

Abstract
Huntington's disease (HD), a genetic neurodegenerative disease caused by a polyglutamine expansion in the Huntingtin (Htt) protein, is accompanied by multiple mitochondrial alterations. Here, we show that mitochondrial fragmentation and cristae alterations characterize cellular models of HD and participate in their increased susceptibility to apoptosis. In HD cells, the increased basal activity of the phosphatase calcineurin dephosphorylates the pro-fission dynamin related protein 1 (Drp1), increasing its mitochondrial translocation and activation, and ultimately leading to fragmentation of the organelle. The fragmented HD mitochondria are characterized by cristae alterations that are aggravated by apoptotic stimulation. A genetic analysis indicates that correction of mitochondrial elongation is not sufficient to rescue the increased cytochrome c release and cell death observed in HD cells. Conversely, the increased apoptosis can be corrected by manoeuvres that prevent fission and cristae remodelling. In conclusion, the cristae remodelling of the fragmented HD mitochondria contributes to their hypersensitivity to apoptosis.
Keywords: apoptosis, cristae remodelling, fission, Huntington's disease, mitochondria
→See accompanying
Closeup by Oliveira and Lightowlers DOI 10.1002/emmm.201000104.
INTRODUCTION
During apoptosis, mitochondria are key organelles to sense and amplify damage, releasing cytochrome c and other cofactors for the effector caspases that dismantle the cell (Danial & Korsmeyer, 2004). This release, tightly controlled by proteins of the Bcl-2 family, is accompanied by fragmentation of the mitochondrial network (Frank et al, 2001) and remodelling of the mitochondrial cristae (Scorrano et al, 2002). Both processes are required for the progression of apoptosis and cristae remodelling is downstream of fragmentation (Germain et al, 2005). During cell life and death, mitochondrial shape is regulated by a growing family of pro-fission (the cytoplasmic dynamin related protein 1, Drp1; and its mitochondrial receptor fission-1, Fis1) and pro-fusion (the large GTPases Optic Atrophy 1, Opa1, in the inner membrane and Mitofusin, Mfn, 1 and 2 in the outer mitochondrial membrane) mitochondria-shaping proteins (Liesa et al, 2009).
Neurons are highly dependent on mitochondria, since they are characterized by high energy demands and are unable to switch to glycolysis when mitochondrial oxidative phosphorylation is impaired. A large number of neurodegenerative diseases are indeed caused by an impairment of mitochondrial function (Bossy-Wetzel et al, 2003). More recently, mutations in the genes coding for mitochondria-shaping proteins have been associated with some genetic neurodegenerative diseases, implicating mitochondrial shape regulation in the health of neurons (Chan, 2007). In addition, considerable interest was recently captured by the role of mitochondrial morphology changes in familial forms of Parkinson's disease (PD) caused by mutations in the PINK1 and PARKIN genes (Poole et al, 2008), be it primary (Lutz et al, 2009) or amplificatory (Morais et al, 2009). Whether mitochondrial morphology plays a role also in Huntington's disease (HD) remains to be elucidated.
HD is an autosomal dominant, neurodegenerative disease caused by the expansion beyond 36 of a CAG repeat in the IT15 gene (4p16.3) (The Huntington's Disease Collaborative Research Group, 1993). HD is characterized clinically by variable age of onset (normally between 40 and 50) and severity that correlate directly with the length and the gene dosage of the CAG repeat number (Duyao et al, 1993). HD patients are affected by neurological (choreoathetosis, psychiatric disturbances and cognitive defects) and extraneurological (wasting, immunological and cardiological defects) alterations and ultimately die in 10–20 years from the onset of the disease (Martin & Gusella, 1986). The key pathological feature of HD is the progressive loss of neurons with atrophy and gliosis of the basal ganglia and the cortex, especially of the GABAergic spiny neurons of the striatum (Ferrante et al, 1991). The IT15 gene encodes for the ubiquitous protein Huntingtin (Htt), and the CAG repeats result in the expansion of an N-terminal polyglutamine trait (Schilling et al, 1995; Sharp et al, 1995). Htt is a large protein of 350 kDa with no homology with other known proteins, located in the cytoplasm and found associated with a variety of subcellular structures, from Golgi to the endoplasmic reticulum, to mitochondria, to the nucleus where it exerts transcriptional effects (De Rooij et al, 1996; Difiglia et al, 1995; Gutekunst et al, 1995; Kegel et al, 2002; Panov et al, 2002). Htt is required during development (Zeitlin et al, 1995) and is subjected to post-translational modifications, including phosphorylation and cleavage, that are important for the pathogenesis of HD (Graham et al, 2006; Gu et al, 2009; Hackam et al, 1998; Pardo et al, 2006; Wellington et al, 2000).
The exact pathobiology of HD remains elusive. Several theories have been put forward to explain how mutated Htt is neurotoxic: they range from altered transcriptional activity (Sugars & Rubinsztein, 2003) to impaired intracellular trafficking (Gunawardena et al, 2003) to the formation of aggregates (Difiglia et al, 1997) that clog the proteasome (Jana et al, 2001) and impede the cargo recognition by autophagosomes (Martinez-Vicente et al, 2010), to the hypersensitivity to excitotoxicity (Fernandes et al, 2007). Irrespective of the apical mechanism, the key feature of HD remains the death of the GABAergic neurons of the striatum, calling for a crucial role of mitochondria in the process. This is substantiated by a number of experimental evidence pointing to altered mitochondrial Ca2+ buffering capacity (Panov et al, 2002), altered mitochondrial bioenergetics (Grunewald & Beal, 1999), increased susceptibility of cells derived from animal models of HD to excitotoxicity by N-methyl-d-aspartate (NMDA) receptor activation (Zeron et al, 2002). These changes are not only observed in cells from HD patients or from animal models of HD, but also recapitulated in vitro or by the expression of mutant Htt (Choo et al, 2004; Panov et al, 2002). The N-terminus of mutant Htt, whose role in inducing striatal dysfunction is well established (Gu et al, 2009), can colocalize with mitochondria and is probably responsible for the retrieval of full length Htt on the organelle (Orr et al, 2008). In addition, administration of the toxin 3-nitropropionic acid (3NPA), a well known inhibitor of complex II, recapitulates the features of HD and is widely used to model the disease in the animal and in vitro (Almeida et al, 2004; Beal et al, 1993). Interestingly, 3NPA has been recently reported to cause mitochondrial morphological abnormalities in primary cortical neurons (Liot et al, 2009) and expression of the N-terminal portion of Htt bearing polyglutamine repeats in HeLa cells causes mitochondrial fragmentation, suggesting that altered mitochondrial dynamics may participate in the pathobiology of HD (Wang et al, 2009). Finally, lymphoblasts from HD patients display striking mitochondrial ultrastructural abnormalities resembling the apoptotic cristae remodelling (Mormone et al, 2006), suggesting that mitochondrial morphology could play a role in HD.
We used a combination of imaging and genetics to investigate if mitochondrial morphological and ultrastructural changes play a role in the increased susceptibility of HD cells to apoptosis.
RESULTS
Mitochondrial fragmentation and cristae disruption in cellular models of HD
Lymphoblasts from HD patients bearing a heterozygous 48 polyglutamine repeat (48Q) display striking abnormalities of mitochondrial ultrastructure (Mormone et al, 2006). We compared morphology of mitochondria in these cells with that of age and gender matched healthy control lymphoblasts. 3D surface rendered confocal z-stacks of a mitochondrially targeted yellow fluorescent protein (mtYFP) revealed fragmentation and clustering of mitochondria in 48Q lymphoblasts. When we extended our analysis to lymphoblasts from a patient with a longer repeat (70Q) or from another patient carrying a homozygous mutation with 45 and 47 repeats (45 + 47Q), we observed a length- and gene dosage-dependent increase in mitochondrial fragmentation (Fig 1A,C). We then turned to two clonal striatal progenitor cell lines isolated from knock-in HdhQ111 mouse embryos bearing a 111 polyglutamine repeat (Q111/0 and Q111/1) (Trettel et al, 2000). Statistically significant mitochondrial fragmentation also characterized these cells, Q111/0 showing 15% more fragmentation than Q111/1 as compared to wt (Fig 1B,D). The morphological defect was retained also when we induced neuronal differentiation (Supporting Information Fig S1). Electron microscopy showed that this increased fragmentation was accompanied by a derangement in the structure of mitochondrial cristae (Fig 1E,F). Thus, mitochondrial fragmentation and disruption of cristae are a hallmark of HD cell lines.
Figure 1. Mitochondrial fragmentation and cristae derangement in HD lymphoblasts and striatal precursors.

- A. Lymphoblasts of the indicated genotype were transfected with mtYFP. Randomly selected confocal, 14 µm deep z axis stacks were acquired, stored, reconstructed and volume rendered. Scale bar: 5 µm.
- B. Striatal precursors of the indicated genotype were transfected with mtYFP. Confocal images of mtYFP from randomly selected cells. Scale bar: 20 µm.
- C. Morphometric analysis. Experiments were as in (A). Data represent mean ± SE of 4 independent experiments where 30 randomly selected, reconstructed and volume rendered z stack series were classified as described. Wt refers to gender-matched control of HD lymphoblasts. (p < 0.05 in a paired Student's t-test between HD samples and their relative control).
- D. Morphometric analysis of mitochondrial morphology. Experiments were as in (B). Data represent mean ± SE of 5 independent experiments where 50 randomly selected images of mtYFP fluorescence were classified as described. p < 0.05 in a paired Student's t-test between HD samples and their relative control.
- E. Representative electron micrographs of wt and Q111 striatal neurons. Cells were fixed and TEM images of randomly selected fields were acquired. Boxed areas represent a 2.3× magnification. Scale bar: 1 µm.
- F. Morphometric analysis. Experiments were performed as in (E). Data represent mean ± SE of 3 independent experiments (n = 50 mitochondria per condition from 15 different neurons of the indicated genotype). Data are normalized to the ratio of wt cells.
- G,H. Equal amounts of proteins (20 µg) from total cell lysates from lymphoblasts (G) and striatal precursors (H) of the indicated genotype were separated by SDS–PAGE and immunoblotted with the indicated antibodies. For MFN1, equal amounts of proteins (50 µg) from mitochondria isolated from lymphoblasts (G) and neurons (H) were separated by SDS–PAGE. For lymphoblasts, wt refers to gender-matched control.
- I,J. Equal amounts of proteins (30 µg) from mitochondria isolated from neurons (I) and lymphoblasts (J) of the indicated genotype were analysed by SDS–PAGE/immunoblotting using the indicated antibodies. Drp1/TOM20 levels in Q111/0 and Q111/1 mitochondria were 5.1 ± 1.03- and 3.05 ± 1.2-fold of wt (n = 3 independent experiments, p < 0.05 in a paired Student's t-test).
Increased Drp1 dephosphorylation and mitochondrial translocation in HD
To understand the molecular basis of the observed fragmentation, we measured levels of mitochondria-shaping proteins in the different HD cellular models, without noticing any relevant change in pro-fusion or pro-fission proteins (Fig 1G,H). We then turned to isolated mitochondria and monitored levels of pro-fission Drp1, that once dephosphorylated translocates to mitochondria (Cereghetti et al, 2008). More Drp1 was associated with HD mitochondria, irrespective of the cellular model they were isolated from, the highest levels retrieved on mitochondria from the Q111/0 clone and the lymphoblasts from the 45 + 47Q patient, which display the most severe fragmentation (Fig 1I,J). Drp1 was also more active, as judged by the abundance of its homo-oligomers following cross-linking (Supporting Information Fig S2) (Zhu et al, 2004). Thus, HD-associated fragmentation can result from increased mitochondrial levels and activity of the pro-fission Drp1. The Ca2+-dependent phosphatase calcineurin can mediate translocation of Drp1 to mitochondria (Cereghetti et al, 2008) and its activation (Cribbs & Strack, 2007). Specific immunoblotting revealed a depletion of Drp1 in the fraction of phosphorylated proteins purified from Q111/0 and Q111/1 cells by affinity chromatography (Cereghetti et al, 2008) (Fig 2A). Accordingly, a specific assay showed that the activity of calcineurin was increased in these HD cells (Fig 2B). Phosphorylated Htt is targeted for degradation (Thompson et al, 2009), suggesting that hyperactive calcineurin could cause an accumulation of Htt. However, Htt was, if anything, slightly reduced in HD lymphoblasts and striatal precursors (not shown and (Trettel et al, 2000)). While levels of the phosphatase (Fig 2C) or of its endogenous inhibitor RCAN1L (Ermak et al, 2009) were comparable (Fig 2D), cyclopiazonic acid releasable intracellular Ca2+ stores were increased (Fig 2E,F), lending support to the observed hyperactivity of calcineurin. Thus, HD cells display increased dephosphorylation of Drp1, endoplasmic reticulum Ca2+ stores and activity of calcineurin.
Figure 2. Hyperactivation of calcineurin and dephosphorylation of Drp1 in HD cells.

- A. Phosphorylated and unphosphorylated proteins were separated from total cell lysates (2.5 mg) from cells of the indicated genotype. Equal amounts (20 µg) of total, phosphorylated and un-phosphorylated proteins were separated by SDS–PAGE and immunoblotted.
- B. Calcineurin enzyme activity measured in total cell extracts from cell lines of the indicated genotype. Data are mean ± SE of three independent experiments.
- C,D. Equal amounts of proteins (40 µg) from total cell lysates from cells of the indicated genotype were separated by SDS–PAGE and immunoblotted with the indicated antibodies. RCAN1L/actin levels (relative to their relative wt): 0.98 ± 0.05 in Q111/0; 0.82 ± 0.08 in Q111/1; 1.28 ± 0.21 in 48Q; 0.91 ± 0.04 in 70Q and 1.22 ± 0.02 in 45 + 47Q lymphoblasts.
- E. Representative traces of Fura-2 ratio of cytosolic Ca2+ ([Ca2+]i) following passive discharge of ER Ca2+ stores by CPA (100 µM) in cells of the indicated genotype.
- F. Quantification of peak and basal [Ca2+]i. Experiments were as in (E). Data represent mean ± SE of eight independent experiments (p < 0.002 by paired Student's t-test).
Mitochondrial fragmentation and cristae disruption in primary striatal YAC128 mouse neurons
To extend our findings to the relevant cell type in HD and to verify if mitochondrial fragmentation and ultrastructural defects were observed in primary striatal HD neurons, we turned to mixed striatal cultures from P0 pups of the established YAC128 mouse model of HD. Three-D reconstruction and volume rendering of mitochondria immunostained with an antibody specific for the outer membrane protein TOM20 revealed fragmentation of the organelle in primary neurons identified using the marker Tubulin III (Fig 4A–C). Electron microscopy also confirmed changes in the shape of the cristae which appeared dilated and disorganized in the YAC128 primary striatal neurons (Fig 4D). Thus, mitochondrial fragmentation and ultrastructural changes are widespread in models of HD and characterize also primary striatal neurons.
Figure 4. Correction of mitochondrial morphology in primary striatal YAC128 neurons.

- Primary neurons of the indicated genotype were immunostained with anti-Tom20 (red) and anti-Tubulin III (green) antibodies. Randomly selected confocal, z axis stacks were acquired, stored, reconstructed and volume rendered. Scale bar: 20 µm.
- 3× magnification of the boxed areas in (A). The bottom panel was turned 90° counter-clockwise.
- Morphometric analysis of mitochondrial shape. Experiments were done as in (A). Data represent mean ± SE of three independent experiments (n = 70 stacks).
- Representative electron micrographs of wt and YAC128 primary striatal neurons. Cells were fixed and TEM images of randomly selected fields were acquired. Boxed areas are magnified 2.4×. Scale bar: 1 µm.
- Cells of the indicated genotype were electroporated with mtRFP and empty vector or the indicated plasmids. Samples were then immunostained with anti-Tubulin III antibody (magenta) and incubated with TUNEL reagent. Images were acquired exactly as in (A). Scale bar: 20 µm.
- Morphometric analysis of mitochondrial morphology. Experiments were performed as in (E). Data represent mean ± SE of three independent experiments (n = 40 stacks).
Genetic and pharmacological correction of mitochondrial fragmentation in HD models
We next verified if we could restore normal mitochondrial morphology by expressing pro-fusion proteins in lymphoblasts from HD patients. The pro-fusion protein Opa1 fully corrected mitochondrial fragmentation in 48Q, 70Q (Fig 3A,D) and in 45 + 47Q (Fig 3B,E) lymphoblasts, as judged by our 3D confocal imaging approach. The correction was also observed in the striatal cell lines Q111/1 and Q111/0 (Fig 3C,F) and in primary striatal neurons (Fig 4E,F). As expected, Opa1 also caused mitochondrial elongation in the lymphoblasts from the corresponding healthy donors and in the striatal cell line from the wt mouse (Figs 3 and 4). We then decided to further explore which other manoeuvres could be devised to correct mitochondrial fragmentation in HD models. Mitochondrial elongation was restored by the efficient expression (Supporting Information Fig S3) of the pro-fusion partner of Opa1, Mfn1 or of a dominant-negative mutant of Drp1 in the 45 + 47Q lymphoblast and in the striatal HD cell lines (Fig 3B–F). Similarly, expression of Mfn1 and dominant negative mutant of Drp1 restored mitochondrial morphology also in YAC128 striatal neurons (Fig 4E,F). Mitochondrial fragmentation was similarly corrected in both the lymphoblast and the striatal cell models of HD by the efficient expression (Supporting Information Fig S3) of a dominant-negative calcineurin mutant that blocks Drp1 mitochondrial translocation (Cereghetti et al, 2008) (Fig. .3). The calcineurin pharmacological inhibitor FK506 similarly ameliorated mitochondrial morphology in the HD striatal models (Fig 3C,F). In sum, these data demonstrate that mitochondrial morphology can be corrected in the HD cell lines and primary neurons by enforcing fusion or by blocking fission. Notably, blockage of calcineurin is one of the approaches that restored mitochondrial elongation, reinforcing the role of this phosphatase in organelle fragmentation.
Figure 3. Correction of mitochondrial morphology in HD cells.

- A,B. Lymphoblasts of the indicated genotype were cotransfected with mtYFP and the indicated plasmids. Experiments were performed exactly as in Fig 1. Scale bar: 5 µm.
- C. Cells of the indicated genotype were cotransfected with mtYFP and the indicated plasmids. When indicated, cells were treated with 1 µM FK506 for 1 h before acquisition of images. Images were acquired exactly as in Fig 1. Scale bar: 20 µm.
- D,E. Morphometric analysis of mitochondrial shape. Experiments were done as in (A) and (B), respectively. Data represent mean ± SE of four independent experiments (n = 30 stacks).
- F. Morphometric analysis of mitochondrial morphology. Experiments were performed as in (C). Data represent mean ± SE of five independent experiments (n = 50 cells).
Increased Opa1-dependent cristae remodelling in HD models
We next sought to determine if the observed mitochondrial fragmentation led to increased susceptibility to cell death by intrinsic stimuli that engage the mitochondrial pathway. To this end, we measured exposure of phosphatidylserine in lymphoblasts, activation of effector caspases in striatal precursors by monitoring cleavage of the caspases substrate PARP, and DNA cleavage in primary striatal neurons by TUNEL assay. Irrespective of the method used, HD lymphoblasts (Fig 5A–C), striatal precursors (Fig 5D,E) and primary striatal neurons (Fig 6) were more sensitive to death by staurosporine as well as by other stimuli tested (hydrogen peroxide, etoposide, in HD lymphoblasts and Q111 cells, not shown). Why are HD cells, displaying fragmented mitochondria, more sensitive to apoptosis? We excluded that this was a consequence of higher levels of proapoptotic (Bax, Bak) Bcl-2 family members (Supporting Information Fig S4A,B). We then verified if the increased death was associated with changes in the mitochondrial pathway of apoptosis. Upon apoptotic stimulation, translocation of Bax to mitochondria (Supporting Information Fig S4C) and activation of Bax and Bak, judged by their retrieval in higher-order oligomers (Wei et al, 2000) (Fig 5F–I), were not increased in the lymphoblast and striatal precursor models of HD. We next compared the rate of cytochrome c release from mitochondria of the striatal precursors in situ. Irrespective of the stimulus used, mitochondria from the cell bearing the 111Q repeat released cytochrome c faster (Fig 5J,K). Similarly, cytochrome c release in response to recombinant BID from mitochondria purified from HD lymphoblasts was faster than that observed in mitochondria isolated from their control counterparts (Fig 5L–N). Interestingly, disruption of the Opa1 containing oligomers that keep cristae junctions in check (Frezza et al, 2006) correlated perfectly with the rate of cytochrome c release in mitochondria isolated from the HD lymphoblasts (Fig 5O). Opa1 oligomers were less represented and more easily disrupted in situ in the HD striatal precursors than in their wt counterpart (Supporting Information Fig S5A). In addition, in the HD striatal cells, following apoptotic stimulation, cleavage of Opa1 to short forms was increased (Supporting Information Fig S5B), leading to an unbalance between the long and the short forms required for the formation of the Opa1 oligomer (Frezza et al, 2006) and for competent mitochondrial fusion (DeVay et al, 2009). Morphometric analysis on electron microscopy images of the shape of mitochondrial cristae in wt, Q111/1 and Q111/0 striatal precursors early (3 h) following induction of apoptosis revealed that the length of the cristae was significantly more reduced in HD cells than in their wt counterparts (Fig 7A–D). These results point to a role for the Opa1-controlled cristae remodelling pathway in the increased susceptibility of HD mitochondria to apoptosis.
Figure 5. Increased cytochrome c release and susceptibility to apoptosis in HD.

- A-C. Lymphoblasts of the indicated genotype were treated with 2 µM staurosporine. At the indicated times, viability was determined cytofluorimetrically. Data represent mean ± SE of seven independent experiments.
- D. Cells of the indicated genotype were treated with 2 µM staurosporine. At the indicated times, cells were lysed and equal amounts of proteins (30 µg) were separated by SDS–PAGE and immunoblotted using the indicated antibodies.
- E. Densitometric analysis of uncleaved/cleaved PARP levels. Experiments were performed as in (D). Data are normalized to the ratio in untreated samples and represent mean ± SE of four independent experiments.
- F,H. Mitochondria isolated from lymphoblasts (F) and neurons (H) of the indicated genotype treated where indicated with 2 µM staurosporine for 4 h were treated with BMH where indicated. Equal amounts (40 µg) of mitochondrial protein were analysed by SDS–PAGE/immunoblotting using an anti-BAX antibody. Asterisks: BAX multimers.
- G. Mitochondria isolated from lymphoblasts of the indicated genotype were treated with cBID for the indicated time and with BMH where indicated. Equal amounts (40 µg) of mitochondrial protein were analysed by SDS–PAGE/immunoblotting using an anti-BAK antibody. Asterisks: BAK multimers.
- I. Mitochondria isolated from cells of the indicated genotype treated where indicated with 2 µM staurosporine for 4 h were incubated with BMH where indicated. Equal amounts (40 µg) of protein were analysed by SDS–PAGE/immunoblotting using an anti-BAK antibody. Asterisks: BAK multimers. In (F–I) immunoblots are representative of three independent experiments.
- J. Representative confocal images of striatal cells of the indicated genotype transfected with mtRFP and immunostained with anti-cytochrome c (green) antibody. When indicated, cells were treated for 2 h with 1 mM H2O2.
- K. Localization index of cytochrome c. Cells of the indicated genotype were treated where indicated for 2 h with 1 mM H2O2 or for 5 h with 0.75 µM staurosporine (STS). Localization index of cytochrome c was determined as described. Data represent mean ± SE of three independent experiments (n = 50 cells per condition in each experiment).
- L-N. Mitochondria isolated from lymphoblasts of the indicated genotype were treated for the indicated times with cBID. The amount of cytochrome c in supernatant and pellet was determined as described. Data represent mean ± SE of four independent experiments.
- O. Mitochondria from lymphoblasts of the indicated genotype were treated with cBID for the indicated times and then crosslinked with EDC. Equal amounts of proteins were analysed by SDS–PAGE/immunoblotting using anti-OPA1 antibody.
Figure 6. Increased susceptibility to apoptosis of striatal YAC128 primary neurons: correction by Opa1 and dominant-negative Drp1.

- Representative confocal images of TUNEL assay of primary striatal neurons. Neurons of the indicated genotype were electroporated with mtRFP, treated where indicated with 500 nM staurosporine for 14 h, immunostained with anti-Tubulin III and processed for TUNEL. Arrowheads, TUNEL positive neurons. Scale bar: 20 µm.
- Quantification of TUNEL assay of primary neurons. Cells of the indicated genotype were transfected as indicated and treated as in (A). Cell death is represented as the percentage of TUNEL positive cells in the mtRPF–Tubulin III double positive population. Data represent mean ± SE of three independent experiments (n = 300 neurons per condition).
Figure 7. Apoptotic ultrastructural changes in HD mitochondria are faster and prevented by OPA1 and blockage of DRP1.
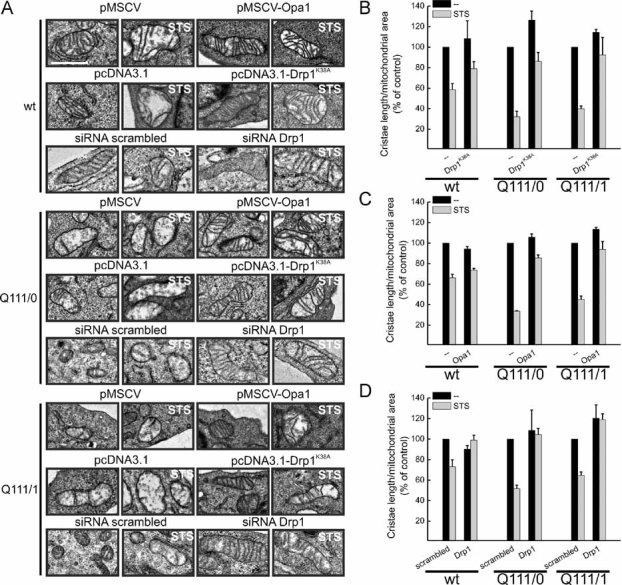
- A. Representative EM fields of neurons of the indicated genotype. Cells were cotransfected with GFP and the indicated plasmids or siRNAs, sorted and GFP-positive cells were seeded and after 18 h treated when indicated with 2 µM staurosporine for 3 h, fixed and processed for EM. Scale bar: 1 µm.
- B-D. Morphometric analysis of cristae shape. Experiments were performed as in (A). Data represent mean ± SE of 3 independent experiments (n = 50 mitochondria from 30 different cells per each condition) and are normalized to the value in empty vector-transfected, untreated cells.
Apoptosis and cristae remodelling in HD are corrected by inhibition of Drp1 and by Opa1, but not by Mfn1
Expression of Opa1 rescued the increased susceptibility to apoptosis of the lymphoblasts from HD patients (Fig 8A) and of the severely affected Q111/0 clone (Fig 8B,G). Interestingly, expression of Mfn1, that also counteracts mitochondrial fragmentation of HD cells (Fig 3), did not correct apoptosis in HD lymphoblasts, or in the Q111/0 clone (Fig 8C,G). Notably, the increased basal levels of apoptosis of primary YAC128 striatal neurons, as well as their greater susceptibility to staurosporine were fully corrected by Opa1, but not by Mfn1 (Fig 6B). Thus, enforced fusion is not sufficient to correct the mitochondrial apoptotic phenotype of HD models, further highlighting the importance of Opa1-dependent cristae shape for their increased apoptosis.
Figure 8. HD cells are more susceptible to apoptosis: correction by Opa1 and inhibition of Drp1.

- A. Lymphoblasts of the indicated genotype transfected as indicated were treated with 2 µM staurosporine. At the indicated times, cell death was determined cytofluorimetrically. Data represent mean ± SE of five independent experiments.
- B-E. Cells of the indicated genotype were cotransfected with GFP and the indicated vectors or siRNAs. After 24 (B–D) or 30 h (E) cells were treated with 2 µM staurosporine for 3 h and 3 × 105 GFP positive cells were sorted and lysed. Proteins were analysed by SDS–PAGE/immunoblotting using the indicated antibodies.
- F. Cells of the indicated genotype were treated where indicated with 1 µM FK506 and 2 µM staurosporine. Cells were lysed and equal amounts of proteins (30 µg) were separated by SDS–PAGE and immunoblotted using the indicated antibodies. (B–F) Western blots are representative of 3 independent experiments.
- G. Densitometric analysis of uncleaved/cleaved PARP levels. Experiments were performed as in (B,D,F). Data are normalized to the ratio in untreated samples and represent mean ± SE of four independent experiments.
Cristae remodelling can be triggered by the activation of Drp1 (Germain et al, 2005), offering a possible explanation for the link between Drp1-dependent fragmentation, cristae alterations and increased Opa1-sensitive apoptosis in HD cells. We tested this hypothesis by addressing if blockage of Drp1 could revert the cristae alterations and the increased cell death in the HD models. The dominant negative mutant of Drp1 protected the homozygous HD lymphoblasts (Fig 8A), the Q111/0 striatal clone (Fig 8D,G) and the primary YAC128 striatal neurons (Fig 6B) from death. Similarly, efficient siRNA-mediated silencing of Drp1 corrected mitochondrial fragmentation (Supporting Information Fig S6) and reduced apoptosis in Q111/0 cells (Fig 8E). Finally, preventing translocation of Drp1 to mitochondria with the calcineurin inhibitor FK506 also protected the striatal Q111 clone (Fig 8F,G). Electron microscopy and morphometric analysis revealed that mitochondrial cristae were rapidly disrupted upon apoptotic stimulation of HD cells and that expression of Opa1 blocked this rapid damage (Fig 7A,C). Interestingly, apoptotic disruption of the cristae was also blunted in HD cells where Drp1 was genetically inhibited or downregulated (Fig 7A–C), substantiating a cross-talk between Drp1 and cristae shape in the context of HD.
DISCUSSION
The role of mitochondria in the pathogenesis of neurodegenerative diseases is under close scrutiny (Chan, 2007). This is not surprising, given their position at the crossroad of energy conversion and integration of apoptotic signalling. Mounting evidence support a role for altered mitochondrial shape in neurodegeneration. For example, the genes mutated in the genetic familial form of PD have been reported to impinge on mitochondrial shape (Poole et al, 2008). Whether this is the case also for HD is less clear. Our data show that mitochondria in cells from HD patients or from a mouse model expressing a pathologic polyQ repeat are fragmented and display changes in the ultrastructure that are causally linked to their increased susceptibility to apoptosis.
Ectopic expression of a 74 polyQ from Htt in HeLa cells was reported to cause mitochondrial fragmentation (Wang et al, 2009). Moreover, evidence of cristae remodelling is one of the hallmarks of lymphoblasts from HD patients (Mormone et al, 2006), a widely employed model to study the mitochondrial alterations in the disease (Panov et al, 2002; Sawa et al, 1999). Mitochondrial fragmentation is a hallmark of HD lymphoblasts, of striatal precursors and differentiated striatal neurons from the Q111 knock-in mouse (Trettel et al, 2000), as well as of primary striatal neurons isolated from the YAC128 mouse model. Mechanistically, fragmentation is associated with increased mitochondrial translocation and activation of Drp1, a process that depends on the cytosolic phosphatase calcineurin (Cereghetti et al, 2008).
The role of calcineurin in the context of HD is controversial. The reduction in the levels of the calcineurin inhibitor RCAN1-1L (Ermak et al, 2009) and the dysregulation of cytosolic Ca2+, the proximal activator of calcineurin (Tang et al, 2005), cooperate to increase the activity of calcineurin in HD. It has been reported that calcineurin inhibition has a protective (Pardo et al, 2006; Xifro et al, 2008) or a worsening (Hernandez-Espinosa & Morton, 2006) effect in models of HD. However, studies in whole animals reported that both FK506 and cyclosporine A, that does not cross the blood–brain barrier, have the same worsening effect on the phenotype of the disease (Hernandez-Espinosa & Morton, 2006), raising the question of whether it could be caused by extraneurological action of these immunomodulators. Our results suggest that another target of calcineurin in the context of HD can be Drp1 and hence mitochondrial morphology. In fact mitochondrial fragmentation can be reverted by genetic or pharmacological inhibition of calcineurin. It will be interesting to verify if the natural history of the disease would be modified in mouse models crossed with conditional models of Drp1 ablation recently described (Ishihara et al, 2009).
How does increased fragmentation of the mitochondria contribute to the progression of the apoptotic cascade? One unifying model suggests that activation of the pro-fission protein Drp1 not only causes the fragmentation of the organelle, but also induces remodelling of the cristae, thereby augmenting the availability of cytochrome c in the intermembrane space of the organelle and hence its release across the outer membrane (Germain et al, 2005). This was reinforced by recent data showing changes in Opa1 pattern in cells where expression of Drp1 was modulated (Mopert et al, 2009). Such a scenario seems plausible also in the HD models tested here. The fragmented mitochondria of HD lymphoblasts and striatal precursors are more susceptible to cytochrome c release and oligomers of Opa1, which control cristae junctions (Cipolat et al, 2006; Frezza et al, 2006; Yamaguchi et al, 2008) are readily destabilized in the HD cells. This occurs despite any measurable increase in the activation of the effector multidomain proapoptotics Bax and Bak in the HD sample, pointing to a role for Drp1 downstream of them. Accordingly, the integrity of mitochondrial cristae during apoptosis is maintained not only by Opa1, but also by the genetic inhibition of Drp1. Both Opa1 and inhibition of Drp1 protect all the HD models tested from cell death, while enforcing fusion by expression of Mfn1 does not. Notably, Mfn1 does not affect shape of the cristae and is dispensable for the anti-apoptotic activity of Opa1 (Frezza et al, 2006). This further substantiates the role of ultrastructural changes in the HD phenotype analysed here.
How mitochondria are affected in HD is a matter of debate. It has been reported that the multiple changes in mitochondrial Ca2+ handling (Panov et al, 2002), metabolism (Damiano et al, 2010), and susceptibility to apoptosis (Sawa et al, 1999) could be related to mitochondrial localization of mutated Htt (Orr et al, 2008) or to transcriptional regulation of the master mitochondrial biogenetic gene PGC1α (Cui et al, 2006). Here we add one potential mechanism of structural alterations, orchestrated by calcineurin-dependent mitochondrial translocation and activation of the pro-fission protein Drp1. Our studies lend the molecular basis for future in vivo studies to verify the contribution of this pathway to the neurological phenotype of the disease and open the possibility to drug this pathway to change the natural history of the disease.
MATERIALS AND METHODS
Plasmids and siRNA
mtRFP, mtYFP, pCB6-MYC-Mfn1, pcDNA3.1-HA-K38A-DRP1, pMSCV-OPA1, pEGFP and pcDNA3.1-H155Q-CnA were previously described (Cereghetti et al, 2008; Cipolat et al, 2004; de Brito & Scorrano, 2008). siRNA against Drp1 (Ambion, Huntington, UK) (5′-UCC GUG AUG AGU AUG CUU Utt-3′) and scrambled control siRNA were used at the same final concentration (200 nM).
Cell culture, transfection, reagents and sorting
EBV immortalized human B lymphoblasts (control and 48 CAG repeats) were described in Mao and Wang (2002). Lymphoblasts from two male (control and 45 + 47 CAG) and two female subjects were from the Coriell Institute for Medical Research. Lymphoblasts were grown in DMEM-F12 (Gibco), 20% fetal bovine serum (FBS), 50 U/ml penicillin, 50 µg/ml streptomycin, 100 µM non-essential amino acids (MEM, Gibco/Invitrogen) and 2 mM glutamine (Gibco) and electroporated using the MicroPorator system (Digital Bio Technology) following manufacturer's instructions.
Q111 and wt clonal striatal cell lines were described previously and cultured as indicated in Trettel et al (2000). Cells were transfected with DNA using Transfectin (Biorad) following manufacturer's instructions and with siRNA by electroporation (Microporator, Digital Bio Technology).
For sorting, 4 × 106 cotransfected cells were analysed by light forward and side scatter and for GFP fluorescence through a 530 nm band pass filter as they traversed the beam of an argon ion laser (488 nm, 100 mW) of a FACSAria (BD). Non-transfected cells were used to set the background fluorescence.
Primary striatal cultures were generated from the offsprings of crosses between heterozygous males YAC128 HD transgenic mice and wild-type females from the same genetic background (FVB/N) (Charles River Laboratories), following a protocol modified from Mao and Wang (2002). Neonatal 1-day-old mice were decapitated, brains extracted and maintained in ice cold PBS supplemented with 3 mg/ml BSA (PBS/BSA). Striata were isolated using a dissecting microscope and digested in 0.08% trypsin (Sigma) in PBS/BSA for 20 min at 37°C. Dissociation of the cells was achieved by gentle pipetting in PBS/BSA supplemented with 0.08 mg/ml DNase I (Roche) and 0.52 mg/ml soybean trypsin inhibitor (Invitrogen). After removing not dissociated tissue the suspension was centrifuged for 7 min at 1000 rpm. Cells were resuspended and cultured in 70% DMEM/Ham's F12 medium, 30% Neurobasal medium (Invitrogen), 1% glucose, 200 mM glutamate and 10 mg/ml penicillin/streptomycin (Invitrogen), 2% B27 (Invitrogen). 1–2 × 105 cells were seeded onto 13 mm round glass coverslips coated with 10 mg/ml poly-d-lysine (Sigma) and analysed after one week in culture. Primary neurons were electroporated before plating using the MicroPorator system (Digital Bio Technology) following manufacturer's instructions.
Analysis of cell death
Lymphoblasts treated as indicated were stained with propidium iodide (PI) and Annexin-V-FITC (Bender MedSystem). Where indicated, cells were cotransfected with pEGFP and the indicated vector. After 24 h cells were treated as described and stained with Annexin-V-PE (Bender MedSystem) according to the manufacturer's protocol. Cell death was measured by flow cytometry (FACSCalibur) as the percentage of Annexin-V-positive events in the GFP-positive population and viability as the percentage of Annexin-V-negative, PI-negative cells for transfected and untransfected cells, respectively.
For TUNEL assays, 1–2 × 105 primary neurons electroporated and seeded onto 13 mm round glass coverslips were treated after 6 days with 500 nM staurosporine for 14 h, fixed, immunostained with anti-Tubulin III antibody (1:200, Sigma) and isotype-matched Alexa-647 secondary antibody, and incubated in TUNEL reagent (Roche) following manufacturer's instructions. Coverslips were mounted on slides with a DAPI-containing Vectashield mounting solution (Vector Laboratories, Inc.). For DAPI, TUNEL, mtRFP and Tubulin III detection, excitation was performed using the Diode 405/30 405 nm, Argon/2 488/514 nm and HeNe 633 nm lasers, respectively. Images were acquired sequentially using four separate colour channels using of a Zeiss LSM Meta using a 40 × 1.3 Oil DIC EC Plan Neofluar objective (Zeiss). Cell death was measured as the percentage of TUNEL positive cells in the mtRPF–Tubulin III positive population. A total of 300 neurons were counted per condition in each independent experiments.
Ca2+ measurements
Cells of the indicated genotype (8 × 105) plated on 24-mm glass coverslips were loaded with 5 µM Fura-2/AM (Invitrogen) at 33°C for 45 min in 1 ml of cell culture medium supplemented with 0.04% pluronic acid and sulphinpyrazone (250 µM). Cells were washed with modified Krebs–Ringer buffer and then bathed in a Ca2+-free medium, containing EGTA (100 µM). Cells were placed on the thermostated (33°C) stage of an Olympus lX-81 microscope equipped with 60X UPLAN FLN oil objective (NA 1.25; Olympus) and 12-bit F-VIEW II camera (Soft Imaging System), alternatively illuminated at 340 and 380 nm, and images (1 ratio image/s) were acquired using the CellR software (Olympus). The data are reported as 340/380 ratio values (R), calculated off-line after background subtraction from each single image.
Calcineurin activity assay and phosphoprotein purification
Calcineurin activity was measured using an in vitro assay kit (Calbiochem) following manufacturer's instructions as previously described (Cereghetti et al, 2008). For phosphorylation studies, total cell lysates were loaded on a phosphoprotein binding column (Qiagen) as previously described (Cereghetti et al, 2008). Flow-through (unphosphorylated) and eluted (phosphorylated) proteins were collected and concentrated and 20 µg of proteins were separated by 4–12% SDS–PAGE.
In vitro mitochondrial assays
Mitochondria were isolated by standard differential centrifugation in isolation buffer (IB) as described in Frezza et al (2006). Cytochrome c release in response to recombinant cBID was determined as described in Ermak et al (2009). p7/p15 recombinant BID was produced, purified, and cleaved with caspase-8 as described in Scorrano et al (2002). Unless noted, it was used at a final concentration of 32 pmol/mg.
Biochemistry
For protein crosslinking, mitochondria were treated with 2 mM EDC (Pierce) in EB (30 min, 37°C). Samples were centrifuged for 10 min at 12,000 × g at 4°C, and the pellets were resuspended in SDS–PAGE sample loading buffer. DTT in the sample buffer quenched the crosslinking reaction. For Opa1 crosslinking in neurons, cells treated as indicated were incubated with 1 mM DSS (Pierce, 30 min, 25°C). The reaction was quenched by 100 mM Tris/HCl buffer (pH 7.4, 15 min, 25°C). For BAX and BAK crosslinking mitochondria were incubated with 2 mM (15 min, 4°C) or 10 mM (30 min, 37°C) BMH (Pierce) and the reaction was quenched with 20 mM β-mercaptoethanol (15 min, 25°C). For immunoblotting, proteins were separated by 7% Tris–acetate, 3–8% Tris–acetate or 4–12% Tris–MOPS SDS–PAGE (NuPage, Invitrogen), transferred onto PVDF membranes (Millipore), probed using the indicated primary antibodies and isotype matched secondary antibodies conjugated to horseradish peroxidase (Amersham) and detected using ECL (Amersham). The following antibodies were employed: mouse anti-OPA1 antibody (1:1500, BD Biosciences); mouse anti-DRP1 (1:1500, BD Biosciences); mouse anti-MFN2 (1:1000, Abnova); rabbit anti-FIS1 (1:1000, Abnova); rabbit anti-TOM20 (1:2000, St. Cruz Biotechnology); mouse anti-actin (1:3000, Chemicon); rabbit anti-PARP (1:1000, Cell Signaling); chicken anti-MFN1 (1:500, Abcam); rabbit anti-calcineurin (1:500, Cell Signaling); rabbit anti-BAX (1:1000, Millipore); rabbit anti-BAK (1:1000, Millipore); rabbit anti-RCAN1 antibody (1:2000) (Ermak et al, 2009) Densitometric quantification of western blot was performed using the Gel Pro analyser 4 software.
The paper explained
PROBLEM
Huntington's disease (HD) is a genetic, progressive, neurodegenerative disease caused by the expansion of a polyglutamine trait in the Huntingtin protein. The main neurological signs and symptoms are caused by the loss of the medium spiny neurons of the striatum. The exact pathogenesis of the disease is unknown but mitochondria, which are positioned at the crossroad of energy production and control of cell death, have been heavily implicated.
RESULTS
Here, we demonstrate that a feed-forward loop of mitochondrial fragmentation and alterations of their ultrastructure play an important role in determining the susceptibility of HD cellular models to apoptosis. This loop is sustained by the activation of dynamin related protein 1, a large GTP hydrolysing enzyme that is a key mediator of mitochondrial fission. Its genetic blockage restores mitochondrial morphology and the response to apoptosis in the HD models tested.
IMPACT
Our findings extend the potential therapeutic targets to interfere with the natural history of HD to mitochondrial morphology.
Transmission electron microscopy and mitochondrial morphometry
Cells were fixed for 1 h at 25°C using glutaraldehyde at a final concentration of 2.5% (v/v) in PBS. Embedding and staining were performed as described in Scorrano et al (2002). Thin sections were imaged on a Tecnai-20 electron microscope (Philips-FEI). Mitochondrial cristae morphology was quantified measuring the length of every cristae in each mitochondrion analysed and normalized for the area of the organelle using the MetaMorph software. For comparative reasons, the ratio in untreated empty vector-transfected cells was set to 100%.
Imaging
For imaging, lymphoblasts of the indicated genotype were transfected as indicated. After 24 h, cells were plated in serum-free medium for 1.5 h to promote adhesion to fibronectin (5 µg/ml, Sigma) coated coverslips. Samples were analysed as described above. For imaging of embryonic striatal neuronal cell lines, 105 cells were seeded onto 24 mm round glass coverslips and transfected as indicated. After 24 h cells were incubated in Hank's Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES and coverslips were placed on the stage of a Zeiss LSM 510 inverted microscope. Cells expressing mtYFP were excited using the 488 nm line of the Argon laser using a 63 × 1.4 NA Plan Apochromat objective (Zeiss). For imaging of mitochondrial network in primary neurons, 1–2 × 105 cells were seeded onto 13 mm round glass coverslips coated with 10 mg/ml poly-d-lysine (Sigma), fixed and immunostained with anti-TOM20 (1:200, St. Cruz Biotechnology) and anti-Tubulin III (1:200, Sigma) and isotype matched TRITC and FITC-conjugated secondary antibody. For mitochondrial morphology correction experiments, primary neurons were transfected with mtRFP and the indicated vector, immunostained with anti-Tubulin III and isotype matched Alexa 647-conjugated antibody (Sigma) and incubated in TUNEL reagent (Roche). Stacks separated by 0.4 µm along the z axis were acquired for each channel using a Zeiss LSM Meta microscope with a 63 × 1.4 NA Plan Apochromat objective (Zeiss). 3D reconstruction and volume rendering were performed using a plug-in of ImageJ (NIH). Quantitative analysis of mitochondria morphology was performed as described. Cells were classified as having fragmented mitochondria when more than 50% of the total cellular organelles displayed a major axis shorter than 5 µm for neurons and 3 µm for lymphoblasts.
Cytochrome c release imunofluorescence experiments were performed on 12 × 104 cells as described in Frezza et al (2006).
Acknowledgments
Luca Scorrano is a Senior Telethon Scientist. This work was supported by Telethon Italy GGP06254A (to LS and WM) and S02016TELU (to LS), SNF 31-118171 (to LS), Fondazione CARIPARO and EC FP7 ERANET (to EC)
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
VC and LS conceived research, analysed data and wrote the manuscript. VC, MG, RH, RL, DL performed experiments and analysed data. GE and KJAD provided novel reagents. EC and WM analysed data.
For more information
National Institute of Neurological Disorders and Stroke, Huntington's Disease Information:
http://www.ninds.nih.gov/disorders/huntington/huntington.htm
National Human Genome Research Institute, Learning About Huntington's Disease:
http://www.genome.gov/10001215
CHDI Foundation, Inc.:
http://www.highqfoundation.org/index.php
Accompanying Closeup by Jorge Oliveira and Lightowlers:
DOI 10.1002/emmm.201000104
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Almeida S, Domingues A, Rodrigues L, Oliveira CR, Rego AC. FK506 prevents mitochondrial-dependent apoptotic cell death induced by 3-nitropropionic acid in rat primary cortical cultures. Neurobiol Dis. 2004;17:435–444. doi: 10.1016/j.nbd.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, de Martins BO, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial dynamics in disease. N Engl J Med. 2007;356:1707–1709. doi: 10.1056/NEJMp078040. [DOI] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- Cipolat S, de Brito OM, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio L, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1[alpha] by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Damiano M, Galvan L, Deglon N, Brouillet E. Mitochondria in Huntington's disease. Biochim Biophys Acta. 2010;1802:52–61. doi: 10.1016/j.bbadis.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- De Rooij KE, Dorsman JC, Smoor MA, Den Dunnen JT, van Ommen GJ. Subcellular localization of the Huntington's disease gene product in cell lines by immunofluorescence and biochemical subcellular fractionation. Hum Mol Genet. 1996;5:1093–1099. doi: 10.1093/hmg/5.8.1093. [DOI] [PubMed] [Google Scholar]
- DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difiglia M, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- Difiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Duyao M, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- Ermak G, Hench KJ, Chang KT, Sachdev S, Davies KJ. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J Biol Chem. 2009;284:11845–11853. doi: 10.1074/jbc.M900639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Frezza C, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RK, C, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Grunewald T, Beal MF. Bioenergetics in Huntington's disease. Ann N Y Acad Sci. 1999;893:203–213. doi: 10.1111/j.1749-6632.1999.tb07827.x. [DOI] [PubMed] [Google Scholar]
- Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci USA. 1995;92:8710–8714. doi: 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Espinosa D, Morton AJ. Calcineurin inhibitors cause an acceleration of the neurological phenotype in a mouse transgenic for the human Huntington's disease mutation. Brain Res Bull. 2006;69:669–679. doi: 10.1016/j.brainresbull.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Ishihara N, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jana NR, Zemskov EA, Wang G, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–1059. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- Kegel KB, et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz AK, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Wang JQ. Interactions between ionotropic and metabotropic glutamate receptors regulate cAMP response element-binding protein phosphorylation in cultured striatal neurons. Neuroscience. 2002;115:395–402. doi: 10.1016/s0306-4522(02)00400-1. [DOI] [PubMed] [Google Scholar]
- Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mopert K, Hajek P, Frank S, Chen C, Kaufmann J, Santel A. Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization. Exp Cell Res. 2009;315:2165–2180. doi: 10.1016/j.yexcr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Morais VA, et al. Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1:1–14. doi: 10.1002/emmm.200900006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG, Piacentini M, Frati L, Malorni W, Squitieri F. Genotype-dependent priming to self- and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with Huntington's disease. J Neurochem. 2006;98:1090–1099. doi: 10.1111/j.1471-4159.2006.03998.x. [DOI] [PubMed] [Google Scholar]
- Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Pardo R, Colin E, Regulier E, Aebischer P, Deglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- Schilling G, Sharp AH, Loev SJ, Wagster MV, Li SH, Stine OC, Ross CA. Expression of the Huntington's disease (IT15) protein product in HD patients. Hum Mol Genet. 1995;4:1365–1371. doi: 10.1093/hmg/4.8.1365. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Sharp AH, et al. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinís R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci USA. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thompson LM, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Wang H, Lim PJ, Karbowski M, Monteiro MJ. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wellington CL, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- Xifro X, Garcia-Martinez JM, del TD, Alberch J, Perez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J Neurochem. 2008;105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem. 2004;279:35967–35974. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.