

Abstract
Infection and systemic inflammation are risk factors for cerebrovascular diseases and poststroke infections impair outcome in stroke patients, although the mechanisms of their contribution are mostly unknown. No preclinical studies have identified how chronic infection affects ischemic brain damage and which key inflammatory mediators are involved. We used a well established model of gut infection (Trichuris muris) to study how chronic infection contributes to brain injury. We show that, in mice, infection that leads to a chronic Th1-polarized immune response dramatically (60%) exacerbates brain damage caused by experimental stroke. Chronic Th1-type infection resulted in systemic upregulation of proinflammatory mediators and profoundly altered stroke-induced early (40 min to 4 h) and late (48 h) inflammation in the brain and peripheral tissues. Using the same infection, we show that a Th1-, but not Th2-polarized response augments brain injury by increasing the Th1 chemokine CCL5 [regulated on activation, normal T-cell expressed and secreted (RANTES)] systemically. This infection-associated response paralleled altered regulatory T-cell response, accelerated platelet aggregation in brain capillaries, and increased microvascular injury and matrix metalloproteinase activation after stroke. Antibody neutralization of RANTES reversed the effect of chronic infection on brain damage, microvascular MMP-9 activation, and cellular inflammatory response. Our results suggest that chronic infection exacerbates ischemic brain damage via a RANTES-mediated systemic inflammatory response, which leads to delayed resolution of inflammation and augmented microvascular injury in the brain.
Introduction
Cerebrovascular disease is profoundly influenced by peripheral immune and inflammatory processes. Comorbidities and known causes of stroke such as atherosclerosis, diabetes, obesity, or systemic infection all involve chronic inflammation, which can directly contribute to ischemic damage and adversely impact on recovery (Emsley and Hopkins, 2008; McColl et al., 2009; Dénes et al., 2010). Infections may trigger up to one-third of ischemic strokes and worsen outcome (Emsley and Hopkins, 2008). In addition to the potentially harmful effects of pathogen-derived factors, the host immune response itself appears to be a major contributor to ischemic brain damage. Recent experimental data highlight the importance of interleukin-1β (IL-1β) or endotoxin-induced systemic inflammation (McColl et al., 2008), T-cell-associated interferon-γ (IFN-γ) (Yilmaz et al., 2006), regulatory T-cells (Liesz et al., 2009), and IL-17-producing γδT-cells (Shichita et al., 2009), which all influence the development of ischemic brain injury.
Peripheral inflammatory processes can induce changes in the kinetics of blood–brain barrier (BBB) disruption, increase in neurovascular gelatinolytic activity, and altered activation of inflammatory cells and cytokines in the brain after stroke (McColl et al., 2007, 2008; Liesz et al., 2009; Shichita et al., 2009). However, we do not know which inflammatory substances mediate the effect of chronic peripheral inflammation on brain damage and how. Furthermore, it is not known how peripheral immune processes relate to changes in neurovascular milieu, such as capillary coagulation, microvascular injury, and impaired blood flow recovery after stroke. Relevant experimental models of comorbidities in stroke provide valuable tools to study some of these mechanisms, but most models are highly complex. Moreover, no study has investigated the effect of infection-induced chronic peripheral inflammation on experimental stroke.
Thus, we used the murine intestinal parasitic whipworm Trichuris muris to induce a chronic, systemic inflammatory response dominated by CD4+ T helper-1 (Th1)/Th17 cells (Cliffe and Grencis, 2004; Hayes et al., 2004; Owyang et al., 2006) to investigate brain and peripheral inflammatory responses after transient middle cerebral artery occlusion (MCAo). In this well established model, low-dose parasite infection results in an IFN-γ-induced class switch to parasite-specific IgG2a, and this response persists to chronicity (Bancroft et al., 2001; Cliffe and Grencis, 2004). This has been confirmed by depletion of IFN-γ, which results in expulsion of the parasite via the development of a protective Th2-type response (Else et al., 1994; Helmby et al., 2001).
Using T. muris, we identify mechanisms whereby chronic, systemic inflammation markedly exacerbates experimental stroke and show that this is mediated by the chemokine CCL5 [regulated on activation, normal T-cell expressed and secreted (RANTES)].
Materials and Methods
Mice and T. muris infection.
Experiments were performed in adult male C57BL/6J mice (Harlan Olac), maintained in individually ventilated cages, under temperature-, humidity-, and light-controlled conditions. Mice were orally infected with ∼20 infective eggs (low dose) when 12–16 weeks of age and incubated for 35 d when chronic Th1-polarized response is well established (Bancroft et al., 1994, 2001; Else et al., 1994) before surgery. Th1-polarized response is dominated by proinflammatory cytokines such as IFN-γ or tumor necrosis factor-α (TNFα), whereas during a Th2-polarized response cytokines such as IL-4, IL-9, IL-10, and IL-13 are expressed. To induce Th2-polarized response, mice were given ∼100 eggs (high dose) followed by 21 d incubation (peak of Th2-polarized infection). No mortality or behavioral changes were observed in infected mice. All animal procedures were performed under an appropriate Home Office license and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986).
Filament MCAo and perfusion.
Focal cerebral ischemia or sham surgery were performed on infected mice and age/weight-matched controls weighing 26–32 g. Anesthesia was induced with isoflurane. During surgery, core temperature was maintained at 37 ± 0.5°C. We investigated whether body temperature was altered in infected mice before surgery or after experimental stroke. No significant difference in rectal temperature was observed between infected and uninfected mice before surgery (36.9 ± 0.2 vs 36.8 ± 0.3°C). Animals were exposed to sham surgery or MCAo for 45 min using an intraluminal filament (180 μm diameter, left side occluded) followed by 4, 24, or 48 h reperfusion. After MCAo, mice developed mild hypothermia, which was normalized by 24 h. No significant difference in body temperature was observed after MCAo between infected and uninfected animals (35.1 ± 0.8 vs 35.1 ± 1.1°C at 3 h reperfusion). Serial blood samples (typically seven times 20 μl/mouse) were obtained from the tail vein before surgery and after MCAo. Before whole-body transcardial perfusion with saline, blood was taken from the heart using 3.8% sodium citrate as an anticoagulant (1:10). The upper body (above the diaphragm) was perfused with 4% paraformaldehyde, whereas unfixed (saline-perfused) liver, spleen, femoral-tibial bone marrow, mesentheric lymph nodes, and cecum were frozen. In separate experiments, saline-perfused brains were collected at various time points after MCAo. Paraformaldehyde-fixed brains were sectioned on a freezing microtome and kept at −20°C in cryoprotectant solution.
Measurement of infarct volume and BBB damage.
The volume of ischemic and BBB damage was measured as described previously (Dénes et al., 2007). Briefly, areas of ischemic damage were identified on cresyl violet-stained sections at eight neuroanatomically defined coronal levels (between 2.9 mm rostral and −4.9 mm caudal to bregma). Digitized images were created, and areas of damage were measured using ImageJ software. The volume of damage was calculated by integration of areas of damage with the distance between coronal levels. Leakage of plasma-derived IgG (BBB damage) was detected with biotinylated horse anti-mouse IgG (1:500) followed by incubation with ABC solution (Vector; 1:500), and the color was developed by diaminobenzidine tetrahydrochloride. Calculation of BBB damage was performed as described above.
Assessment of neurological deficit.
Neurological status was assessed blinded to drug treatment and according to a neurological grading score of increasing severity of deficit (Bederson et al., 1986): 0, no observable deficit; 1, torso flexion to right; 2, spontaneous circling to right; 3, leaning/falling to right; 4, no spontaneous movement.
Systemic neutralization of RANTES.
Monoclonal antibody recognizing RANTES [clone R6G9 (Glass et al., 2004)] or control IgG were administered intraperitoneally, 24 h before and immediately before MCAo (two times 0.25 mg/mouse in 200 μl of sterile PBS).
Cytokine measurements with cytometric bead array and ELISA.
Liver, spleen, and ipsilateral brain hemisphere samples were homogenized as described previously (Chapman et al., 2009). Protein concentrations were calculated using BCA assay (Pierce/Thermo Fisher Scientific). Lymph node cells were isolated and restimulated in vitro with T. muris excretory/secretory (ES) antigen. Sixteen key inflammatory cytokines [granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), IFN-γ, IL-1β, IL-10, IL-13, IL-17A, IL-2, IL-4, IL-5, IL-6, IL-9, KC, MCP-1, TNFα, RANTES] were measured in all tissues examined using appropriate cytometric bead array (CBA) Flex Sets (BD Biosciences) according to the manufacturer's protocol. GM-CSF, IL-2, IL-4, IL-5, and IL-17A were not routinely measured all the time because of their low or tissue-specific expression found in pilot experiments. Parasite-specific IgGs were measured by ELISA as described previously (Else et al., 1993).
Flow cytometry.
Spleen and bone marrow cells were isolated and stained with Gr1-APC, CD4-FITC, CD3-PerCP, CD11b-PE, Foxp3-APC (BD Biosciences) alone or in combination. Cells were acquired on a FACSCalibur (BD Biosciences). Acquisition and analysis of data were performed using CellQuest Pro software (BD Biosciences).
Immunohistochemistry and immunofluorescence.
Immunostaining was performed on free-floating brain sections using various combinations of primary antibodies: rat anti-mouse CD45 (Serotec), goat anti-Iba1 (Abcam), rabbit anti-SJC (kindly provided by Drs. Daniel Anthony and Sandra Campbell, University of Oxford, Oxford, UK) (Anthony et al., 1998), goat anti-RANTES (R&D Systems), goat anti-MMP-9 (R&D Systems), rat anti-mouse CD41 (BD Biosciences), mouse anti-NeuN (Millipore), and rabbit anti-cleaved caspase-3 (Cell Signaling). Sections were incubated in primary antibody (diluted in PBS and 0.3% Triton X-100) for 12–48 h at 4°C. For fluorescent detection, adequate fluorochrome (Alexa 594, Alexa 488)-conjugated antibodies (Invitrogen) were used. Cell nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). Biotinylated tomato lectin (Sigma-Aldrich) was visualized with streptavidin-Alexa 350 conjugate (Invitrogen). Peroxidase staining was developed with diaminobenzidine tetrahydrochloride alone or in the presence of nickel ammonium sulfate.
Microscopy.
Images were collected on a Zeiss Axioskop or an Olympus BX51 microscope using a Coolsnap ES camera (Photometrics) through MetaVue software (Molecular Devices). Fluorescent images were processed using ImageJ and Adobe Photoshop softwares.
Quantitative analysis of neuronal death and immunostaining for inflammatory markers.
Quantification was performed on parallel series of coronal brain sections using a stereological approach (typically 8–12 sections per hemisphere were counted rostrocaudally, 400 μm apart). In each brain (n = 8–10), two to three randomly selected fields of the ischemic striatum or cerebral cortex were viewed on each section in a live CCD camera window and cells positive to NeuN and/or cleaved caspase-3 were counted. SJC-, RANTES-, CD41 (n = 6–10)-, and MMP-9 (n = 5–7)-positive profiles were quantified as above. Data were expressed as the average number of objects per square millimeter or area in the given brain region.
Randomization, quantification, and statistical analysis.
Animals were randomized for experiments where it was possible (e.g., anti-RANTES treatment). All quantitative assessments were performed in a blinded manner. For two groups, Student's t test (two-tailed), and for three or more groups, one-way or two-way ANOVA followed by Bonferroni's post hoc multiple-comparison or paired comparison were used. For correlation analysis, p value (two-tailed) and R2 were determined.
Results
Chronic infection exacerbates ischemic brain damage
First, we examined the effect of chronic peripheral infection on ischemic brain damage, by using low-dose parasite infection to induce a chronic, Th1-polarized immune response. The chronic Th1-polarized response resulted in a 60% increase in brain ischemic damage (p < 0.01), which paralleled augmented BBB damage (p < 0.001) compared with uninfected mice, 48 h after MCAo (Fig. 1A–C). Neuronal loss was significantly increased (p < 0.01) in the ipsilateral hemisphere in infected mice (Fig. 1D,E). This was attributable mainly to necrotic cell death, as cleaved caspase-3-positive apoptotic neurons in the core of the infarct were also significantly reduced (p < 0.01) in mice with chronic Th1-type response, corresponding to low numbers of surviving cells (Fig. 1E,F). To investigate the loss of neuronal processes after MCAo, expression of NK1 [Substance P receptor, which is distributed widely on striatal GABAergic interneurons and cholinergic projection neurons (Gerfen, 1991; Stumm et al., 2001; Blomeley et al., 2009)] was assessed by immunohistochemistry. MCAo upregulated neuronal NK1, whereas marked degeneration of NK1-positive processes and boutons was observed in the core of the infarct in infected mice compared with uninfected animals (Fig. 1G; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Chronic Th1-type peripheral immune response augments ischemic brain damage. Ischemic (A) and BBB (B) damage are significantly larger in infected mice compared with uninfected mice at 48 h reperfusion after 45 min MCAo, as identified by cresyl violet staining (C) and leakage of plasma-derived IgG (t test, two-tailed). Quantitative analysis of NeuN (D) and cleaved caspase-3 (F) immunofluorescence (E) shows augmented neuronal loss after chronic infection (ANOVA, followed by Bonferroni's multiple-comparison test; #p < 0.001 sham vs stroke; **p < 0.01). G, Hematoxylin and eosin staining shows significant loss of neurons in the ipsilateral striatum at 48 h reperfusion after 45 min MCAo, which is more pronounced in infected mice. Chronic peripheral infection causes marked degeneration and loss of NK1-positive cell bodies (arrowheads) and neuronal processes after MCAo. Measurement of parasite-specific IgG1 (H) and IgG2a (I) indicates the development of a Th1-polarized response. J, Correlation analysis indicates significant negative correlation between anti-parasitic IgG1 (dominant in Th2-type response) levels in the plasma and the volume of ischemic damage. Data are expressed as means ± SEM. LV, Lateral ventricle. Scale bars: E, 10 μm; G, 100 μm. Data shown are representative of 10 mice in each group corresponding to two independent experiments.
Next, we investigated whether parasite-specific IgG levels in the plasma, which reflect infection status and polarization of the immune response, show correlation with the size of infarct after MCAo. Elevated parasite-specific IgG2a levels in the plasma after both MCAo and sham surgery (Fig. 1I) and parasite-specific IgG1 (Fig. 1H) demonstrated the predicted development of a chronic Th1-type response in this model (Else et al., 1993, 1994; Bancroft et al., 2001; Cliffe and Grencis, 2004). Th1 cytokines, such as IFN-γ, can promote class switching to IgG2a antibody production in B-cells (Snapper et al., 1997), which leads to elevated serum IgG2a levels. Antiparasitic IgG1, which is dominant in the Th2-type response, during which IgG2a is not elevated (Bancroft et al., 1994; Cliffe and Grencis, 2004), showed a negative correlation (p < 0.05; R2 = 0.66) with the volume of ischemic brain damage in infected mice (Fig. 1J). This indicated that the exacerbation of the ischemic brain damage is caused by the Th1-polarized response induced by chronic infection.
Chronic type 1 cytokine-dominated infection results in systemic inflammation
We looked at different organs known to be involved in the generation of the systemic inflammatory response to reveal whether chronic infection had systemic effects and to see how stroke-induced inflammatory changes were altered by chronic infection. In the mesenteric lymph node (the major draining lymph node of the gut), infected animals expressed elevated IFN-γ, TNFα, RANTES, and IL-17 (proinflammatory cytokines) compared with uninfected controls after both sham and MCAo surgery (Fig. 2A), indicating a well established chronic Th1-polarized response. In infected mice, induction of KC (CXCL1) was observed in the mesenteric lymph node in response to MCAo, which was not seen in uninfected animals (Fig. 2A). In spleen homogenates, IFN-γ and MCP-1 were increased in infected mice before surgery (Fig. 2B). MCAo caused induction of splenic KC and G-CSF within 4 h (Fig. 2B; supplemental Fig. 2, available at www.jneurosci.org as supplemental material) in both infected and uninfected mice. In contrast, splenic IL-9 and IL-13 (type 2 cytokines) expression was blunted in infected animals at 48 h reperfusion after MCAo, but proinflammatory cytokines were not different (Fig. 2C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In the liver of infected mice, RANTES was elevated at most time points examined, compared with uninfected mice (Fig. 2D). Other liver cytokines were not significantly different at 4 h (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) or 48 h after stroke (data not shown). Importantly, only RANTES was significantly increased in the plasma of infected mice up to 48 h after sham surgery (Fig. 2E) compared with uninfected animals of the 16 key inflammatory cytokines examined.
Figure 2.

Chronic Th1-type infection results in organ-specific proinflammatory activation and systemic upregulation of RANTES. A, Mesenteric lymph node cells isolated 48 h after sham surgery or MCAo were restimulated with parasite ES antigens in vitro. CBA analysis was performed on spleen homogenates of mice without surgery (0 h), at 4 h reperfusion (B), and after 48 h reperfusion (C). D, Liver homogenates harvested at various time points show chronically elevated RANTES in infected mice. E, Plasma samples show elevated RANTES in sham mice at 48 h reperfusion. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Bonferroni's multiple-comparison test. Data are expressed as means ± SEM. Data shown are representative of 6–10 mice in each group from two independent experiments.
Chronic infection alters the acute phase response to stroke and leads to sustained inflammation in the brain
Plasma IFN-γ and TNFα were moderately elevated (2 ± 2 vs 12 ± 4 and 6 ± 1 vs 24 ± 6 pg/ml, respectively; p < 0.05) in infected mice compared with uninfected animals before surgery (data not shown). After the induction of ischemia, circulating cytokines MCP-1, KC, IL-6, and G-CSF increased significantly (20- to 200-fold) within 40–120 min and returned to baseline 24–48 h after reperfusion (Fig. 3A). In infected mice, the increases in MCP-1, KC, and G-CSF in response to ischemia were attenuated, whereas RANTES was elevated.
Figure 3.
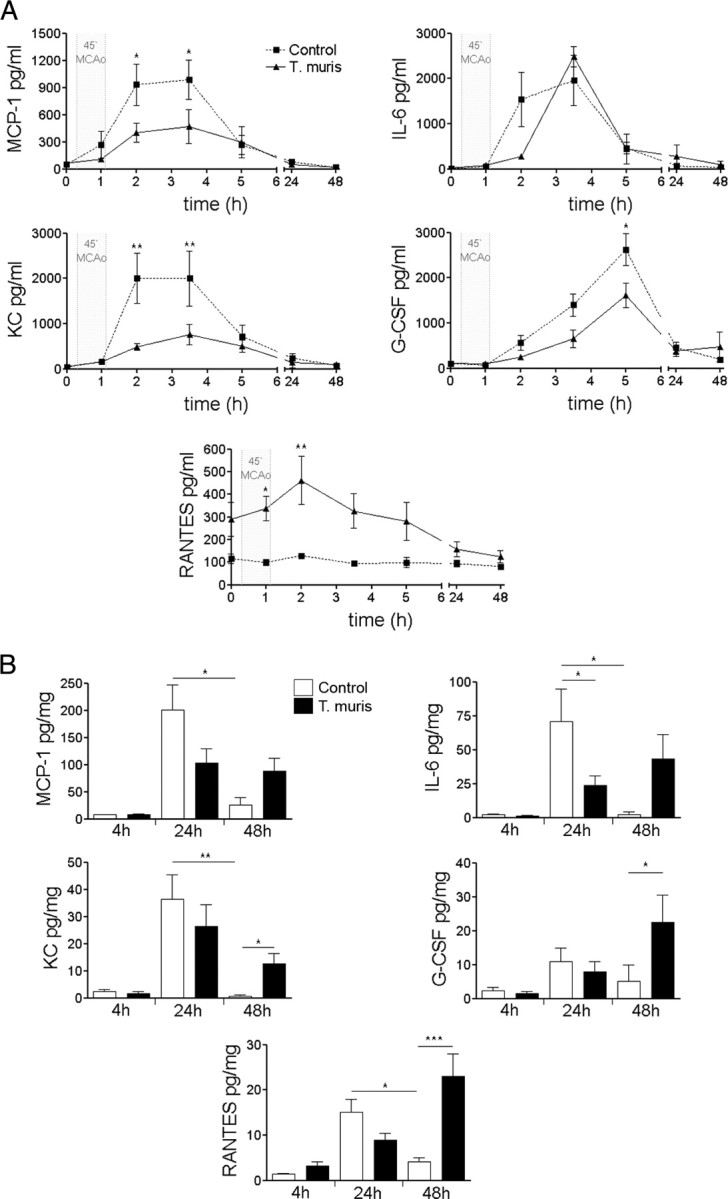
Chronic infection alters MCAo-induced acute phase response in the plasma and the brain. A, Serial plasma samples. B, Ipsilateral brain hemisphere homogenates. *p < 0.05, **p < 0.01, two-way ANOVA followed by Bonferroni's posttest. Data are expressed as means ± SEM. Data shown are representative of two independent experiments.
In the brain, induction of inflammatory cytokines in uninfected mice peaked 24 h after MCAo (Fig. 3B), and MCP-1, KC, IL-6, and RANTES levels had declined by 48 h. In uninfected mice, brain IL-6 levels were significantly higher 24 h after stroke compared with infected animals. In contrast, MCP-1, KC, and IL-6 showed sustained upregulation, whereas G-CSF and RANTES exhibited additional increases by 48 h in infected mice (Fig. 3B), indicating slower resolution of inflammation in the brain after stroke.
Infection-induced systemic RANTES is associated with microvascular dysfunction and increased platelet aggregation in the brain after stroke
Immunohistochemistry revealed strong vascular RANTES labeling after MCAo (in both small-medium size blood vessels and in microvessels) in the ischemic hemisphere of infected mice (Fig. 4A,B), but not in uninfected controls. RANTES immunostaining was most marked in the ipsilateral thalamus and hippocampus, which were ischemic in 90% of T. muris-infected mice, but showed only marginal damage (in 40–50% of mice) or were not affected in uninfected mice. These areas are especially sensitive to remote damage after stroke in rodents and primates (Dihné et al., 2002; Ling et al., 2009; Bihel et al., 2010). In infected mice, an increased number of RANTES-positive platelets (average diameter, 2.4 μm) was found in capillaries (Fig. 4A, I, inset). Glial RANTES expression was also markedly enhanced in these mice 48 h after stroke (Fig. 4A, III–IV; C). A population of these cells was CD45-positive microglia/macrophages that coexpressed Iba1 (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Immunofluorescence revealed CD41-positive platelets in the areas of vascular RANTES deposition, mostly in microvessels. Platelet aggregates occluding capillaries were numerous in the ischemic hemisphere of infected mice after stroke (Fig. 4D; supplemental Fig. 4B, available at www.jneurosci.org as supplemental material) compared with uninfected mice. We found no significant differences in cerebral blood flow recovery after MCAo between infected and uninfected mice as measured by laser Doppler flowmetry.
Figure 4.

Chronic Th1-polarized response increases RANTES, platelet aggregation, leukocyte recruitment, and matrix metalloproteinase activity in the ischemic hemisphere. A, Immunohistochemistry reveals increased RANTES in microvessels (top inset, arrow) and platelets (bottom inset, arrowhead) in Th1-polarized mice (I), but not in uninfected animals (II) after MCAo. Glial RANTES expression was augmented in the affected hippocampus (III) of infected but not in uninfected mice (IV). Quantification of vascular (B) and glial (C) RANTES staining 48 h after MCAo (infected vs uninfected: p < 0.001, two-way ANOVA; Bonferroni's posttest, *p < 0.05). D, Vascular deposition of RANTES (green) and platelet aggregates (CD41, red) are increased in infected mice. E, Blood cell lysates contain elevated RANTES at 4 h reperfusion in infected mice compared with uninfected animals, which is reduced by 48 h reperfusion (**p < 0.01, one-way ANOVA followed by Bonferroni's multiple-comparison test). F, Vascular RANTES deposition was associated with leukocyte recruitment (CD45; red). G, Granulocyte numbers (SJC) were increased in the ipsilateral hemisphere in infected mice (infected vs control: p < 0.01, two-way ANOVA). H, Vascular MMP-9 (green) expression was enhanced after MCAo in the ischemic striatum of Th1-polarized mice. Scale bars: D, F, H, 10 μm; A, I, II, 20 μm; A, III, IV, 100 μm. Data are expressed as means ± SEM. Data shown are representative of 6–10 mice in each group from two independent experiments.
RANTES was also upregulated in pelleted blood cell and platelet lysates in infected mice before surgery and up to 48 h after sham surgery compared with uninfected mice (p < 0.01). Blood cells contained elevated RANTES in infected mice 4 h after MCAo, which reduced by 48 h, indicating altered production or enhanced release during the formation of ischemic damage (Fig. 4E). Compared with uninfected mice, infected mice displayed elevated RANTES in the brain after sham surgery (0.7 ± 0.2 vs 3.6 ± 1.5 pg/mg, respectively; p < 0.05) or without surgery (data not shown), indicating cerebrovascular inflammation before the induction of experimental stroke. RANTES deposition on cerebral blood vessels in the ipsilateral hemisphere was also associated with increased vascular adhesion of CD45-positive leukocytes in Th1-polarized mice (Fig. 4F) after MCAo. Granulocytes as identified by SJC immunostaining were more numerous in the ischemic brains of infected mice after MCAo compared with uninfected mice (p < 0.01) (Fig. 4G). This was accompanied by increased number and proportion of Gr1-positive (a marker of neutrophils and monocytes) leukocytes in the spleen (4.4 ± 0.3% in uninfected vs 9.1 ± 2% in infected mice; p < 0.05).
To investigate whether chronic infection modified neurovascular gelatinase expression in the brain after MCAo, we used immunostaining to MMP-9. MMP-9 is augmented in the brain by acute systemic inflammatory challenge after experimental stroke, which is associated with increased BBB permeability (McColl et al., 2008). Microvessels showed markedly increased MMP-9 immunostaining within the ischemic hemisphere in infected mice compared with uninfected animals (Fig. 4H), after MCAo. It indicated that chronic Th1-polarized response to infection was associated with augmented vascular injury in the brain after experimental stroke, corresponding to increased ischemic and BBB damage observed in infected mice (Fig. 1B).
Systemic neutralization of RANTES reverses the effect of chronic infection on brain injury
We investigated whether systemic blockade of RANTES has an effect on ischemic brain damage and inflammation after MCAo in uninfected and infected mice, using a specific neutralizing antibody (Glass et al., 2004). Anti-RANTES treatment prevented the infection-driven exacerbation of ischemic damage (p < 0.01) (Fig. 5A) but had no significant effect in uninfected mice. After systemic RANTES neutralization, analysis of mesenteric lymph nodes showed successful inhibition of RANTES (1005 ± 63 pg/ml after control IgG vs 630 ± 57 pg/ml after anti-RANTES; p < 0.01) and KC (718 ± 110 pg/ml after control IgG vs 383 ± 121 pg/ml after anti-RANTES; p = 0.02), whereas a trend was observed for reduced TNFα (630 ± 202 pg/ml after control IgG vs 404 ± 42 pg/ml after anti-RANTES). IFN-γ markedly increased in the plasma after anti-RANTES treatment compared with infected, control IgG-treated mice. Inhibition of stroke-induced G-CSF by chronic Th1-type infection was reversed (Fig. 5B), whereas levels of neutralized RANTES increased. Infected, control IgG-treated mice displayed increased vascular MMP-9 immunostaining in the ischemic hemisphere (p < 0.05), whereas the number of MMP-9/CD45-positive cells was not significantly different. Anti-RANTES treatment inhibited vascular MMP-9 expression (p < 0.01) in infected mice (Fig. 5C).
Figure 5.

Systemic neutralization of RANTES reverses the effects of Th1-type infection on brain damage. A, The exacerbation of the ischemic brain damage is prevented by RANTES inhibition. *p < 0.05, **p < 0.01, one-way ANOVA followed by Bonferroni's multiple-comparison test. B, Effect of RANTES neutralization on plasma cytokine levels. Two-way ANOVA followed by Bonferroni's posttest. Infection versus control: p < 0.01 for IFN-γ, G-CSF, RANTES, two-way ANOVA (T. muris anti-RANTES vs *control anti-RANTES, #control IgG, ‡T. muris IgG). C, Vascular MMP-9 immunostaining, expressed as the number of MMP-9-positive blood vessels (bv) per square millimeter, was significantly increased in infected, control IgG-treated mice in the ipsilateral hemisphere after MCAo, compared with uninfected animals. This was reversed in response to anti-RANTES treatment. The number of CD45/MMP-9-positive cells was not significantly different (right). Fluorescent micrographs showing MMP-9, CD45, and lectin triple-immunofluorescent staining in the ipsilateral striatum (left). D, Flow cytometric analysis shows that Th1-polarized mice have reduced CD4+/Foxp3+ (Treg) cells in the spleen 48 h after MCAo. E, The effect of systemic neutralization of RANTES on effector Th cells, Treg, and Gr1-positive leukocytes in the spleen. Scale bar, 50 μm. Data are expressed as means ± SEM. Data shown are representative of two independent experiments.
We investigated whether infection and systemic neutralization of RANTES influenced regulatory T-cells (Treg), which play a key protective role in cerebral ischemia (Liesz et al., 2009) and increase in number after stroke in the spleen (Offner et al., 2006). Infected mice exhibited reduced numbers of Treg in the spleen (Fig. 5D) after MCAo compared with uninfected mice, whereas a similar ratio of effector CD4-positive T-cells was found. Anti-RANTES treatment reduced splenic effector Th cells but had no effect on Treg. Anti-RANTES also reversed increased Gr1-positive leukocyte numbers in infected mice (Fig. 5E).
Th2-polarized peripheral immune response after T. muris infection does not modify ischemic brain damage
We also investigated whether the T. muris effect was specific to the parasite infection or dependent on the induced Th1 response. We infected mice with a high dose of T. muris before MCAo. This regimen activates a Th2-polarized immune response against the intestinal parasite and results in expulsion of worms from the host (Bancroft et al., 2001; Cliffe and Grencis, 2004). Low levels of parasite-specific IgG2a levels confirmed the development of a Th2-polarized response (Fig. 6A,B). Eosinophil granulocyte numbers increased significantly in the bone marrow in Th2-polarized mice compared with uninfected and Th1-polarized mice (Fig. 6C). The ischemic brain damage was not exacerbated in Th2-polarized mice (Fig. 6D) compared with uninfected animals but was significantly smaller (p < 0.05) than that of Th1-polarized mice (29.8 ± 2.8, 28.9 ± 6.0, and 49.6 ± 5.3 mm3 in uninfected, T. muris high-dose-, and T. muris low-dose-infected animals, respectively). Neurological deficit was significantly augmented in Th1-polarized mice compared with Th2-polarized and uninfected mice (Fig. 6E), after MCAo. The extent of brain edema was also significantly smaller in Th2-polarized mice compared with Th1-polarized animals (Fig. 6F). Proinflammatory cytokines were not increased in mesenteric lymph nodes (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material) after high-dose infection. Stroke-induced changes in G-CSF and KC levels in the plasma were not different from low-dose infection. Importantly, plasma RANTES was not elevated at any time point before or after MCAo after high-dose (Th2-type) infection compared with uninfected mice (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material).
Figure 6.

T. muris high-dose infection results in Th2-polarized immune response, which does not exacerbate brain damage. A, B, Antiparasitic IgG levels in the plasma of T. muris high-dose (HD)-infected (Th2-polarized) mice. C, Eosinophil granulocytes are elevated in the bone marrow in T. muris HD mice compared with uninfected (control) and T. muris low-dose (LD)-infected (Th1-polarized) mice. D, Representative cresyl violet-stained brain sections from uninfected, T. muris HD, and T. muris LD mice after 45 min MCAo and 48 h reperfusion. E, Neurological deficit is significantly exacerbated in T. muris LD mice compared with uninfected and T. muris HD mice. F, Brain edema is larger in T. muris LD mice compared with T. muris HD animals. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Bonferroni's posttest. Data are expressed as means ± SEM. Data shown are representative of two independent experiments (n = 10).
Discussion
We show for the first time that chronic peripheral infection in mice, dominated by type 1 cytokines, exacerbates ischemic brain damage, which is associated with delayed resolution of brain inflammation and augmented microvascular dysfunction after stroke. We identify RANTES as a key mediator of these events, whereby chronic peripheral inflammation affects brain injury. In light of clinical studies, which usually fail to distinguish between infections that trigger stroke and an aggravation of stroke pathophysiology, our data confirm that preexisting infections can exacerbate infarct evolution if the ischemic event occurs.
No experimental studies have implicated RANTES as a mediator of chronic inflammation, which impacts on remote injury or specifically exacerbates ischemic brain damage. Recent preclinical data indicate reduced ischemic brain damage in RANTES-deficient mice (Terao et al., 2008). In contrast, the present study shows that RANTES neutralization did not modify brain damage in uninfected mice. However, we show for the first time that the exacerbation of ischemic damage caused by chronic infection is prevented by RANTES inhibition. The clinical relevance of our findings is supported by several reports, which implicate RANTES as an atherogenic chemokine in vascular disease and in comorbidities associated with stroke. Inhibition of RANTES-dependent pathways is protective in experimental models of atherosclerosis (Zernecke et al., 2006; Koenen et al., 2009). In peripheral tissues, platelet-derived RANTES is deposited on the inflamed endothelium where it triggers monocyte arrest (von Hundelshausen et al., 2001; Schober et al., 2002). Our results show for the first time that vascular RANTES is increased in the brain in response to chronic infection after stroke. Human aortic aneurysms release high levels of RANTES, which recruits neutrophils to the intraluminal thrombi (Houard et al., 2009). In patients, RANTES plasma levels correlate with acute-phase proteins C-reactive protein (CRP) and fibrinogen in coronary artery diseases (Koh et al., 2009). RANTES gene polymorphism is associated with relative risk of coronary arteriosclerosis (Simeoni et al., 2004) and cerebral ischemia (Um and Kim, 2009). Therefore, blood RANTES may be a potential biomarker to identify patients at risk of stroke by indicating the presence of high level of chronic inflammation.
T. muris is a model for a human pathogen, Trichuris trichiura. To our knowledge, gastrointestinal helminth infections are not associated with negative outcome after stroke in clinical practice, although these types of infection are relatively rare in well developed countries where the incidence of stroke is high. In addition, although nematodes favor to polarize the immune response away from Th2 dominance, protection against nematodes mostly leads to a Th2-type response, and a well established infection model (using appropriate strain, dose, and timing) is needed to establish a chronic Th1-polarized infection in this case. Interestingly, a comprehensive set of data link Chagas' disease, caused by a parasite, Trypanosoma cruzi, to increased stroke prevalence, which is an emerging health problem in Europe and the United States after Latin America (Carod-Artal and Gascon, 2010). Therefore, other parasitic diseases may have previously unrecognized role in stroke. Our aim here was to use T. muris infection as a well characterized model in mice to induce a chronic peripheral inflammation (which is an important contributor to poor outcome in patients) by modulating the immune response rather than modeling certain known comorbidities in stroke, which are highly complex. We used well established markers to characterize the immune state of the animal such as antigen-specific IgG subclasses or key inflammatory cytokines, which also correlate with outcome in stroke patients. For example, elevated serum Chlamydia pneumoniae- or Helicobacter pylori-specific IgGs in patients are predictive of cerebrovascular and cardiovascular events and extent of brain injury, similarly to infection burden and elevated serum CRP (Corrado et al., 2006; Smith et al., 2006). Our results indicate that the chronic Th1-polarized immune response, but not T. muris infection itself or the protective Th2-type response is detrimental. Although high-dose-infected (Th2-polarized) mice mostly expelled their parasites by 21 d after infection, low-dose-infected (Th1-polarized) mice had adult worms in the cecum after anti-RANTES treatment, whereas their ischemic brain damage was not different from uninfected mice. Our data also show that excess plasma IgG1 levels, which indicate more pronounced resistance to infection and Th2-type regulation (Bellaby et al., 1996; Perrigoue et al., 2009), correlate negatively with infarct size, whereas the Th2-polarized response to T. muris did not worsen brain damage.
Chronic Th1-polarized response was also associated with increased platelet aggregation in the brain after stroke. Some observations link chronic inflammation to a procoagulant state, including the effect of proinflammatory mediators on platelet activation, aggregation, or reduction of anticoagulant factors (Levi et al., 2004). These mechanisms are involved in diabetes (Stegenga et al., 2008), obesity, and hypertension (Alessi and Juhan-Vague, 2008), which are individual risk factors in stroke. RANTES may not mediate platelet aggregation or adhesion alone (Shenkman et al., 2004), but interaction among multiple cytokines and pathways is likely to occur in inflammation and stroke. For example, IL-6, which was reduced in the first 24 h after MCAo in the brain of type 1-polarized mice, is protective against coagulatory and hemostatic disturbance (Inoue et al., 2004). G-CSF, which showed blunted early (4 h) production, but delayed (48 h) upregulation after MCAo in type 1-polarized mice, is a wide-action neuroprotectant in transient experimental stroke models if administered within 4 h (England et al., 2009). One other example of the complex interactions between systemic infection and stroke in our model is the downregulation of infection-induced proinflammatory cytokines such as MCP-1 and IFN-γ in the spleen within 4 h or RANTES in the plasma and blood cells between 4 and 48 h, in response to MCAo. We think that either enhanced release, deposition and proteolytic cleavage, or reduced production because of stroke-induced immunosuppression (Prass et al., 2003) are possible mechanisms. Nevertheless, these data argue for the need of precise understanding of these interactions to deliver appropriate medicines for patients with different comorbidities.
RANTES contributes to the proinflammatory response to IFN-γ (Ma et al., 2005), which is controlled by Treg. These cells protect against focal cerebral ischemia (Liesz et al., 2009). Interestingly, obesity, a major risk factor for stroke, leads to profound and site-specific loss of Treg cells in the adipose tissue and highly upregulated RANTES expression (Feuerer et al., 2009). Low-dose-infected mice had low worm burden in the cecum, which may account for less pronounced regulation of the ongoing chronic inflammatory response. This is clearly indicated by reduced Treg (but not effector Th cells) in the spleen of infected compared with uninfected mice. The present study did not investigate whether a causal role exists for Treg in inflammation-induced brain damage after stroke or not. It is also unclear whether increased leukocyte recruitment, microvascular injury, and BBB disruption contribute to brain damage in infected, Th1-polarized mice or simply reflect larger infarcts in these animals. In addition to reducing brain injury, the effect of anti-RANTES treatment on splenic inflammatory cells indicates an important role for this chemokine in the modulation of infection-induced leukocyte responses after stroke.
Additional investigation is required to understand how systemic inflammation and Th1 prepolarization of the immune system interact with stroke-induced immune changes in the late phases of infarct evolution and affect recovery. As the ischemic brain injury itself can polarize the immune system toward a Th2 state, induce immunosuppression, and lead to poststroke infections (Prass et al., 2003), it is possible that this response is altered by preexisting infection or chronic inflammation. Recent data indicate that IL-17-producing γδT-cells (Shichita et al., 2009) or Treg (Liesz et al., 2009), which play an important role in multiple infectious diseases, can also influence delayed brain injury after experimental stroke. Therefore, it is important for future studies to establish how these inflammatory pathways interact and affect damage formation or recovery in stroke patients with comorbidities such as infection or chronic inflammatory disease.
Together, our results demonstrate for the first time that chronic peripheral infection exacerbates ischemic brain damage by systemic upregulation of RANTES. Targeted modulation of RANTES-dependent pathways may be therapeutically useful in several known comorbidities that augment the effects of stroke and impair outcome. Moreover, given ∼1 billion people infected with similar intestinal nematodes worldwide, these data extend our understanding of the immunomodulatory capacity of such pathogens and their potential effects in vascular diseases.
Footnotes
This work was supported by the Medical Research Council (MRC) (N.R.) (MRC Research Professorship), European Union's Seventh Framework Programme (FP7/2008-2013) under Grant Agreements 201024 and 202213 (European Stroke Network) (N.R., A.D.), and the Wellcome Trust (N.H., R.G.). We thank Dr. Allison Bancroft and Dr. Kelly Millar for help with the T. muris model and cytometric bead array.
References
- Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost. 2008;99:995–1000. doi: 10.1160/TH07-11-0682. [DOI] [PubMed] [Google Scholar]
- Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K. CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol. 1998;8:923–926. doi: 10.1016/s0960-9822(07)00373-9. [DOI] [PubMed] [Google Scholar]
- Bancroft AJ, Else KJ, Grencis RK. Low-level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur J Immunol. 1994;24:3113–3118. doi: 10.1002/eji.1830241230. [DOI] [PubMed] [Google Scholar]
- Bancroft AJ, Else KJ, Humphreys NE, Grencis RK. The effect of challenge and trickle Trichuris muris infections on the polarisation of the immune response. Int J Parasitol. 2001;31:1627–1637. doi: 10.1016/s0020-7519(01)00281-8. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Bellaby T, Robinson K, Wakelin D. Induction of differential T-helper-cell responses in mice infected with variants of the parasitic nematode Trichuris muris. Infect Immun. 1996;64:791–795. doi: 10.1128/iai.64.3.791-795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihel E, Pro-Sistiaga P, Letourneur A, Toutain J, Saulnier R, Insausti R, Bernaudin M, Roussel S, Touzani O. Permanent or transient chronic ischemic stroke in the non-human primate: behavioral, neuroimaging, histological, and immunohistochemical investigations. J Cereb Blood Flow Metab. 2010;30:273–285. doi: 10.1038/jcbfm.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomeley CP, Kehoe LA, Bracci E. Substance P mediates excitatory interactions between striatal projection neurons. J Neurosci. 2009;29:4953–4963. doi: 10.1523/JNEUROSCI.6020-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carod-Artal FJ, Gascon J. Chagas disease and stroke. Lancet Neurol. 2010;9:533–542. doi: 10.1016/S1474-4422(10)70042-9. [DOI] [PubMed] [Google Scholar]
- Chapman KZ, Dale VQ, Dénes A, Bennett G, Rothwell NJ, Allan SM, McColl BW. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab. 2009;29:1764–1768. doi: 10.1038/jcbfm.2009.113. [DOI] [PubMed] [Google Scholar]
- Cliffe LJ, Grencis RK. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv Parasitol. 2004;57:255–307. doi: 10.1016/S0065-308X(04)57004-5. [DOI] [PubMed] [Google Scholar]
- Corrado E, Rizzo M, Tantillo R, Muratori I, Bonura F, Vitale G, Novo S. Markers of inflammation and infection influence the outcome of patients with baseline asymptomatic carotid lesions: a 5-year follow-up study. Stroke. 2006;37:482–486. doi: 10.1161/01.STR.0000198813.56398.14. [DOI] [PubMed] [Google Scholar]
- Dénes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27:1941–1953. doi: 10.1038/sj.jcbfm.9600495. [DOI] [PubMed] [Google Scholar]
- Dénes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Dihné M, Grommes C, Lutzenburg M, Witte OW, Block F. Different mechanisms of secondary neuronal damage in thalamic nuclei after focal cerebral ischemia in rats. Stroke. 2002;33:3006–3011. doi: 10.1161/01.str.0000039406.64644.cb. [DOI] [PubMed] [Google Scholar]
- Else KJ, Entwistle GM, Grencis RK. Correlations between worm burden and markers of Th1 and Th2 cell subset induction in an inbred strain of mouse infected with Trichuris muris. Parasite Immunol. 1993;15:595–600. doi: 10.1111/pim.1993.15.10.595. [DOI] [PubMed] [Google Scholar]
- Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med. 1994;179:347–351. doi: 10.1084/jem.179.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley HC, Hopkins SJ. Acute ischaemic stroke and infection: recent and emerging concepts. Lancet Neurol. 2008;7:341–353. doi: 10.1016/S1474-4422(08)70061-9. [DOI] [PubMed] [Google Scholar]
- England TJ, Gibson CL, Bath PM. Granulocyte-colony stimulating factor in experimental stroke and its effects on infarct size and functional outcome: a systematic review. Brain Res Rev. 2009;62:71–82. doi: 10.1016/j.brainresrev.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. Substance P (neurokinin-1) receptor mRNA is selectively expressed in cholinergic neurons in the striatum and basal forebrain. Brain Res. 1991;556:165–170. doi: 10.1016/0006-8993(91)90563-b. [DOI] [PubMed] [Google Scholar]
- Glass WG, Hickey MJ, Hardison JL, Liu MT, Manning JE, Lane TE. Antibody targeting of the CC chemokine ligand 5 results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. J Immunol. 2004;172:4018–4025. doi: 10.4049/jimmunol.172.7.4018. [DOI] [PubMed] [Google Scholar]
- Hayes KS, Bancroft AJ, Grencis RK. Immune-mediated regulation of chronic intestinal nematode infection. Immunol Rev. 2004;201:75–88. doi: 10.1111/j.0105-2896.2004.00193.x. [DOI] [PubMed] [Google Scholar]
- Helmby H, Takeda K, Akira S, Grencis RK. Interleukin (IL)-18 promotes the development of chronic gastrointestinal helminth infection by downregulating IL-13. J Exp Med. 2001;194:355–364. doi: 10.1084/jem.194.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houard X, Touat Z, Ollivier V, Louedec L, Philippe M, Sebbag U, Meilhac O, Rossignol P, Michel JB. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res. 2009;82:532–541. doi: 10.1093/cvr/cvp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Takano H, Yanagisawa R, Sakurai M, Shimada A, Morita T, Sato M, Yoshino S, Yoshikawa T, Tohyama C. Protective role of interleukin-6 in coagulatory and hemostatic disturbance induced by lipopolysaccharide in mice. Thromb Haemost. 2004;91:1194–1201. doi: 10.1160/TH03-09-0595. [DOI] [PubMed] [Google Scholar]
- Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- Koh SJ, Kim JY, Hyun YJ, Park SH, Chae JS, Park S, Kim JS, Youn JC, Jang Y, Lee JH. Association of serum RANTES concentrations with established cardiovascular risk markers in middle-aged subjects. Int J Cardiol. 2009;132:102–108. doi: 10.1016/j.ijcard.2007.10.038. [DOI] [PubMed] [Google Scholar]
- Levi M, van der Poll T, Büller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–2704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Ling L, Zeng J, Pei Z, Cheung RT, Hou Q, Xing S, Zhang S. Neurogenesis and angiogenesis within the ipsilateral thalamus with secondary damage after focal cortical infarction in hypertensive rats. J Cereb Blood Flow Metab. 2009;29:1538–1546. doi: 10.1038/jcbfm.2009.76. [DOI] [PubMed] [Google Scholar]
- Ma B, Kang MJ, Lee CG, Chapoval S, Liu W, Chen Q, Coyle AJ, Lora JM, Picarella D, Homer RJ, Elias JA. Role of CCR5 in IFN-gamma-induced and cigarette smoke-induced emphysema. J Clin Invest. 2005;115:3460–3472. doi: 10.1172/JCI24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Allan SM, Rothwell NJ. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. 2009;158:1049–1061. doi: 10.1016/j.neuroscience.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, Artis D. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrigoue JG, Zaph C, Guild K, Du Y, Artis D. IL-31-IL-31R interactions limit the magnitude of Th2 cytokine-dependent immunity and inflammation following intestinal helminth infection. J Immunol. 2009;182:6088–6094. doi: 10.4049/jimmunol.0802459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106:1523–1529. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- Shenkman B, Brill A, Brill G, Lider O, Savion N, Varon D. Differential response of platelets to chemokines: RANTES non-competitively inhibits stimulatory effect of SDF-1 alpha. J Thromb Haemost. 2004;2:154–160. doi: 10.1111/j.1538-7836.2004.00527.x. [DOI] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Simeoni E, Winkelmann BR, Hoffmann MM, Fleury S, Ruiz J, Kappenberger L, März W, Vassalli G. Association of RANTES G-403A gene polymorphism with increased risk of coronary arteriosclerosis. Eur Heart J. 2004;25:1438–1446. doi: 10.1016/j.ehj.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Emsley HC, Vail A, Georgiou RF, Rothwell NJ, Tyrrell PJ, Hopkins SJ. Variability of the systemic acute phase response after ischemic stroke. J Neurol Sci. 2006;251:77–81. doi: 10.1016/j.jns.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Snapper CM, Marcu KB, Zelazowski P. The immunoglobulin class switch: beyond “accessibility.”. Immunity. 1997;6:217–223. doi: 10.1016/s1074-7613(00)80324-6. [DOI] [PubMed] [Google Scholar]
- Stegenga ME, van der Crabben SN, Blümer RM, Levi M, Meijers JC, Serlie MJ, Tanck MW, Sauerwein HP, van der Poll T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood. 2008;112:82–89. doi: 10.1182/blood-2007-11-121723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm R, Culmsee C, Schafer MK, Krieglstein J, Weihe E. Adaptive plasticity in tachykinin and tachykinin receptor expression after focal cerebral ischemia is differentially linked to GABAergic and glutamatergic cerebrocortical circuits and cerebrovenular endothelium. J Neurosci. 2001;21:798–811. doi: 10.1523/JNEUROSCI.21-03-00798.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao S, Yilmaz G, Stokes KY, Russell J, Ishikawa M, Kawase T, Granger DN. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke. 2008;39:2560–2570. doi: 10.1161/STROKEAHA.107.513150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JY, Kim HM. Polymorphisms of RANTES and IL-4 genes in cerebral infarction. J Mol Neurosci. 2009;37:1–5. doi: 10.1007/s12031-008-9047-9. [DOI] [PubMed] [Google Scholar]
- von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, Weber C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103:1772–1777. doi: 10.1161/01.cir.103.13.1772. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zernecke A, Liehn EA, Gao JL, Kuziel WA, Murphy PM, Weber C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. 2006;107:4240–4243. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]