

Abstract
Accurate functional annotation of regulatory elements is essential for understanding global gene regulation. Here, we report a genome-wide map of 827,000 transcription factor binding sites in human lymphoblastoid cell lines, which is comprised of sites corresponding to 239 position weight matrices of known transcription factor binding motifs, and 49 novel sequence motifs. To generate this map, we developed a probabilistic framework that integrates cell- or tissue-specific experimental data such as histone modifications and DNase I cleavage patterns with genomic information such as gene annotation and evolutionary conservation. Comparison to empirical ChIP-seq data suggests that our method is highly accurate yet has the advantage of targeting many factors in a single assay. We anticipate that this approach will be a valuable tool for genome-wide studies of gene regulation in a wide variety of cell types or tissues under diverse conditions.
A central challenge in modern genomics is to identify all the functional elements in genomes and, ultimately, to understand their individual roles. While the annotation of human protein-coding sequences is now fairly comprehensive, the identification of regulatory sequences remains difficult (The ENCODE Project Consortium 2007; Siepel et al. 2007). Accurate maps of regulatory sites will be essential for a comprehensive understanding of gene regulation and circuitry. Moreover, genetic variation in regulatory elements is a key driver of evolution and disease (Wray 2007; Amit et al. 2009; Nicolae et al. 2010), yet our limited knowledge of which locations in the genome are involved in gene regulation often makes it difficult to predict the functional impact of noncoding variants.
One fundamental mechanism of gene regulation is provided by transcription factors (TFs), many of which bind DNA preferentially at characteristic sequence motifs, thereby providing the sequence specificity required to direct complex programs of gene regulation (Lemon and Tjian 2000). In recent years, chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) has become the gold-standard method for genome-wide detection of the binding locations for individual TFs (Johnson et al. 2007; Robertson et al. 2007). However, ChIP assays are limited in that each experiment profiles just one TF, making it a substantial undertaking to profile more than a few different TFs in any given tissue, let alone across different conditions. Indeed, at this time, TF binding has not been characterized for more than a handful of different transcription factors in any mammalian cell or tissue type. Given that several hundred TFs may be active simultaneously in a given tissue (Vaquerizas et al. 2009), our knowledge of genome-wide TF binding remains rudimentary.
As an alternative, a variety of computational methods have been developed to predict transcription factor binding sites (e.g., Elemento and Tavazoie 2005; Tompa et al. 2005; Xie et al. 2009; Ernst et al. 2010; Won et al. 2010). These methods typically make use of sequence-specific binding motifs of individual factors, either obtained from databases such as TRANSFAC or JASPAR (Wingender et al. 1996; Sandelin et al. 2004), or estimated de novo. However, only a small fraction of genomic locations matching such motifs are actually bound by TFs. By incorporating external information such as evolutionary conservation and experimental data, recent studies have made substantial progress at predicting TF-bound sites; however, error rates remain considerable, and the predictions are generally not specific to particular cell types or conditions (see Supplemental material).
Here, we describe a new algorithm, named CENTIPEDE, that combines genome sequence information with cell-specific experimental data to map bound TF binding sites in a specific sample. Recent work has shown that several genome-wide assays correlate with TF binding, including measures of chromatin accessibility as measured by DNase I sensitivity or FAIRE (formaldehyde-assisted isolation of regulatory elements) assays (Boyle et al. 2008; Gaulton et al. 2010), protection of the actual TF binding site from DNase I cleavage (Fu et al. 2008; Hesselberth et al. 2009; Chen et al. 2010), and ChIP-seq against specific combinations of histone modifications (Heintzman et al. 2007; Ernst et al. 2010) or coactivator protein p300 (Visel et al. 2009). Furthermore, it has long been known that each type of protein–DNA interaction can produce a characteristic DNase I footprint that reflects the specific properties of that interaction (Galas and Schmitz 1978). Here, we show that a model integrating these sources of information agrees very closely with empirical ChIP-seq measurements of TF binding at candidate motif sites.
Like other computational methods for inferring TF binding sites, our approach requires the presence of a DNA sequence motif at the binding site. Presence of a binding motif makes it possible to connect the information from a generic assay such as DNase I to particular TFs (or in some cases, sets of TFs with similar binding motifs). The strength of CENTIPEDE is its ability to identify binding sites for many factors from a single experimental assay. In contrast, ChIP-seq can provide more exhaustive information about individual TFs, including the identification of binding sites without a standard motif (e.g., sites with noncanonical motifs) (Johnson et al. 2007) or sites at which the TF associates with the DNA indirectly via a binding partner (Jothi et al. 2008; Gordân et al. 2009). Hence, we see the two approaches as complementary: ChIP-seq is the tool of choice for in-depth study of limited numbers of factors of particular interest; meanwhile, methods such as CENTIPEDE promise to be useful for rapid profiling of many factors across diverse cell types or experimental conditions. Here, we illustrate the use of CENTIPEDE by creating a map of 827,000 inferred TF binding sites in human lymphoblast cell lines (LCLs). Single nucleotide polymorphisms (SNPs) or copy number variations (CNVs) that disrupt these sites should be of particular interest in disease association studies. The map and software are available at http://centipede.uchicago.edu.
Results
Our method starts by scanning the genome for all positions with substantial similarity to a known sequence motif or position weight matrix (PWM). We then use an unsupervised Bayesian mixture model to infer which candidate sites for each motif are likely to be bound by a TF. Our method does this by assuming that sites that are bound will tend to differ in multiple ways from sites that are not bound. For example, sites that are bound are more likely to be associated with open chromatin (inferred from DNase I data), are often associated with active histone marks, and are more likely to show evolutionary sequence conservation. The mixture modeling approach uses these kinds of data to cluster motif-match sites into two different classes, which we interpret as “bound” and “unbound,” and to compute the posterior probability that a given site belongs to each class.
For each candidate binding site, we separate the available information into two components: G, which refers to genomic information that is independent of cell type or experimental conditions (e.g., a sequence conservation score or PWM match score); and D, which refers to cell-specific experimental data (such as the number of DNase I or histone-mark reads around the candidate site). For each site, we regard the genomic information G as “prior information” that reflects the general propensity of a site to be bound. We model the prior probability that a site is bound, P(Bound|G), as a logistic function of the genomic information G (for details, see Methods). We then model the likelihood of the experimental data as P(D|Bound) or P(D|Not Bound), depending on whether the site is inferred to be bound, or not bound, respectively. The functional form for the likelihood depends on the details of the data, and we discuss this in more detail below.
The likelihood of the experimental data at a single motif site can be written as:
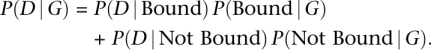 |
This likelihood depends on the coefficients of the logistic prior and on the parameters for the data distributions for bound and unbound sites, which are not known in advance. We use a standard expectation-maximization algorithm to maximize the product of the likelihoods across all candidate sites for a given motif with respect to these parameters. Finally, given the maximum likelihood parameter estimates, we use Bayes' rule to compute the posterior probability, P(Bound|D, G), that each motif site is bound by a TF (Methods). We perform the computation separately for each motif of interest, since different motifs will likely be characterized by different distributions of the genomic and experimental factors.
Figures 1 and 2 illustrate the use of this method to infer binding sites of the transcription factor REST (also known as NRSF) in lymphoblastoid cell lines. We considered three types of prior information: PWM match score; proximity to the nearest transcription start site (TSS); and evolutionary sequence conservation (Pollard et al. 2010). Additionally, we used experimental data on seven histone modifications and DNase-seq data, all from LCLs. These include publicly available data generated by the Bernstein and Crawford labs for the ENCODE project (The ENCODE Project Consortium 2007; McDaniell et al. 2010), plus additional DNase-seq data from our group (for details, see Methods). In a window around each REST candidate binding site, we obtained the total number of ChIP-seq reads for chromatin enriched for binding of five activating histone marks (H3K4me1, H3K4me2, H3K4me3, H3K9ac, and H3K27ac) and for two repressing marks (H3K27me1 and H4K20me1), as well as the number and locations of DNase-seq reads.
Figure 1.

Overview of the CENTIPEDE approach using the factor REST as an example. Each row in the image represents a genomic location that matches the primary REST binding motif. For each motif instance, we extracted prior information (PWM score, TSS proximity, and conservation score) and experimental data (histone marks and DNase-seq reads). Rows are ordered by the posterior probability given by the model (bound sites at the top) and, for validation only, compared to REST ChIP-seq reads extracted in a 400-bp window surrounding the motif (last column). Darker blue coloring indicates, in each column, respectively, higher PWM score; motif closer to the TSS; motif site more conserved; more histone ChIP-seq reads in 400-bp windows around each site; more DNase I cuts per site; higher CENTIPEDE posterior probability of binding; larger number of REST ChIP-seq reads per site. Notice that a 22-bp segment at the center of high posterior sites is protected from DNase I cleavage indicating DNA–protein binding. The plot shows 200 randomly selected motif sites with posterior probability > 0.5, and 200 with posterior < 0.5, respectively; this sampling procedure increases the fraction of bound sites displayed.
Figure 2.

Model learned by the CENTIPEDE approach for the transcription factor REST. (A) Empirical density plots for key aspects of the data for sites inferred by CENTIPEDE to be bound (green lines, CENTIPEDE posterior probabilities >0.95) and unbound (red lines, probabilities < 0.5), respectively. The right-hand column shows comparison to REST ChIP-seq data. (B, upper plot) The distribution of CENTIPEDE posterior probabilities for ChIP-seq positives (motif inside a ChIP-seq peak, as reported by ENCODE using MACS) (Zhang et al. 2008) and for ChIP-seq negatives (fraction of reads from the control experiment is lower or equal compared to that from the ChIP experiment). (Lower plot) ROC curves for four methods of ranking binding sites. In decreasing order of performance, these are CENTIPEDE with DNase I data; CENTIPEDE with DNase I data and histone marks; number of DNase I reads within 200 bp; phastCons conservation score.
We modeled the numbers of sequencing reads originating from each experiment (ChIP-seq for active marks, repressive marks, and DNase-seq reads) with negative binomial distributions, using independent sets of parameters for each data type and independently in the bound and unbound classes. Additionally, we observed that the spatial distribution of DNase-seq reads (i.e., the number of reads obtained at each position in the window, given the total number) is highly informative about binding. We also found that this spatial distribution varies widely from factor to factor, reflecting the specific interactions of each factor with DNA (Fig. 4, below; Supplemental Fig. S8; Boyle et al. 2008; Hesselberth et al. 2009). Hence, we modeled the spatial distribution of DNase-seq reads with a multinomial distribution (fixed to be uniform in the unbound case reflecting the lack of protein binding; see Fig. 2). By jointly modeling both the regional DNase I sensitivity and the exact positional distribution of reads surrounding the motif, our method captures both the chromatin-accessibility information and the base-by-base cleavage pattern of DNase I (the “footprint”) that is characteristic of each factor.
Figure 4.

Footprint strength as a measure of TF occupancy. For REST and GABPA, respectively, we plot the posterior log odds from the CENTIPEDE model against the square-root transformed count of ChIP-seq reads in the 400-bp region surrounding each motif. Additionally, we plot the DNase I footprint for motif sites falling into seven quantile ranges of the ChIP-seq data. In each plot, the data are colored according to the quantile range in the ChIP-seq data. For both factors, there is a clear correlation between CENTIPEDE posteriors and the number of ChIP-seq reads. Furthermore, there is a clear gradient in the footprints going from the most pronounced footprint in the highest ChIP-seq quantiles to virtually no footprint in the lowest quantiles.
When fitting the mixture model for REST (Fig. 2), CENTIPEDE learns that, compared to unbound sites, TF-bound sites tend to be more conserved, are enriched near the TSS, have a slight increase in repressive histone marks (REST is a repressor), no change in activating histone marks (Supplemental Fig. S10), an increase in chromatin accessibility to DNase I cleavage, and a distinctive spatial distribution (i.e., footprint) of DNase I cutsites that is specific for REST (see Supplemental Fig. S8 for other TFs). Even though no data type is fully informative taken alone, by combining information, CENTIPEDE is able to confidently infer whether each motif site is bound or unbound, and these estimates agree extremely closely with REST ChIP-seq data from the Myers group (The ENCODE Project Consortium 2007).
Figure 3 shows a systematic comparison of CENTIPEDE and ChIP-seq, using publicly available ENCODE ChIP-seq data from LCLs. For all six factors, the two methods show remarkable agreement in classifying PWM matches as bound or unbound (see Supplemental Fig. S2 and Supplemental Table S4 for similar results in K562 cells, and Supplemental material for more details). Overall, we achieved the best model performance using CENTIPEDE with the prior genomic data plus DNase-seq data (mean area under the curve [AUC] = 98.11%) (Supplemental Table S5). Several recent studies have shown that specific combinations of histone modifications are associated with active enhancer and promoter elements (Heintzman et al. 2007, 2009; Won et al. 2010). Our results support this conclusion, as the histone data are highly informative when used in the CENTIPEDE model (average AUC = 96.52%) (Supplemental Table S5). However, the histone data do not provide additional predictive power for TF binding when DNase I data are included in the model (Supplemental Table S4), especially if a low FPR is desired. Thus, in the rest of this paper, we apply the CENTIPEDE model without histone marks.
Figure 3.

CENTIPEDE performance for six TFs for which ChIP-seq data are available in LCLs. (A) Individual ROC curves for each TF using the CENTIPEDE model with prior information and full DNase I distribution. (B) Performance across all six TFs in terms of the average sensitivity that can be achieved with a 1% false-positive rate (FPR). For both panels, motif instances are defined as ChIP-seq positives if motifs fall inside a ChIP-seq peak called using MACS, or ChIP-seq negatives if the fraction of reads from the control experiment is less than or equal to that from the ChIP treatment (for details, see Supplemental material).
It is notable that a naive use of DNase I read-depth alone is also very informative about which motif sites are bound by a given TF, as measured by ChIP-seq (Fig. 3; Supplemental Table S5). This implies that, at least for the factors with available ChIP-seq data, most accessible genomic regions containing a suitable binding site for a given factor are bound by that factor. That said, CENTIPEDE provides significantly higher sensitivity for TF binding than does DNase I read depth alone; for example, CENTIPEDE reduces the false-negative rate by about twofold at a 1% false-positive rate (Fig. 3B). To illustrate this, Supplemental Figure S12 shows that in nearly all cases in which a candidate binding site for CTCF lies in a hypersensitive site, but there is no ChIP-seq signal, CENTIPEDE agrees with the ChIP data. The improved performance of CENTIPEDE is largely due to the extra information provided by the precise locations of DNase I cuts (i.e., the footprint).
Thus far we have focused on classifying motif sites as bound or unbound, based on ChIP-seq data. However, it is becoming clear that, in practice, TF binding is quantitative: i.e., corresponding to the fraction of cells that have TF binding at a particular site at any given time (MacArthur et al. 2009; Bradley et al. 2010). Although our model is formulated as a binary mixture of bound and unbound sites, we hypothesized that the degree to which a site matches the expectations for the bound state (summarized by the posterior log odds) might reflect the level of TF occupancy. As shown in Figure 4, this appears to be the case. Taking the number of ChIP-seq reads around each site as an (noisy) estimate of TF occupancy, we find a substantial correlation between ChIP-seq read depth and the posterior log odds of binding from CENTIPEDE. Across the different TFs we tested, these correlations are substantially higher than those between the control reads and the posterior odds (see Supplemental Fig. S15; Supplemental Table S4). Figure 4 also shows that the strength of the DNase I footprint increases steadily with ChIP-seq read depth, again suggesting that DNase I can provide quantitative information about TF occupancy. The accuracy of this approach is likely to improve as advances in DNA sequencing enable ever-increasing numbers of sequence reads. In the remainder of this paper, we focus on sites with a high posterior probability of binding; this likely corresponds to sites with high average TF occupancy.
Application of CENTIPEDE to many motifs
Having validated our computational model for TFs with available ChIP data, we next considered 756 PWMs available from the TRANSFAC and JASPAR databases. For most of the corresponding TFs, ChIP-seq data were unavailable. We would expect that only a fraction of these PWMs correspond to TFs that are both expressed and active in human LCLs, and it was necessary to identify the subset of PWMs for which CENTIPEDE detects a genuine signal of binding. We reasoned that, for active TFs, the inferred binding sites should, on average, show more sequence conservation than the inferred unbound sites (Xie et al. 2005; Pollard et al. 2010). Hence, when we applied CENTIPEDE to these motifs, we held out the sequence conservation data to allow independent validation. For each PWM, we then computed a conservation Z-score that measures whether sequence conservation correlates with the posterior probability of binding from CENTIPEDE (see Supplemental material). Applying this procedure to random PWMs and to PWMs of TFs that are not expressed in LCLs, we found that the distribution of Z-scores is approximately standard normal (Fig. 5A). In contrast, the remaining PWMs showed a very strong enrichment of high conservation Z-scores. We identified 239 PWMs with a Z-score > 6.25 (false discovery rate [FDR] = 1.52%). The most conserved binding sites are for those motifs recognized by CTCF, MAZ, NFYA (CCAAT-box), and SP1 (GC-box).
Figure 5.

Application of the CENTIPEDE model across 756 known PWMs and 17,224 10-mers enriched in DNase I–sensitive regions. (Left panel) The distribution of the conservation Z-score for PWM motifs (A) and 10-mer derived motifs (B). (C) This panel shows that NRF1 binding sites are recovered from both the PWM and 10-mer-based approach. (D) Two examples that appear to be novel binding site motifs. For these motifs, locations inferred to be bound are strongly conserved, show a clear DNase I footprint, and show minimal overlap with known PWMs.
We next attempted to identify the binding sites of unknown or poorly characterized TFs that are not well represented by PWMs in TRANSFAC or JASPAR (Fig. 5B; Methods). We ran CENTIPEDE on 17,224 10-mer sequences (“words”) that were significantly enriched in DNase I–sensitive sites. Of this set, 735 words had a conservation Z-score > 6.25 (FDR = 10%, based on comparison to matched control words). We calculated the CENTIPEDE posterior probabilities for all locations in the genome that were at most 1 mismatch away from the original 10-mers, and the analyses below consider the sites with posterior probability >99% of being bound.
Most of the top-scoring words show strong overlap with known PWMs: The most frequently observed matches are for CTCF, NRF1 (Fig. 5C), ZNF143 (also known as STAF), and SP1 (GC-box). However, 49 of the enriched words show low overlap (<10% of high-posterior sites) with any PWM in TRANSFAC or JASPAR, and hence these seed-words may represent previously unrecognized or poorly characterized TF binding sites (Fig. 5D). In fact, two of the novel words (TCTCGCGAGA and AGGAGGAGGA) have been recently characterized as regulatory motifs (Guo et al. 2008; Frietze et al. 2010). For some of these words, recent protein-binding microarray data (Bulyk et al. 2001) may provide clues as to the identity of their binding partners (see Supplemental material).
We combined these analyses to construct a genome-wide map consisting of 826,896 putative binding sites, i.e., locations estimated to be bound by factors recognizing at least one PWM or word. Of these locations, 431,724 were detected using PWMs and 574,567 using novel words (with 179,395 recovered from both analyses). For many sites, the likely binding partner is somewhat ambiguous: For example, the E-box family of TFs (e.g., MYC, MAX, USF1, CLOCK, ARTNL) all share some overlap in their predicted binding locations due to their shared DNA sequence preferences (i.e., the canonical E-box motif, CACGTG). Altogether, the inferred binding sites span slightly less than 0.5% of the genome.
We next used this map to study the properties of the inferred binding sites (Fig. 6). There is great variation in the numbers of inferred binding sites among transcription factors, ranging from a few hundred (e.g., REST and SRF) to tens of thousands (e.g., CTCF and SP1), as well as variation in the extent to which binding sites occur near transcription start sites. For example, 70% and 93% of GC-box and TCTCGCGAGA sites are within 1 kb of a TSS, respectively, compared to just 8% of binding sites for the insulator CTCF. For each motif, most TF-bound sites fall near genes that are enriched for a specific function. Indeed, 98% of the motifs have at least one significantly enriched Gene Ontology (GO) category (Falcon and Gentleman 2007) after Bonferroni correction. Virtually every motif shows significant enrichment for nearby binding sites of other motifs, even when we account for positional biases with respect to the TSS. Moreover, 39% of motifs show enrichment for nearby binding sites of the same motif (FDR < 5%). These patterns are consistent with the notion of combinatorial action of specific TFs in the promoters of eukaryotic genes (Supplemental Figs. S6, S15; Pilpel et al. 2001; Zhu et al. 2005).
Figure 6.

Characteristics of the binding sites for 41 selected motifs. For each motif, we show the total number of inferred active sites (posterior probability > 0.99); the percentage of active sites that are within 1 kb of the nearest TSS; the most enriched GO category of the genes with a TSS within 5 kb of an active site; and the most enriched nonoverlapping element within 100 bp of the motif; the average shift in mean expression for genes containing an active binding site of each element in their promoter region (5 kb from TSS) in the linear model; the difference from average expression of the putative TF targets across nine tissues (Su et al. 2004); a Z-score measuring the enrichment/depletion of seven histone modification marks in the 400-bp region around the bound instances of each motif relative to unbound instances.
Additionally, we find that the presence of many of these binding sites is predictive of gene expression levels and by using a linear model, after performing variable selection, we identified 96 motifs that could, together, explain 38% of the variance in gene expression levels across all genes (for details, see Supplemental material). Finally, we used the Novartis Gene Expression Atlas (Su et al. 2004) to investigate the expression profile of genes that are putative targets of each TF in LCLs (Fig. 6). For example, genes that lie close to REST binding sites are enriched for neural GO categories (Johnson et al. 2007) and, on average, are broadly repressed except in neural tissues (Fig. 6; Supplemental Fig. S13). More generally, we find that the putative target genes show (1) increased expression levels in LCLs for most TFs, (2) high expression levels across all tissues for a few TFs (e.g., SRF and the novel motif TCTCGCGAGA), and (3) increased expression in lymphoblasts and closely related dentritic cells for key lymphoid-related factors such as E2F4, STAT1, PAX5, SPI1, and EBF1 (DeKoter and Singh 2000; Matthias and Rolink 2005; Nutt and Kee 2007).
Discussion
We have shown that an integrative approach can infer binding locations of hundreds of TFs simultaneously using cell-type-specific assays of chromatin accessibility. We anticipate that this kind of approach will be a valuable tool for mapping functional regulatory elements across a broad range of tissues and experimental conditions.
We see ChIP-seq and CENTIPEDE as being complementary tools. ChIP-seq can provide exhaustive information about binding for factors of special interest, including sites that may be missed by CENTIPEDE as they contain no recognizable motif. Additionally, ChIP-seq can avoid the ambiguity of motif-based approaches when multiple factors share a similar motif. However, since ChIP-seq is applied to one factor at a time, it currently does not scale well to studying large numbers of factors under varying conditions.
In contrast, approaches like CENTIPEDE can accurately profile many factors using a single experimental assay. The CENTIPEDE predictions provide precise resolution of binding locations and potentially quantitative measurement of binding occupancy. One important direction for future work is to explore whether the specificity of the DNase-footprint profile can be used to infer which factor(s) are present at a particular location when a motif site can be bound by multiple factors.
Of course, maps of transcription factor binding sites are but a first step toward understanding the architecture of gene regulation. Further experimental work is required to determine which inferred binding sites are functional and which genes they regulate. In the foreseeable future, we can anticipate high-resolution maps of regulatory sites for many different cell types—this will represent one important step toward a better understanding of how DNA sequence information encodes the information required for gene regulation.
Methods
Data
Candidate binding sites were identified using either pre-estimated position weight matrices (PWMs) from TRANSFAC and JASPAR databases or words that we determined to be enriched in hypersensitive sites. For a given PWM or word, we scanned the human genome sequence (hg18) for all matches above a specified threshold and considered each match to be a candidate binding site. For each candidate, we extracted genomic information that would be included in the model prior: sequence conservation (Pollard et al. 2010); quality of the PWM match; and distance to the nearest transcription start site; as well as experimental data in a 200–400-bp window around the site to be used in the likelihood—DNase I sensitivity and ChIP-seq data on seven histone modifications, all from LCLs. The experimental data were publicly available from the ENCODE Project (The ENCODE Project Consortium 2007; McDaniell et al. 2010) and, in the case of the DNase I data, supplemented with additional data from our group. See Supplemental material for further details.
The CENTIPEDE model
We use a probabilistic framework known as a hierarchical mixture model, which is described briefly here, and in greater detail in Supplemental material. The likelihood for a motif match l is written:
where Dl and Gl represent the observed experimental data and the prior information around the motif match. The data Dl are assumed to be generated from one of two underlying distributions that form the mixture model. One distribution corresponds to the bound state of transcription factors (Zl = 1), while the other distribution corresponds to the unbound state (Zl = 0).
For each potential binding location l, we calculate a prior probability πl = P(Zl = 1|Gl) that the site is bound by a TF. This prior probability is modeled using a logistic function:
 |
Here, “PWM Score” is a log-likelihood ratio of the probability of a given sequence under the PWM model, compared to a random sequence model. The “Cons. Score” is the average phastCons conservation score for the nucleotides within the motif match (Pollard et al. 2010). “TSS proximity” is the inverse of the distance to the nearest TSS in kilobases plus one.
As experimental data Dl, CENTIPEDE can combine multiple types of experiments 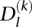 (here k indexes different experiments, such as read counts from DNase-seq and from histone modification ChIP-seq). For example, with three experiments,
(here k indexes different experiments, such as read counts from DNase-seq and from histone modification ChIP-seq). For example, with three experiments,
The underlying assumption is that the different experiments can be considered conditionally independent given that the underlying state Zl is known. We next specify the distribution to be used for each data type 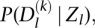 each with its own set of parameters for different k and state, Zl = 0 and Zl = 1.
each with its own set of parameters for different k and state, Zl = 0 and Zl = 1.
For a given experimental data type (e.g., DNase-seq), the collection of reads in a region (200 bp) around the motif matches l can be represented by an L × S matrix X = {Xls}. Each row Xl,. = (Xl,1,…, Xl,S) corresponds to motif-match location l, and each column s indexes the DNase I cut position relative to the center and strand of this motif match. The total number of reads in the region is defined as
The total number of reads is modeled with negative binomial distributions,
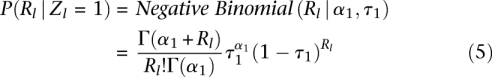 |
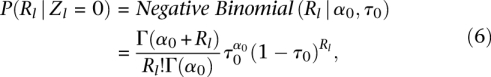 |
which depend on α1, τ1 for the bound class and α0, τ0 for the unbound class. While Poisson distributions may seem like the natural choice for the underlying process, the two-parameter negative binomial distribution allows us to more accurately model the variance in sequence read rate (Supplemental Fig. S7). With these two distributions, we can capture open versus closed chromatin in DNase I hypersensitivity assays or enrichment of certain histone modifications associated with enhancers or repressors measured by ChIP-seq assays. If the positional distribution P(Xl,·|Rl, Zl) is not important (or not very informative), we can leave it unspecified (i.e., any configuration is equally likely). This is the option we chose for the histone modification ChIP-seq assays based on preliminary analysis showing that the read locations were only weakly informative for these data (Supplemental Fig. S11). In contrast, for DNase I the positional information can be very informative as DNase I leaves a distinctive cleavage pattern (footprint) when Zl = 1 (Fig. 4; Supplemental Fig. S8). The spatial distribution of reads surrounding the binding site is modeled with a multinomial distribution
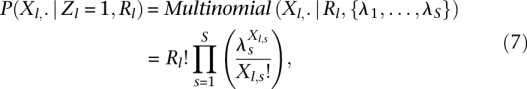 |
where the λl gives the probability that a read is obtained from position index s and Rlλs is the expected value of Xl,s given Rl. For Zl = 0, the TF is not bound, so no specific footprint is expected. In this case, we find it works well to simply model the cut-site distribution as uniform (λs = 1/S).
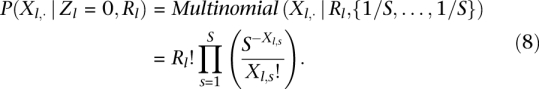 |
The parameters of the CENTIPEDE model 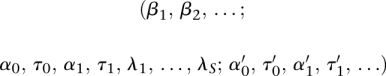 are estimated by maximizing the likelihood function using an expectation maximization (EM) algorithm (for details, see Supplemental material). Once the model has converged, the posterior probability pl is used to infer whether a TF is bound at location l. The form of this probability, pl, can be more easily interpreted in terms of the posterior odds:
are estimated by maximizing the likelihood function using an expectation maximization (EM) algorithm (for details, see Supplemental material). Once the model has converged, the posterior probability pl is used to infer whether a TF is bound at location l. The form of this probability, pl, can be more easily interpreted in terms of the posterior odds:
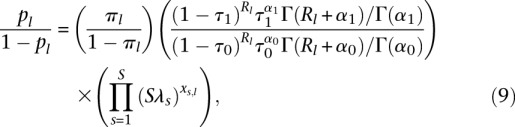 |
illustrating that the posterior odds are equal to the product of the prior odds (given by the logistic model) and the likelihood ratios (LRs) for the models corresponding to each type of data observed. This easily extends to multiple independent types of experimental data, each with its independent set of parameters as described in Supplemental material.
Validation of predicted binding sites
We downloaded publicly available ChIP-seq data from the ENCODE project corresponding to six transcription factors. Receiver operation curves (ROCs) were used for assessing the accuracy of prediction performance for each motif instance. The set of ChIP-seq positives was formed by those motif instances that fall inside a ChIP-seq peak, and the set of ChIP-seq negatives by those containing a lower or equal fraction of mappable reads from the ChIP-seq as compared to the “Control” experiment. For REST, SRF, and GABPA, we used the ChIP-seq peaks as reported by ENCODE, while for the other three factors (CTCF, JUND, and MAX), ChIP-seq peaks were re-extracted using MACS (the same peak-calling algorithm that was used for the initial three; for details, see Supplemental material). We note that, in order to draw an ROC curve, motif instances that are neither ChIP-seq positives nor ChIP-seq negatives are not taken into consideration because otherwise the “gold-standard” data would become contaminated with potentially misclassified borderline instances. For this reason, we also considered a correlation approach that takes into account all locations (except those for which >20% of the possible DNase reads in the surrounding 200 bp would not map uniquely—i.e., motifs in or near repetitive regions). For each motif instance, we extracted the total number of reads from the ChIP-seq and the “control” experiments and measured the Pearson correlation between the square root of the total number of reads and the CENTIPEDE posterior log-odds.
For motifs where ChIP data were unavailable, we used sequence conservation to assess whether the model was correctly detecting TF binding. For this, we withheld the phastCons score when fitting our model and defined a test statistic (conservation Z-score) that measured the significance of the logistic regression of the phastCons score of the motif on the posterior probability of binding (for full details, see Supplemental material).
Novel motif discovery
To identify novel DNA motifs with evidence of protein binding, we examined 10-mers that are enriched in the most DNase I–sensitive regions of the genome. To identify the most sensitive regions, we considered a 200-bp window centered on every single base pair in the genome and selected positions with more than 200 DNase-seq reads in the window. In total, 6.4 Mb (0.21% of the human genome) met this criterion, and on average each 10-mer occurred 12.2 times within this region (where a k-mer and its reverse complement are combined). We defined an “enriched” set of 10-mers as being those words that occurred more than 50 times in these DNase I–sensitive regions (corresponding to the top three percentile of the distribution). In addition, we constructed a “control” set of 20,000 10-mers that occurred six or fewer times (corresponding to the bottom 50 percentile) in these regions.
For each word in the enriched set, we ran the CENTIPEDE model on all the matches of the word in the genome. For the control words, we identified all locations in the genome matching at least 9 bp of the original 10-mer. We then used a rejection sampling strategy to match the distribution of DNase I HS to that for the enriched words. This sampling procedure was used to control for the correlation of DNase I regions with functional elements.
Additional downstream analyses
Several techniques were used to analyze the regulatory map composed of the predicted binding sites for all the motifs (for more details, see Supplemental material). We used hierarchical clustering to identify motifs whose predicted binding sites overlap substantially and most likely describe the same TF, or a TF family that shares sequence preference. For each pair of motifs that do not overlap, we also tested for colocalization using a two-sample Poisson test and controlling for the potential overrepresentation of motifs near TSSs. To evaluate the potential impact of the predicted binding sites on gene regulation, we considered that a gene was the target of a TF if it contained a high posterior binding site within 5 kb of its annotated TSS. The sets of genes that were targets of the same TF were analyzed using Gene Ontology, and the impact of TFs on gene expression was evaluated using a linear regression model. We also calculated general trends of enrichment/depletion of histone modification at predicted TF binding locations using a logistic regression model.
Acknowledgments
This work was supported by grants from the National Institutes of Health (Y.G., J.K.P.), by the Howard Hughes Medical Institute, by the Chicago Fellows Program (R.P.R.), by the American Heart Association (A.A.P.), and by the NIH Genetics and Regulation Training grant (A.A.P., J.F.D.). We thank the ENCODE Project, supported by NHGRI, for making data available prepublication (in particular, the Bernstein, Crawford, Myers, and Snyder groups and the UCSC Genome Browser); Greg Crawford for assistance in constructing DNase I libraries; and John Marioni, Matthew Stephens, and other members of the Pritchard, Przeworski, and Stephens labs and the anonymous reviewers for helpful comments or discussions.
Footnotes
[Supplemental material is available for this article. The regulatory map for lymphoblast cell lines and the source code for CENTIPEDE are available at http://centipede.uchicago.edu.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.112623.110.
References
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. 2009. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326: 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle A, Davis S, Shulha H, Meltzer P, Margulies E, Weng Z, Furey T, Crawford G 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R, Li X, Trapnell C, Davidson S, Pachter L, Chu H, Tonkin L, Biggin M, Eisen M 2010. Binding site turnover produces pervasive quantitative changes in transcription factor binding between closely related Drosophila species. PLoS Biol 8: e1000343 doi: 10.1371/journal.pbio.1000343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulyk M, Huang X, Choo Y, Church G 2001. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc Natl Acad Sci 98: 7158–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Hoffman MM, Bilmes JA, Hesselberth JR, Noble WS 2010. A dynamic Bayesian network for identifying protein-binding footprints from single molecule-based sequencing data. Bioinformatics 26: i334–i342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKoter R, Singh H 2000. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288: 1439–1441 [DOI] [PubMed] [Google Scholar]
- Elemento O, Tavazoie S 2005. Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol 6: R18 doi: 10.1186/gb-2005-6-2-r18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Plasterer H, Simon I, Bar-Joseph Z 2010. Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome Res 20: 526–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23: 257–258 [DOI] [PubMed] [Google Scholar]
- Frietze S, Lan X, Jin V, Farnham P 2010. Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J Biol Chem 285: 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sinha M, Peterson C, Weng Z 2008. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4: e1000138 doi: 10.1371/journal.pgen.1000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galas D, Schmitz A 1978. DNAse footprinting a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5: 3157–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K, Nammo T, Pasquali L, Simon J, Giresi P, Fogarty M, Panhuis T, Mieczkowski P, Secchi A, Bosco D, et al. 2010. A map of open chromatin in human pancreatic islets. Nat Genet 42: 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordân R, Hartemink A, Bulyk M 2009. Distinguishing direct versus indirect transcription factor–DNA interactions. Genome Res 19: 2090–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Bauer S, Hecht J, Schulz M, Busche A, Robinson P 2008. A short ultracon-served sequence drives transcription from an alternate FBN1 promoter. Int J Biochem Cell Biol 40: 638–650 [DOI] [PubMed] [Google Scholar]
- Heintzman N, Stuart R, Hon G, Fu Y, Ching C, Hawkins R, Barrera L, Van Calcar S, Qu C, Ching K, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318 [DOI] [PubMed] [Google Scholar]
- Heintzman N, Hon G, Hawkins R, Kheradpour P, Stark A, Harp L, Ye Z, Lee L, Stuart R, Ching C, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth J, Chen X, Zhang Z, Sabo P, Sandstrom R, Reynolds A, Thurman R, Neph S, Kuehn M, Noble W, et al. 2009. Global mapping of protein–DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Mortazavi A, Myers R, Wold B 2007. Genome-wide mapping of in vivo protein–DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Jothi R, Cuddapah S, Barski A, Cui K, Zhao K 2008. Genome-wide identification of in vivo protein–DNA binding sites from ChIP-Seq data. Nucleic Acids Res 36: 5221–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon B, Tjian R 2000. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev 14: 2551–2569 [DOI] [PubMed] [Google Scholar]
- MacArthur S, Li X, Li J, Brown J, Chu H, Zeng L, Grondona B, Hechmer A, Simirenko L, KerŁnen S, et al. 2009. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80 doi: 10.1186/gb-2009-10-7-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias P, Rolink A 2005. Transcriptional networks in developing and mature B cells. Nat Rev Immunol 5: 497–508 [DOI] [PubMed] [Google Scholar]
- McDaniell R, Lee B, Song L, Liu Z, Boyle A, Erdos M, Scott L, Morken M, Kucera K, Battenhouse A, et al. 2010. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328: 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ 2010. Trait-associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genet 6: e1000888 doi: 10.1371/journal.pgen.1000888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt S, Kee B 2007. The transcriptional regulation of B cell lineage commitment. Immunity 26: 715–725 [DOI] [PubMed] [Google Scholar]
- Pilpel Y, Sudarsanam P, Church G 2001. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet 29: 153–159 [DOI] [PubMed] [Google Scholar]
- Pollard K, Hubisz M, Rosenbloom K, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657 [DOI] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engstrom P, Wasserman W, Lenhard B 2004. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32: D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Diekhans M, Brejov B, Langton L, Stevens M, Comstock C, Davis C, Ewing B, Oommen S, Lau C, et al. 2007. Targeted discovery of novel human exons by comparative genomics. Genome Res 17: 1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su A, Wiltshire T, Batalov S, Lapp H, Ching K, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey T, Church G, De Moor B, Eskin E, Favorov A, Frith M, Fu Y, Kent W, et al. 2005. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23: 137–144 [DOI] [PubMed] [Google Scholar]
- Vaquerizas J, Kummerfeld S, Teichmann S, Luscombe N 2009. A census of human transcription factors: Function, expression and evolution. Nat Rev Genet 10: 252–263 [DOI] [PubMed] [Google Scholar]
- Visel A, Blow M, Li Z, Zhang T, Akiyama J, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R 1996. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res 24: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K, Ren B, Wang W 2010. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol 11: R7 doi: 10.1186/gb-2010-11-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray G 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8: 206–216 [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas E, Golub T, Mootha V, Lindblad-Toh K, Lander E, Kellis M 2005. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Rigor P, Baldi P 2009. MotifMap: A human genome-wide map of candidate regulatory motif sites. Bioinformatics 25: 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer C, Eeckhoute J, Johnson D, Bernstein B, Nusbaum C, Myers R, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137 doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Shendure J, Church G 2005. Discovering functional transcription-factor combinations in the human cell cycle. Genome Res 15: 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]