenylation, PABPN1 binds to nascent poly(A) tails and has two roles: stimulation of poly(A) polymerase, and control of poly(A) tail length (Kühn & Wahle, 2004; Wahle, 1991). Despite the general and ubiquitous function of PABPN1, OPMD affects particular muscles and the disease is characterized by eyelid drooping, swallowing difficulties and proximal limb weakness (Brais, 2003). OPMD is a dominant and progressive genetic disorder that has a late onset. As in polyglutamine expansion diseases, the mutant protein aggregates and forms insoluble nuclear inclusions, in that case in affected muscles. Because nuclear inclusions are characteristic of the disease, alanine expansion contributes in some way to the formation of the inclusions. However, normal PABPN1 oligomerizes during its function in polyadenylation. Moreover, aggregation of the normal protein has been observed in rat hypothalamus neurons (Berciano et al, 2004). In animal and cell models of OPMD, overexpression of normal PABPN1 or PABPN1 deleted for the polyalanine tract also leads to the formation of nuclear inclusions (Chartier et al, 2006; Fan et al, 2001; Tavanez et al, 2005). Therefore, a possibility could be that the polyalanine tract extension modifies the turnover or properties of the protein, leading to increased accumulation and/or accelerated inclusion formation. OPMD nuclear inclusions contain poly(A) ribonucleic acid (RNA), ubiquitin, proteasome subunits and chaperone proteins such as heat-shock protein 70 (HSP70) and human DNAJ homologue-1 (HDJ-1) (Abu-Baker et al, 2003; Calado et al, 2000). In cell models of OPMD, reducing aggregation with drugs or by overexpressing HSP70 decreases cell death (Bao et al, 2004). This suggests that either the presence of PABPN1 inclusions and/or the process of aggregation itself contribute to the pathology. Consistent with this, we found that overexpression of HSP70 is a suppressor of muscular defects in the Drosophila model of OPMD (Chartier et al, 2006). Moreover, anti-aggregation drugs doxycycline and trehalose reduce the number of nuclear inclusions and muscle weakness in a mouse model of OPMD (Davies et al, 2005, 2006).
6-Aminophenanthridine (6AP) and guanabenz acetate (GA) were isolated as active against yeast prions using a yeast-based assay and appeared to be also active against mammalian prion (Bach et al, 2003; Tribouillard et al, 2006; Tribouillard-Tanvier et al, 2008a). The fact that 6AP and GA (as well as other antiprion drugs) are active against different prion proteins, in both yeast and mammalian systems, strongly suggests that these compounds interfere with evolutionary conserved prion-controlling mechanisms that may be involved in protein folding. We therefore hypothesized that these compounds could be beneficial for other protein folding associated diseases, including Huntington's disease. Consistent with this, we recently found that 6AP and GA, as well as several other antiprion drugs isolated using the yeast-based assay, were able to reduce aggregates formed by mutated huntingtin in cell-based assays (European patent: ‘Use of chlorine guanabenz derivatives for treating polyglutamine expansion associated diseases’ A. Bertolotti and M. Blondel, CNRS, no. 06291547.6).
Interestingly, a specific cellular target of both 6AP and GA was recently identified to be the ribosome-borne protein folding activity of the large ribosomal RNA (Tribouillard-Tanvier et al, 2008b). Although they are structurally unrelated (Fig 1A), both drugs interact with the ribosome in an RNA-dependent manner and bind to common specific positions in domain V of the large ribosomal RNA. Strikingly, these interactions have no effect on the peptidyl transferase activity of the ribosome or on global translation, however, they specifically inhibit the ability of the ribosome to assist protein folding in vitro, another largely unexplored activity of the ribosome (Tribouillard-Tanvier et al, 2008b). Importantly, inactive 6-aminophenanthridine (6APi), a close chemical derivative of 6AP, that is inactive against both yeast and mammalian prions, is also unable to interact with domain V of the large ribosomal RNA and to inhibit its protein folding activity. Together, these results imply that the activity of the ribosome in protein folding may play a role in prion propagation and aggregate formation.
Figure 1. 6AP, 6APi and GA structures, and toxicity of drugs fed to larvae.
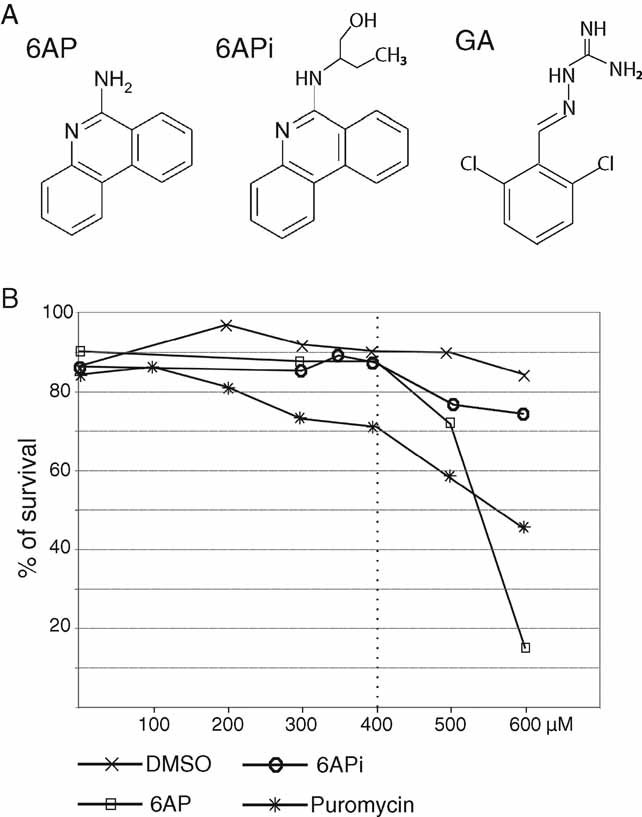
- The molecular structures of 6AP, of its less active chemical derivative 6APi and of GA are shown.
- Toxicity of the drugs fed to larvae was determined by transferring 80 wild-type first instar larvae into vials containing 5 ml of instant Drosophila medium reconstituted either with increasing concentrations of drug in DMSO or with DMSO alone. Puromycin was diluted in water. The graph shows the percentage of flies reaching adulthood at 25°C (n = 120–360). Drugs provided to adults were found to be non-toxic, as determined by the lack of mortality of adults fed with drug-supplemented medium during the course of the experiment.
We previously developed a Drosophila model of OPMD that recapitulates the characteristics of the disease including progressive muscle degeneration with muscle defects proportional to the number of alanines in the tract and formation of PABPN1 nuclear inclusions (Chartier et al, 2006). Here, we show that 6AP and GA are beneficial for OPMD in the Drosophila model. Feeding animals with both drugs decreases muscle degeneration and reduces the size of nuclear inclusions. We observe a synergistic effect of 6AP with deletions of the ribosomal DNA locus, suggesting that the ribosomal RNA is a conserved cellular target of 6AP in Drosophila. Moreover, domain V of the large ribosomal RNA, that bears the protein folding activity and is targeted by 6AP and GA, accelerates fibril formation of the alanine expanded N-terminal domain of PABPN1 in vitro. Together, these data strongly suggest that the protein folding activity of the large ribosomal RNA plays a role in PABPN1 aggregation in OPMD, and strengthen the hypothesis that ribosomal RNA has a general function in the formation of disease-related amyloid fibers. These data also highlight 6AP and GA as general anti-aggregation drugs that might be beneficial for major protein folding diseases. Because GA is routinely used as a treatment against hypertension without major side-effects, our results reveal GA as a strong candidate in potential treatments for OPMD and other protein aggregation disorders.
RESULTS
The Drosophila model of OPMD was generated by expressing mammalian alanine expanded PABPN1, PABPN1-17ala, with the upstream activation sequence (UAS)/GAL4 transcription factor (Gal4) system (Brand & Perrimon, 1993; Chartier et al, 2006). Expression was driven in muscles using the myosin heavy chain (Mhc)-Gal4 driver (Schuster et al, 1996). PABPN1-17ala expression with Mhc-Gal4 leads to progressive wing position defects, progressive muscle degeneration visualized in thoracic indirect flight muscles (IFMs), and formation of PABPN1 nuclear inclusions that were analyzed in larval and adult thoracic muscles.
6AP decreases the wing position phenotypes induced by expression of PABPN1-17ala in muscles
Because OPMD pathology involves protein aggregation, we tested whether drugs known to have anti-aggregation properties could be effective for OPMD using the Drosophila model. Drugs were provided in the food from larval stages to adulthood. The toxicity of the drugs was assessed by measuring the viability of individuals raised on drug-supplemented medium from larval stages (Fig 1B, Supplementary Figs 1 and 2). Both doxycycline and trehalose have a beneficial effect in a mouse model of OPMD (Davies et al, 2005, 2006), and ibuprofen reduces PABPN1 aggregation in a cell model of OPMD (Wang et al, 2005). We found that none of these drugs were effective in the Drosophila model (Supplementary Fig 1). We next tested the effect of 6AP, another anti-aggregation drug with antiprion activity. 6APi, a close chemical derivative of 6AP that was described as inactive or less effective in antiprion assays both in yeast and mammals, was used as a negative control (Fig 1A). Toxicity tests showed substantial survival of individuals fed with up to 400 µM of drugs. In the presence of 600 µM of 6AP, development was delayed and viability was dramatically decreased (Fig 1B, Supplementary Fig 2). We therefore used 400 µM as the highest concentration to feed larvae in further studies.
Expression of PABPN1-17ala with Mhc-Gal4 leads to progressive defects in wing position (wings up or down). At 18°C, abnormal wing position appears at day 3 of adulthood with less than 20% of individuals showing defects, and this percentage reaches 90% at day 6 (Chartier et al, 2006). We analyzed the effect of feeding OPMD larvae and adults with increasing concentrations of 6AP, ranging from 250 to 400 µM. Wing position defects were scored at day 6 of adulthood, when 90% of individuals show defects in the absence of the drug (Fig 2A). A dose-dependent beneficial effect of 6AP was visible. The strongest effect was obtained in the presence of 400 µM 6AP, the percentage of individuals with wing position defects decreased to 50%. An identical experiment performed in the presence of the control molecule 6APi showed that this molecule had no effect (Fig 2A). We found that 400 µM of 6AP was still beneficial, although to a lesser extent, when provided during larval stages only, or from third instar larval to adult stages (Supplementary Fig 3). We verified that the positive effect of 6AP did not result from reduced levels of PABPN1-17ala in thoracic muscles of individuals fed with 6AP (Fig 2B).
Figure 2. 6AP reduces the wing position defects resulting from expression of alanine-expanded PABPN1 in muscles.
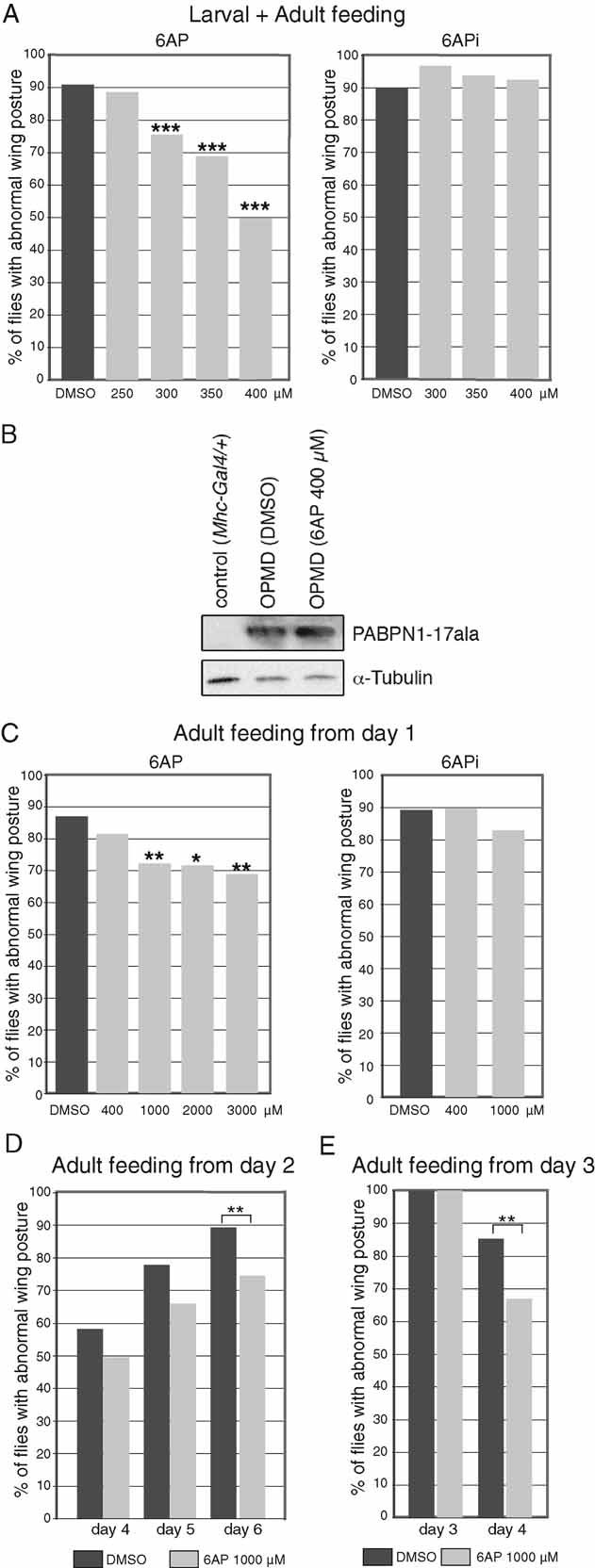
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ first instar larvae were raised at 18°C on instant Drosophila medium supplemented with increasing concentrations of 6AP, 6APi or DMSO alone. The day of birth (day 1), adult males were transferred onto fresh medium with the same concentration of drug and scored for wing position at day 6 (n = 120–320). Oral administration of 6AP reduces the percentage of wing position defects in a dose-dependent manner (left panel), whereas 6APi shows no effect (right panel).
- Western blot of thorax protein extracts from UAS-PABPN1-17ala/+; Mhc-Gal4/+ individuals at day 6, fed from larval stages with 400 µM of 6AP or DMSO alone. The blot was probed with anti-PABPN1, α-tubulin was used as a loading control.
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ embryos were raised on regular Drosophila medium at 18°C. The day of birth (day 1), adult males were transferred onto fresh instant Drosophila medium supplemented with 6AP, 6APi or DMSO alone. Wing position defects were scored at day 6 (n = 40–120). Because of the shorter period of exposure to the drug and the reduced taking of food by adults compared to larvae, concentrations of 6AP higher than 400 µM were found to be non-toxic.
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ adults were transferred to 6AP or DMSO alone-supplemented medium at day 2. Wing position defects were scored at days 4, 5 and 6 (n = 112–119).
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ adults with wing position defects were selected at day 3 and transferred immediately to 6AP or DMSO alone-supplemented medium, wing position defects were scored at day 4 (n = 100). ***p-value <10−4, **p-value < 0.01 and *p-value < 0.05 using the χ2 test.
We next determined whether 6AP could also have a protective effect when provided at later stages. Expression of PABPN1-17ala with Mhc-Gal4 does not affect IFM development as these muscles do not appear altered in early adulthood (Chartier et al, 2006). We therefore provided the drug to OPMD adults at 18°C starting the day of birth (day 1) and scored wing position defects at day 6. Feeding adults with 400 µM of 6AP led to a weak decrease in the percentage of flies showing wing position defects (Fig 2C). We used higher concentrations of 6AP, from 1 to 3 mM, which were found to be non-toxic when provided to adults only. With these concentrations, the percentage of affected flies decreased significantly to 70%. A plateau was reached from 1 mM of 6AP, as the beneficial effect of the drug did not improve with higher concentrations. Therefore, 6AP provided to adults was also beneficial and its protective effect was dose-dependent. We found that 6AP at 1 mM remained beneficial when provided to adults at day 2 (Fig 2D).
To address whether 6AP could have a curative effect on individuals already showing wing position defects, we selected adults with affected wing position at day 3, fed them with 1 mM of 6AP and analyzed their wing position at day 4 (after 24 h). Because wing position defects are not stabilized at day 3, 15% of individuals showed normal wing position at day 4 when fed with dimethyl sulphoxide (DMSO) alone. However, the percentage of flies with normal wing position at day 4 significantly increased to 33% in individuals fed with 6AP (Fig 2E).
Together, these results show that the antiprion compound 6AP is effective to reduce the phenotype of OPMD, a protein aggregation disease that is distinct from prion-based diseases. The protective role of 6AP is dose-dependent and the drug also shows some extent of curative effect.
GA decreases OPMD wing posture phenotypes when fed to adults
Guanabenz acetate is another antiprion drug which appears to have the same mode of action as 6AP (i.e. binding to ribosomal RNA and specific inhibition of its protein folding activity), we thus tested its potential efficiency in OPMD. GA is known to be an unstable molecule. It was reported to have a half-live of a few hours in the human body (Holmes et al, 1983). We therefore provided GA to OPMD adults only and supplied fresh GA-supplemented medium at several time points during the experiment. No toxicity of GA was observed when provided to adults (94% of survival at day 6 (n = 87), when 3 mM GA was provided fresh every day). In a first experiment, adults were fed with GA at day 1 and fresh GA-supplemented medium at the same concentration was provided at days 3 and 5. Wing position defects were recorded at day 6. A dose-dependent beneficial effect of GA was observed, with a decrease to 50% of individuals showing wing position defects when fed with 3 mM of GA (Fig 3A). We then analyzed the effect of the drug when fresh GA-supplemented medium was supplied every day and the treatment started at later stages. Consistent with the short half-life of the drug, the positive effect of GA was increased in these conditions with a reduction to 49% of individuals showing wing position defects at day 6 in the presence of 2 mM of GA (Fig 3B). This effect occurred when GA was provided from day 2 of adulthood. If the drug was provided at an even later stage, from day 3 of adulthood, the 2 mM dose was not significantly effective, however, a significant decrease to 48% of individuals having wing position defects at day 6 was observed with 3 mM GA (Fig 3C).
Figure 3. GA reduces the wing position defects of OPMD individuals when provided to adults.
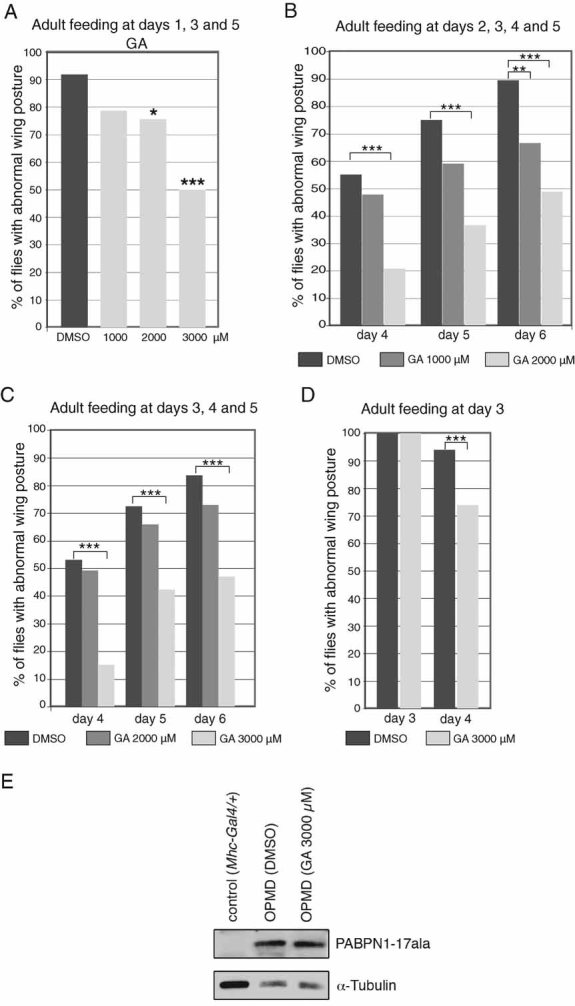
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ embryos were raised on regular medium at 18°C, adult males were transferred to GA or DMSO supplemented medium at day 1 and transferred to fresh supplemented medium at days 3 and 5. Wing position defects were scored at day 6 (n = 49).
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ adult males were transferred to GA or DMSO supplemented medium at day 2 and transferred to fresh supplemented medium every day. Wing position defects were scored at days 4, 5 and 6 (n = 49).
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ adult males without wing position defects were selected at day 3 and transferred to GA or DMSO supplemented medium. They were transferred to fresh supplemented medium every day. Wing position defects were scored at days 4, 5 and 6 (n = 50–80).
- UAS-PABPN1-17ala/+; Mhc-Gal4/+ adult males showing wing position defects were selected at day 3 and immediately transferred to GA or DMSO supplemented medium. Wing position defects were scored at day 4 (n = 65–84). ***p-value <10−3, **p-value < 0.01 and *p-value <0.05 using the χ2 test.
- Western blot of thorax protein extracts from UAS-PABPN1-17ala/+; Mhc-Gal4/+ individuals at day 6, fed with 3 mM GA or DMSO alone from day 2 of adulthood, transferred to fresh medium every day. The blot was probed with anti-PABPN1, α-tubulin was used as a loading control.
We used GA at the concentration of 3 mM, supplied fresh every day, to analyze a potential curative effect on individuals showing wing position defects at day 3. A significant reduction in the percentage of flies with wing position defects at day 4 was observed in the presence of 3 mM of GA compared to DMSO alone, indicating a curative effect of GA (Fig 3D).
We verified that the beneficial effect of GA was not due to a reduction of PABPN1-17ala protein levels in thoracic muscles (Fig 3E).
These results demonstrate a dose-dependent beneficial effect of GA in reducing the appearance of OPMD phenotypes when provided to adults, even at late stages. In addition, GA also had a curative effect on individuals already showing defects.
6AP and GA decrease muscle degeneration induced by expression of PABPN1-17ala
Expression of PABPN1-17ala with Mhc-Gal4 leads to progressive muscle degeneration as visualized by polarized light on thoracic IFMs, which are involved in flight and wing posture (dorso-longitudinal muscles: DLMs or dorso-ventral muscles: DVMs) (Chartier et al, 2006, 2009). We analyzed muscle degeneration with polarized light after expression of PABPN1-17ala with Mhc-Gal4 at day 11, by scoring the number of affected muscles (six DLMs and seven DVMs per thorax). In the absence of drug, defects were visible in most IFMs which appeared thin or were lacking (Fig 4A–E). In the presence of either 400 µM of 6AP provided from larval stages or 3 mM of GA provided to adults and supplied fresh every day, muscle degeneration was strongly reduced (Fig 4A–E). Because in the presence of the drugs, not all flies had normal wing position, we scored muscle defects separately in flies with normal (rescue) and affected (no rescue) wing posture. Indeed, normal wing posture requires both normal muscle structure and function. Therefore, we reasoned that the level of rescue in the presence of drugs might be higher if we analyzed muscle morphology rather than muscle function (i.e. wing position). Strikingly, muscle degeneration was largely prevented in flies with normal wing posture and this beneficial effect was also visible in flies with affected wing posture, although it was less pronounced in these flies (Fig 4D and E). This indicates that after drug treatment, flies that still have an abnormal wing posture can show some rescue of muscle morphology.
Figure 4. 6AP and GA reduce muscle degeneration induced by expression of PABPN1-17ala.

- A. Dorso-longitudinal (DLM) and dorso-ventral (DVM) adult thoracic muscles visualized with polarized light. Examples of muscles at day 11, from wild type (wt) raised on DMSO. Six DLMs (1–6) and six DVMs (I1, I2, II1, II2, III1, III2) are visible; DVM I3 is out of focus (*). Anterior is to the left, dorsal is up.
- B. DLMs and DVMs at day 11, of OPMD (UAS-PABPN1-17ala/+; Mhc-Gal4/+) flies fed with DMSO alone or 400 µM 6AP from larval stages. Arrows indicate very thin or missing muscles. Muscles with a normal appearance from flies fed with 6AP are shown (OPMD (6AP)).
- C. DLM and DVM at day 11, of OPMD flies fed with DMSO alone or 3 mM GA from day 2 of adulthood and transferred to fresh supplemented medium every day. Arrows indicate very thin or missing muscles. Muscles with a normal appearance from flies fed with GA are shown (OPMD (GA)).
- D, E. Percentages of affected muscles either in the absence (DMSO), or in the presence of drugs (6AP or GA). Six DLMs and seven DVMs were scored per thorax (n = 48–169 DLM and 70–322 DVM). Muscles were also scored separately, in subgroups showing or not wing position defects after drug treatment. ‘6AP or GA rescue’ indicates OPMD flies raised on drug-supplemented medium, which have normal wing posture, ‘6AP or GA no rescue’ indicates flies raised on drug-supplemented medium, which still have an affected wing posture. ***p-value <10−3 using the χ2 test.
These data confirm that both 6AP and GA are beneficial for OPMD. Both drugs reduce the main defect in the pathology which corresponds to muscle weakness and degeneration.
6AP and GA reduce the size of PABPN1 nuclear aggregates
An important aspect of OPMD pathology is the formation of salt extraction-resistant nuclear aggregates containing PABPN1 in muscle fibers from OPMD patients. We have shown that the Drosophila model of OPMD reproduces the presence of these aggregates which are very similar to aggregates observed in patients, both in composition and in structure. In Drosophila, PABPN1-17ala-containing nuclear inclusions are resistant to 1 M KCl extraction and recruit HSP70 and ubiquitin. When visualized by electron microscopy (EM), these inclusions are composed of unbranched tubular filaments closely resembling the filaments observed in nuclear inclusions in OPMD patients (Chartier et al, 2006). The 6AP and GA molecules were reported to have anti-aggregation properties. We tested whether the protective effect of these drugs for OPMD correlates with an ability to reduce aggregation of PABPN1-17ala. 6AP and 6APi were provided from larval stages at 400 µM and nuclei containing an inclusion were quantified in thoracic muscles expressing PABPN1-17ala at day 11. These nuclei represent 18% of nuclei when larvae were fed with DMSO alone (Fig 5A). The percentage of nuclei with a nuclear inclusion was not reduced when larvae were fed with 6APi or 6AP (Fig 5A). However, 6AP treatment had a significant and specific effect in reducing the size of nuclear inclusions (Fig 5B and E). No significant difference in the size of the nuclear inclusions was observed between individuals fed with 6AP that had either a rescued or a non-rescued wing posture. However, it is important to note that flies with affected wing position showed some rescue of muscle morphology (Fig. .4). Therefore, there was a correlation between the reduced size of nuclear inclusions and the rescue of muscle morphology.
Figure 5. 6AP and GA reduce the size of PABPN1-17ala nuclear inclusions in Drosophila muscles.

- A. Percentages of nuclear inclusions in OPMD (UAS-PABPN1-17ala/+; Mhc-Gal4/+) thoracic muscles. Larvae were raised in the presence or absence of drug, and adult thoracic muscles were dissected at day 11 and stained with anti-PABPN1 and DAPI. Nuclear inclusions were visualized and scored using both staining.
- B. Quantification of nuclear inclusion areas. Each nuclear inclusion was delimited in a focal plan and the surface area was calculated using Image J. Mean values of the surface areas are shown in arbitrary units (top). Distribution of nuclear inclusion surface areas are shown as box plots (bottom). The boxes represent 50% of the values, horizontal lines correspond to the medians (50% of the values on each side of the line), vertical bars correspond to the range. Extreme values are in open circles. *** Distributions are different using the Student's t-test, p-value <10−4, ns: non-significant. After drug treatment, quantifications are indicated both regardless of the wing posture (6AP), and in subgroups showing or not wing position defects (6AP no rescue and 6AP rescue). ‘no rescue’ indicates flies which still have an affected wing posture, ‘rescue’ indicates flies with normal wing posture.
- C, D. Percentages of nuclear inclusions (C) and quantification of nuclear inclusion areas (D) in OPMD thoracic muscles either in the presence or absence of 3 mM of GA provided fresh every day, from day 2 of adulthood. Nuclear inclusions were scored at day 6. Legend is as in (A, B).
- E. Confocal images of nuclear inclusions visualized with anti-PABPN1 staining. DNA was revealed with DAPI. In OPMD individuals fed with DMSO alone, nuclear inclusions are large and can occupy most of the nucleus (inclusion). An example of nucleus without inclusion is shown for comparison (no inclusion). Nuclear inclusions were smaller when flies were raised on drug-supplemented medium. Examples of small inclusions at day 11 in the presence of 400 µM of 6AP from larval stages (OPMD (6AP)), and at day 6 in the presence of 3 mM of GA from day 2 of adulthood, provided fresh every day (OPMD (GA)).
To analyze the effect of GA, the drug was provided to adults at 3 mM, supplied fresh every day, and PABPN1-17ala nuclear inclusions in thoracic muscles were scored at day 6. Thirteen percent of nuclei contain an inclusion when adults were fed with DMSO alone (Fig 5C). The lower number of nuclear inclusions at day 6 compared to day 11 is consistent with our previous data showing that the number of inclusions increases with the age of the flies (Chartier et al, 2006). As it was the case for 6AP, the percentage of nuclei with an inclusion was similar in the presence of GA or DMSO alone (Fig 5C). However, nuclear inclusions were significantly smaller after treatment with GA than in the control (Fig 5D and E).
We conclude that both 6AP and GA interfere with the aggregation process of alanine-expanded PABPN1, eventually leading to the formation of smaller aggregates.
6AP acts synergistically with deletions of the ribosomal DNA locus to reduce OPMD phenotypes
The ribosomal RNA has been identified as a specific cellular target of both 6AP and GA. Both drugs bind specifically to several common positions in domain V of the large ribosomal RNA and these interactions prevent the ribosomal RNA-mediated activity in protein folding, as assessed in vitro and in an in vivo test in bacteria (Tribouillard-Tanvier et al, 2008b). If the beneficial effect of 6AP and GA on OPMD in Drosophila also results from a reduced activity of the large ribosomal RNA, a similar beneficial effect might be observed with mutants affecting ribosomal RNA activity. The ribosomal DNA locus, called bobbed (bb) in Drosophila, is composed of 120–240 tandem repeats on each of chromosomes X and Y. These repeats encode the 18S, 5.8S, 2S and 28S ribosomal RNA (Long & Dawid, 1980). We used bbN, a weak allele of bb, in which a small number of repeats is deleted, and Df(1)17-87, a complete deletion of the region including the whole bb locus, to determine whether genetically decreasing the copy number of ribosomal DNA repeats would affect OPMD phenotypes. We verified that none of the bb mutant combinations showed abnormal wing posture independently of OPMD. Each of the bb deletions alone had no significant effect on the percentage of OPMD flies with wing posture defects. However, when both deletions (bbN/Df(1)17-87) were combined in OPMD flies, we observed a significant decrease in the number of these flies showing wing position defects (Fig 6B). To determine whether the beneficial effect of 6AP on OPMD could occur through the modulation of ribosomal RNA activity, we analyzed if 6AP could act synergistically with a reduction of the copy number of ribosomal DNA repeats. Neither heterozygous bbN/+ and Df(1)17-87/+ mutations, nor 6AP at 250 µM had an effect on OPMD wing position defects (Fig 2A, 6B). However, the combination of both bbN/+ or Df(1)17-87/+ and 250 µM of 6AP strongly decreased the percentage of OPMD flies with wing posture defects (Fig 6C). This shows that 6AP and deletions of the ribosomal DNA locus act synergistically and strongly suggests that the ribosomal RNA is also a cellular target of 6AP in Drosophila. It should be noted that, because the survival of bbN/Df(1)17-87 flies raised on DMSO alone or 400 µM of 6AP was very low (<10%), wing position was not scored in these conditions.
Figure 6. The ribosomal RNA is a cellular target of 6AP.

- Percentages of wing position defects in OPMD flies at 18°C, in the presence or absence of bb mutations. Note that because the bb locus is on chromosome X, females were scored in these experiments (n = 140–220). The percentage of abnormal wing position in OPMD females (73–77%) is lower than in males (90%, Fig 2).
- Synergy between bb mutants and 6AP. Percentages of wing position defects in OPMD flies at 18°C at day 6, either in the presence or absence of bb mutations and 250 µM of 6AP. OPMD females were scored (n = 70–110). ***p-value <10−4 using the χ2 test.
- Western blot of protein extracts from adult female thoraxes at day 6, showing that the expression of PABPN1-17ala is not affected in bb mutants. The blot was probed with anti-PABPN1, α-tubulin was used as a loading control.
- Percentages of wing position defects in OPMD flies at 18°C in the presence or absence of puromycin provided from first instar larval to adult stages. OPMD males were scored (n = 50–190).
It has been shown that the interaction of 6AP with the large ribosomal RNA does not affect protein synthesis, both in vivo in yeast and in in vitro translation system based on rabbit reticulocyte lysate (Tribouillard-Tanvier et al, 2008b). We verified that the decrease of OPMD phenotypes in bbN/Df(1)17-87 mutant flies did not result from a lower production of PABPN1-17ala in this context (Fig 6D). To confirm that the beneficial effect of 6AP on the Drosophila model of OPMD did not depend on a global decrease in protein synthesis, we studied the effect on OPMD phenotypes of puromycin, a general protein synthesis inhibitor. The toxicity of this drug allowed its utilization up to 600 µM (Fig 1B). Wild-type flies fed from larval stages with puromycin and raised at 25°C were small and had a delayed development, starting from 300 µM, consistent with reduced protein synthesis. However, up to 600 µM of this drug had no effect on the number of OPMD flies showing abnormal wing posture (Fig 6E). Together, these results suggest that the beneficial effect of 6AP in OPMD does not result from reduced protein synthesis, either global or specifically for PABPN1-17ala.
Ribosomal RNA is involved in PABPN1-17 aggregation and fibril formation
We propose that 6AP might have a positive effect on OPMD by interfering with the protein folding activity of ribosomal RNA and that this activity of ribosomal RNA would play a role in PABPN1-17ala aggregation. A prediction of this model is that PABPN1-17ala nuclear inclusions would be altered in the genetic background of ribosomal DNA deletions that led to a decrease in wing position defects (Fig 6B). Quantification of the percentage and size of PABPN1-17ala nuclear inclusions showed that, similarly to what was observed after oral treatments with 6AP or GA, the number of nuclear inclusions was not affected, but their size was significantly reduced in bbN/Df(1)17-87 deleted flies, compared to the wild-type ribosomal DNA background (Fig 7A–C).
Figure 7. The ribosomal RNA is involved in PABPN1-17ala aggregation and in fibril formation of its N-terminal domain in vitro.
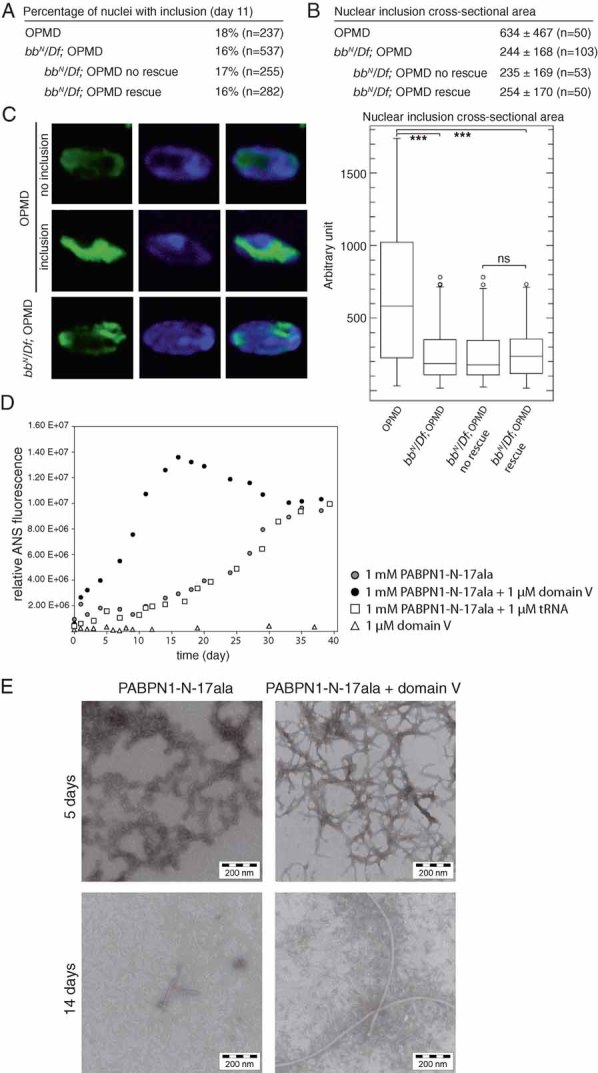
- Percentages of nuclear inclusions in OPMD thoracic muscles at day 11, either in the presence or absence of bb deletions. In the presence of bb deletions, quantifications are indicated both regardless of the wing posture (bbN/Df; OPMD), and in subgroups showing or not wing posture defects (bbN/Df; OPMD no rescue, and bbN/Df; OPMD rescue)‘rescue’ indicates OPMD flies which have normal wing posture, ‘no rescue’ indicates flies which still have an affected wing posture.
- Quantification of nuclear inclusion areas. Each nuclear inclusion was delimited in a focal plan and the surface area was calculated using Image J. Mean values of the surface areas are shown in arbitrary units (top). Distribution of nuclear inclusion surface areas are shown as box plots (bottom). Legend is as in Fig 5B.
- Confocal images of nuclear inclusions visualized with anti-PABPN1 staining. DNA was revealed with DAPI. Examples of nuclei without an inclusion (no inclusion), and with a large inclusion (inclusion) in OPMD thoracic muscles. Example of a small inclusion in an OPMD thoracic muscle in the presence of bb deletions.
- Kinetics of fibril formation of the PABPN1-17ala N-terminal domain, either in the presence or absence of ribosomal RNA domain V, monitored by ANS fluorescence. tRNA was used as a negative control.
- Electron micrographs of oligomers or fibrils of PABPN1-N-17ala formed in vitro, either in the presence or absence of ribosomal RNA domain V.
We next analyzed a potential direct effect of the ribosomal RNA on PABPN1-17ala aggregation using in vitro assays. We previously used the N-terminal domain of PABPN1 (PABPN1-N: residues 1-125 of 306) to monitor fibril formation in vitro. Because full-length PABPN1 forms amorphous deposits, the soluble N-terminal domain, which contains the polyalanine tract, was selected for the biophysical analyses (Scheuermann et al, 2003). Importantly, this domain has no activity in RNA binding (Kuhn et al, 2003). PABPN1 N-terminal domain was shown to form fibrils, which have the characteristics of amyloid fibers, and a polyalanine expansion in this domain (PABPN1-N-17ala) strongly accelerates fibril formation (Scheuermann et al, 2003). The protein folding activity of ribosomal RNA is borne by the domain V of the large ribosomal RNA. We therefore analyzed the capacity of this RNA domain to influence fibril formation of PABPN1-N-17ala in vitro. PABPN1-N-17ala protein samples were incubated at 1 mM, either in the presence or the absence of 1 µM of Drosophila domain V RNA, and analyzed for binding to 1-anilino-8-naphtalensulfonate (ANS), as a measure of fibril formation. In the absence of domain V RNA, ANS signals rose after a lag phase of 10 days (Fig 7D). Strikingly, the addition of domain V RNA led to an immediate increase in ANS fluorescence (Fig 7D). This effect was observed neither when PABPN1-N-17ala was mixed with 1 µM transfer RNA (tRNA), nor when ribosomal RNA domain V was incubated alone (Fig 7D).
The presence of fibrils was confirmed using EM. After 5 days of incubation, PABPN1-N-17ala alone only formed oligomers, which then started to convert into short fibrils after 14 days of incubation (Fig 7E, left panels). When PABPN1-N-17ala was incubated in the presence of ribosomal RNA domain V, fibrils had already formed after 5 days, and they had converted into long, unbranched fibrils after 14 days (Fig 7E, right panels). We conclude that the domain V of the large ribosomal RNA accelerates the formation of PABPN1-N-17ala fibrils in vitro.
It should be noted that the effects of 6AP and GA could not be analyzed in these in vitro assays as both drugs precipitated in the protein incubation buffer, and DMSO alone affected ANS fluorescence.
Taken together, these in vivo and biophysical data strongly support an important role of the ribosomal RNA in the aggregation process of PABPN1-17ala and its assembly as nuclear inclusions in vivo.
DISCUSSION
Our results using the Drosophila model of OPMD demonstrate that the antiprion drugs 6AP and GA have a protective and, to some extent, a curative effect for OPMD. We further show that these drugs reduce the aggregation load of alanine expanded PABPN1. When larvae and adults are fed with 6AP and GA, respectively, PABPN1-17ala nuclear inclusions are smaller. Together with the reduced muscle degeneration, these results suggest that both drugs interfere with the aggregation process, potentially slowing down a conformational change that would be involved in both aggregate formation and impairment of PABPN1 function.
We have recently shown that 3F5, an intrabody against PABPN1, prevents OPMD phenotypes when co-expressed with PABPN1-17ala in Drosophila muscles. Interestingly, 3F5 has an effect similar to that of 6AP or GA on PABPN1-17ala nuclear inclusions. When 3F5 is co-expressed with PABPN1-17ala, the number of muscle nuclei showing an inclusion is similar to that observed without the intrabody, but the sizes of the nuclear inclusions are smaller (Chartier et al, 2009). The relationships between the formation of nuclear inclusions and the disease process have not been clarified. In other words, it is not yet known whether a specific conformer of PABPN1-17ala is toxic and participates in the disease symptoms, or whether the mutation leads to two independent phenomena: protein misfunction and aggregation, aggregates being a read-out of, but not directly involved in, the disease process.
We have shown previously that the normal function of PABPN1 and more specifically its activity in RNA binding is required for OPMD pathology (Chartier et al, 2006). Moreover, we have also reported that muscle defects can be uncoupled from the presence of aggregates, for example when a deleted form of PABPN1 without the polyalanine tract is expressed in muscles (Chartier et al, 2006). Together, these data led us to propose that the early defects in OPMD would depend on PABPN1 function and that aggregate formation might contribute to pathogenesis during later stages when aggregates reach a large portion of nuclei, thus altering nuclear function.
Both the 3F5 intrabody and the 6AP and GA compounds have strong effects in decreasing muscle degeneration and also reduce the PABPN1-17ala aggregation load. This indicates that at least one step is common to the aggregation process and the pathology. It could be a conformational change of PABPN1-17ala that might affect the function of the protein and would eventually lead to the formation of aggregates.
Recent data on polyglutamine diseases also reveal an essential role of normal protein functions in pathogenesis (Duvick et al, 2010; Nedelsky et al, 2010). These data led to a model in which the normal protein exists in at least two different conformations that would be responsible for two different normal functions. Extension of the polyglutamine tract would favour one conformer, thus disturbing the equilibrium and leading to both the gain of function of one conformer and the lack of function of the other (Kratter & Finkbeiner, 2010). One possibility could be that the conformer that is stabilized upon mutation could eventually form aggregates. This model is particularly suited for OPMD as the normal protein has been shown to assemble in aggregates under specific physiological conditions (Berciano et al, 2004).
A major piece of information presented here is the fact that GA has a strong effect in preventing OPMD pathology in the Drosophila model. Drosophila has proven to be useful to identify drugs with a good efficacy in mammalian models for specific diseases, in particular neurodegenerative disorders (Marsh & Thompson, 2004). GA is used routinely in the clinic as an antihypertensive drug (Holmes et al, 1983). It has been administrated for many years to patients on a daily basis without major side-effects. Therefore, our results identify GA as a strong candidate to be evaluated in potential treatments for OPMD.
Another important aspect of our results is that they support the function of the large ribosomal RNA in propagation of protein aggregation in diseases. In addition to its central function in protein synthesis, an ability of the ribosome to assist protein folding has been described (Argent et al, 2000; Chattopadhyay et al, 1996; Pal et al, 1999; Samanta et al, 2008; Sanyal et al, 2002). More specifically, this activity is borne by the domain V of the large ribosomal RNA. 6AP and GA have antiprion properties both in yeast and in mammalian systems (Bach et al, 2003; Tribouillard et al, 2006; Tribouillard-Tanvier et al, 2008a). These compounds bind to specific positions in domain V of the large ribosomal RNA and prevent its activity in protein folding, without affecting its function in protein synthesis (Tribouillard-Tanvier et al, 2008b). Together, this set of data suggests that the large ribosomal RNA has a function in prion propagation, and that this function is prevented by direct interactions with 6AP or GA, thus leading to reduced prion stability or propagation. Here, we show the benefit of 6AP and GA in vivo on a physiological defect, namely muscle degeneration. We further find that reduced copy number of ribosomal DNA repeats has a similar beneficial effect on OPMD phenotypes and that 6AP acts synergistically with reduced copy number of ribosomal DNA repeats to decrease OPMD phenotypes. This provides genetic evidence that the ribosomal RNA is a conserved cellular target of 6AP in Drosophila. More importantly, these data strongly suggest that the ribosomal RNA plays a role in PABPN1-17ala aggregation in OPMD, and that the beneficial effect of 6AP on OPMD results from the inhibition of this specific function of the ribosomal RNA. Consistent with this, we find that deletions of ribosomal DNA repeats result in the reduced size of PABPN1-17ala nuclear aggregates. Moreover, we show that the domain V of ribosomal RNA is able to accelerate fibril formation of the N-terminal domain of PABPN1-17ala in vitro. This identifies, for the first time, a direct effect of the domain V of ribosomal RNA on the conformational transition of a protein domain implicated in a disease.
Because the ribosomal RNA is highly conserved between species, it is very likely that its function in protein folding and in aggregate propagation in diseases is also conserved. 6AP and GA specifically prevent the protein folding activity of the large ribosomal RNA and are unique in targeting this general cellular function (Tribouillard-Tanvier et al, 2008b; Voisset et al, 2008). This could be the basis for the widespread effect of 6AP and GA. The antiprion activity of both drugs was evidenced with different, unrelated prion proteins, both in terms of sequence and function, and in different organisms ranging from yeast to mammals (Bach et al, 2003; Tribouillard et al, 2006; Tribouillard-Tanvier et al, 2008a). In addition, we demonstrate here the protective effect of 6AP and GA on a different protein aggregation disease, and in Drosophila, yet another model system. Because of the specific function of PABPN1 in RNA metabolism (Benoit et al, 1999, 2005; Lemay et al, 2010; Wahle, 1991), we cannot exclude an additional role of the ribosomal RNA in OPMD pathogenesis that could also be modified by 6AP and GA. However, our biophysical approach shows a direct role of domain V of ribosomal RNA on the conformational change of a PABPN1 domain that is not involved in RNA binding.
This study, in addition to the widespread antiprion effect of 6AP and GA, suggests that 6AP and GA might have a general role in preventing protein aggregation and that they might be beneficial for other major protein aggregation neurodegenerative disorders.
MATERIALS AND METHODS
Fly stocks, drug supplemented medium and analysis of drug toxicity
The Gal4 driver was Mhc-Gal4 (Schuster et al, 1996), which induces expression in muscles. UAS-PABPN1-17ala has been described previously (Chartier et al, 2006). The crosses were performed at 18°C to allow a slow progression of OPMD phenotypes (Chartier et al, 2006). OPMD males were scored, except in the experiments with the bobbed (bb) deletions. w1118 stock was used as the wild-type control. bbN is a weak allele of bb (FlyBase: http://flybase.org/) and Df(1)17-87 corresponds to a deficiency including the bb locus (Schalet & Lefevre, 1973). Drug supplemented food was prepared as follows. Instant Drosophila medium (Carolina Biological Supply Company) was reconstituted in each vial with a solution of 1% yeast in water, supplemented with either increasing concentrations of drug diluted in DMSO (Sigma D2650), or DMSO alone. Puromycin was diluted in water. Except for 6AP and 6APi, the drugs were purchased (Sigma) as follows: puromycin dihydrochloride (P8833), doxycycline hydrate (D9891), d-(+)-trehalose dihydrate (T9531), ibuprofen sodium salt (I1892), guanabenz acetate salt (G110). Each vial contained 5 or 2 ml of reconstituted medium, to raise larvae or adults, respectively. When individuals were fed with the drugs from larval stages, 70–80 first instar larvae were transferred per vial and developed in the same vial up to adulthood; adults were transferred in a new vial with the same concentration of drug. To determine drug toxicity, 80 first instar larvae were transferred into vials containing drug- or DMSO alone-supplemented medium. Individuals reaching adulthood were scored.
Analysis of wing position and adult musculature
Abnormal wing position and visualization of thorax muscles under polarized light were determined as described previously (Chartier et al, 2006).
Immunohistochemistry and western blots
Western blots and immunostaining of adult IFMs were performed as reported previously (Chartier et al, 2006). Rabbit anti-PABPN1 antibody was used at dilution 1:1000. Thoracic muscles of 10–15 adults were stained per experiment and nuclei were scored for the presence or absence of nuclear inclusions; scoring was performed from two independent staining experiments.
The paper explained
PROBLEM
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant, late onset, muscular dystrophy that usually starts in the fifth or sixth decade and is characterized by progressive eyelid drooping (ptosis), swallowing difficulties (dysphagia), and proximal limb weakness. OPMD is a rare disease that is caused by extension of a polyalanine tract in PABPN1. Insoluble nuclear aggregates form in diseased muscles. There is no pharmacological treatment for this disease at the moment.
RESULTS
Using a Drosophila model of OPMD that recapitulates the features of the disorder, the authors of this report aim to identify drugs that might be beneficial for OPMD. Because aggregates are formed in OPMD, they tested drugs with anti-aggregation properties. They find that 6AP and guanabenz are beneficial for OPMD in the Drosophila model. Both drugs reduce muscle degeneration and weakness, as well as the formation of aggregates. The cellular target of these drugs has been identified as the large ribosomal RNA and its activity in protein folding. Using a genetic approach, they show that deletions of the ribosomal DNA locus reduce OPMD phenotypes and act synergistically with sub-effective dose of 6AP. In a complementary biophysical approach, they show that ribosomal RNA accelerates in vitro fibril formation of PABPN1 N-terminal domain. These results suggest a conserved role of the protein folding activity of ribosomal RNA in OPMD.
IMPACT
This preclinical study shows that guanabenz has a strong effect in preventing OPMD pathology in the Drosophila model. Guanabenz is used routinely in medicine as as a treatment against hypertension. It has been administrated for many years to patients on a daily basis, without major side-effects. Therefore, this report identifies guanabenz as a strong candidate to be evaluated in potential treatments for OPMD.
Fibril analysis by ANS fluorescence and electron microscopy
Recombinant gene expression and protein purification of the N-terminal domain of PABPN1 containing seven additional alanine residues, PABPN1-N-17ala was performed as published for CspB fusions (Sackewitz et al, 2008) except that heat precipitation was performed for 5 min at 80°C. PABPN1-N-17ala was dissolved to a final concentration of 1 mM and incubated at 37°C in 5 mM KH2PO4, 100 mM NaCl at pH 7.5, containing 1 µg/ml NaN3 and 0.8 U/µl RiboLock™ RNase Inhibitor (Fermentas, Germany), either in the presence or absence of RNA. Domain V (747 nt) of the Drosophila 28S ribosomal RNA was produced using in vitro transcription (Fermentas, Germany) and a PCR fragment amplified with primers 5′TAATACGACTCACTATAGGGGATATTAGACCTCGGTTTGGTATCG (T7 promoter sequence is underlined) and 5′TGTGCCATTGGTCCGTACCTGCGGG, as a template. The RNA was phenol-chloroform extracted, precipitated and resuspended in H2O. The control experiment with an irrelevant RNA was performed with tRNA (Applied Biosystems, USA). ANS fluorescence was determined as described previously (Sackewitz et al, 2008). For EM analysis, carbonized copper grids (Plano, Wetzlar, Germany) were prepared as published (Lodderstedt et al, 2007) and visualized with a Zeiss EM 900 EM operating at 80 kV.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique (UPR1142), the GIS ‘Maladies Rares’ (# 35), the ANR-Maladies Rares (ANR-06-MRAR-035-01), the ANR Genopat (ANR-09-GENO-025-01), the FRM ‘Equipe FRM 2007’ and the European Commission (TRI-EX QLG2-CT-2001-01673 and PolyALA LSHM-CT-2005-018675) to M. S., by the ANR Blanche (ANR-06-BLAN-0056-01) to M. B. and by the German Research Foundation (DFG, SFB 610) to E.S. N. B. held a salary from the EC (PolyALA LSHM-CT-2005-018675) and ANR-Maladies Rares. A. C. held a salary from Association Française contre les Myopathies and ANR-Maladies Rares. Y. B. held a salary from the EC (PolyALA LSHM-CT-2005-018675). C. V. held an INSERM ‘junior’ contract. A. B. was funded through a stipend of the Graduiertenkolleg GRK1026 (Conformational transitions in macromolecules).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
NB, AC and YB designed and performed the experiments and analyzed the data. AB designed, performed and analyzed the biophysical experiments. CV, HG and MB provided material. MB, ES and MS analyzed the data. MS designed the study and wrote the paper. All authors discussed the results and commented on the manuscript.
For more information
OMIM: Oculopharyngeal muscular dystrophy
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Abu-Baker A, Messaed C, Laganiere J, Gaspar C, Brais B, Rouleau GA. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet. 2003;12:2609–2623. doi: 10.1093/hmg/ddg293. [DOI] [PubMed] [Google Scholar]
- Argent RH, Parrott AM, Day PJ, Roberts LM, Stockley PG, Lord JM, Radford SE. Ribosome-mediated folding of partially unfolded ricin A-chain. J Biol Chem. 2000;275:9263–9269. doi: 10.1074/jbc.275.13.9263. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bach S, Talarek N, Andrieu T, Vierfond JM, Mettey Y, Galons H, Dormont D, Meijer L, Cullin C, Blondel M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat Biotechnol. 2003;21:1075–1081. doi: 10.1038/nbt855. [DOI] [PubMed] [Google Scholar]
- Bao YP, Sarkar S, Uyama E, Rubinsztein DC. Congo red, doxycycline, and HSP70 overexpression reduce aggregate formation and cell death in cell models of oculopharyngeal muscular dystrophy. J Med Genet. 2004;41:47–51. doi: 10.1136/jmg.2003.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit B, Nemeth A, Aulner N, Kühn U, Simonelig M, Wahle E, Bourbon HM. The Drosophila poly(A)-binding protein II is ubiquitous throughout Drosophila development and has the same function in mRNA polyadenylation as its bovine homolog in vitro. Nucleic Acids Res. 1999;27:3771–3778. doi: 10.1093/nar/27.19.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit B, Mitou G, Chartier A, Temme C, Zaessinger S, Wahle E, Busseau I, Simonelig M. An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev Cell. 2005;9:511–522. doi: 10.1016/j.devcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Berciano MT, Villagra NT, Ojeda JL, Navascues J, Gomes A, Lafarga M, Carmo-Fonseca M. Oculopharyngeal muscular dystrophy-like nuclear inclusions are present in normal magnocellular neurosecretory neurons of the hypothalamus. Hum Mol Genet. 2004;13:829–838. doi: 10.1093/hmg/ddh101. [DOI] [PubMed] [Google Scholar]
- Brais B. Oculopharyngeal muscular dystrophy: a late-onset polyalanine disease. Cytogenet Genome Res. 2003;100:252–260. doi: 10.1159/000072861. [DOI] [PubMed] [Google Scholar]
- Brais B, Bouchard J-P, Xie Y-G, Rochefort DL, Chrétien N, Tomé FMS, Lafrenière RG, Rommens JM, Uyama E, Nohira O, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altered cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E, Carmo-Fonseca M. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein II aggregates which sequester poly(A) RNA. Hum Mol Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- Chartier A, Benoit B, Simonelig M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 2006;25:2253–2262. doi: 10.1038/sj.emboj.7601117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier A, Raz V, Sterrenburg E, Verrips CT, van der Maarel SM, Simonelig M. Prevention of oculopharyngeal muscular dystrophy by muscular expression of Llama single-chain intrabodies in vivo. Hum Mol Genet. 2009;18:1849–1859. doi: 10.1093/hmg/ddp101. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S, Das B, Dasgupta C. Reactivation of denatured proteins by 23S ribosomal RNA: role of domain V. Proc Natl Acad Sci USA. 1996;93:8284–8287. doi: 10.1073/pnas.93.16.8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Davies JE, Wang L, Garcia-Oroz L, Cook LJ, Vacher C, O'Donovan DG, Rubinsztein DC. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med. 2005;11:672–677. doi: 10.1038/nm1242. [DOI] [PubMed] [Google Scholar]
- Davies JE, Sarkar S, Rubinsztein DC. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet. 2006;15:23–31. doi: 10.1093/hmg/ddi422. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, Orr HT. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Dion P, Laganiere J, Brais B, Rouleau GA. Oligomerization of polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death. Hum Mol Genet. 2001;10:2341–2351. doi: 10.1093/hmg/10.21.2341. [DOI] [PubMed] [Google Scholar]
- Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS. Guanabenz: a review of its pharmacodynamic properties and therapeutic efficacy in hypertension. Drugs. 1983;26:212–229. doi: 10.2165/00003495-198326030-00003. [DOI] [PubMed] [Google Scholar]
- Kratter IH, Finkbeiner S. PolyQ disease: too many Qs, too much function. Neuron. 2010;67:897–899. doi: 10.1016/j.neuron.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn U, Wahle E. Structure and function of poly(A) binding proteins. Biochim Biophys Acta. 2004;1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Kuhn U, Nemeth A, Meyer S, Wahle E. The RNA binding domains of the nuclear poly(A)-binding protein. J Biol Chem. 2003;278:16916–16925. doi: 10.1074/jbc.M209886200. [DOI] [PubMed] [Google Scholar]
- Lemay JF, D'Amours A, Lemieux C, Lackner DH, St-Sauveur VG, Bahler J, Bachand F. The nuclear poly(A)-binding protein interacts with the exosome to promote synthesis of noncoding small nucleolar RNAs. Mol Cell. 2010;37:34–45. doi: 10.1016/j.molcel.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Lodderstedt G, Hess S, Hause G, Scheuermann T, Scheibel T, Schwarz E. Effect of oculopharyngeal muscular dystrophy-associated extension of seven alanines on the fibrillation properties of the N-terminal domain of PABPN1. FEBS J. 2007;274:346–355. doi: 10.1111/j.1742-4658.2006.05595.x. [DOI] [PubMed] [Google Scholar]
- Long EO, Dawid IB. Repeated genes in eukaryotes. Annu Rev Biochem. 1980;49:727–764. doi: 10.1146/annurev.bi.49.070180.003455. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Can flies help humans treat neurodegenerative diseases. Bioessays. 2004;26:485–496. doi: 10.1002/bies.20029. [DOI] [PubMed] [Google Scholar]
- Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z, Neale G, Taylor JP. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron. 2010;67:936–952. doi: 10.1016/j.neuron.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Chandra S, Chowdhury S, Sarkar D, Ghosh AN, Gupta CD. Complementary role of two fragments of domain V of 23 S ribosomal RNA in protein folding. J Biol Chem. 1999;274:32771–32777. doi: 10.1074/jbc.274.46.32771. [DOI] [PubMed] [Google Scholar]
- Sackewitz M, von Einem S, Hause G, Wunderlich M, Schmid FX, Schwarz E. A folded and functional protein domain in an amyloid-like fibril. Protein Sci. 2008;17:1044–1054. doi: 10.1110/ps.073276308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D, Mukhopadhyay D, Chowdhury S, Ghosh J, Pal S, Basu A, Bhattacharya A, Das A, Das D, DasGupta C. Protein folding by domain V of Escherichia coli 23S rRNA: specificity of RNA-protein interactions. J Bacteriol. 2008;190:3344–3352. doi: 10.1128/JB.01800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal SC, Pal S, Chowdhury S, DasGupta C. 23S rRNA assisted folding of cytoplasmic malate dehydrogenase is distinctly different from its self-folding. Nucleic Acids Res. 2002;30:2390–2397. doi: 10.1093/nar/30.11.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalet A, Lefevre GJ. The localisation of “ordinary” sex-linked genes in Section 20 of the polytene X chromosome of Drosophila melanogaster. Chromosoma. 1973;44:183–202. doi: 10.1007/BF00329116. [DOI] [PubMed] [Google Scholar]
- Scheuermann T, Schulz B, Blume A, Wahle E, Rudolph R, Schwarz E. Trinucleotide expansions leading to an extended poly-L-alanine segment in the poly(A) binding protein PABPN1 cause fibril formation. Protein Sci. 2003;12:2685–2692. doi: 10.1110/ps.03214703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. I. Fasciclin II controls synaptic stabilization and growth. Neuron. 1996;17:641–654. doi: 10.1016/s0896-6273(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- Tavanez JP, Calado P, Braga J, Lafarga M, Carmo-Fonseca M. In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. Rna. 2005;11:752–762. doi: 10.1261/rna.7217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Tribouillard D, Bach S, Gug F, Desban N, Beringue V, Andrieu T, Dormont D, Galons H, Laude H, Vilette D, et al. Using budding yeast to screen for anti-prion drugs. Biotechnol J. 2006;1:58–67. doi: 10.1002/biot.200500001. [DOI] [PubMed] [Google Scholar]
- Tribouillard-Tanvier D, Beringue V, Desban N, Gug F, Bach S, Voisset C, Galons H, Laude H, Vilette D, Blondel M. Antihypertensive drug guanabenz is active in vivo against both yeast and mammalian prions. PLoS One. 2008a;3:e1981. doi: 10.1371/journal.pone.0001981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribouillard-Tanvier D, Dos Reis S, Gug F, Voisset C, Beringue V, Sabate R, Kikovska E, Talarek N, Bach S, Huang C, et al. Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs. PLoS One. 2008b;3:e2174. doi: 10.1371/journal.pone.0002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisset C, Thuret JY, Tribouillard-Tanvier D, Saupe SJ, Blondel M. Tools for the study of ribosome-borne protein folding activity. Biotechnol J. 2008;3:1033–1040. doi: 10.1002/biot.200800134. [DOI] [PubMed] [Google Scholar]
- Wahle E. A novel poly(A)-Binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- Wahle E. Poly(A) tail length control is caused by termination of processive synthesis. J Biol Chem. 1995;270:2800–2808. doi: 10.1074/jbc.270.6.2800. [DOI] [PubMed] [Google Scholar]
- Wang Q, Mosser DD, Bag J. Induction of HSP70 expression and recruitment of HSC70 and HSP70 in the nucleus reduce aggregation of a polyalanine expansion mutant of PABPN1 in HeLa cells. Hum Mol Genet. 2005;14:3673–3684. doi: 10.1093/hmg/ddi395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.