

Abstract
Hepatitis C virus (HCV) is remarkably efficient at evading host immunity to establish chronic infection. During chronic HCV infection, interleukin-12 (IL-12) produced by monocytes/macrophages (M/Mφ) is significantly suppressed. Programmed death-1 (PD-1), an inhibitory receptor on immune cells, plays a pivotal role in suppressing T-cell responses during chronic viral infection. To determine whether PD-1 regulates IL-12 production by M/Mφ during chronic HCV infection, we examined the expressions of PD-1, its ligand PDL-1, and their relationship with IL-12 production in M/Mφ from HCV-infected, HCV-resolved, and healthy subjects by flow cytometry. Toll-like receptor (TLR) -mediated IL-12 production by M/Mφ was selectively suppressed, while PD-1/PDL-1 expressions were up-regulated, in HCV-infected subjects compared with HCV-resolved or healthy subjects. Up-regulation of PD-1 was inversely associated with the degree of IL-12 inhibition in HCV infection. Interestingly, the reduced response of M/Mφ from HCV-infected individuals to TLR ligands appeared not to be the result of a lack of the ability to sense pathogen, but to an impaired activation of intracellular janus kinase/signal transducer and activator of transfection (STAT) pathway as represented by inhibited STAT-1 phosphorylation in M/Mφ from HCV-infected individuals compared with HCV-negative subjects. Successful HCV treatment with pegylated interferon/ribavirin or blocking PD-1/PDL-1 engagement ex vivo led to reduced PD-1 expression and improved IL-12 production as well as STAT-1 activation in M/Mφ from HCV-infected individuals. These results suggest that the PD-1 inhibitory pathway may negatively regulate IL-12 expression by limiting STAT-1 phosphorylation in M/Mφ during chronic HCV infection.
Keywords: hepatitis C virus infection, interleukin-12, Immune regulation, macrophages, programmed death-1
Introduction
Hepatitis C virus (HCV) infection is a serious and growing health threat affecting approximately 3% (around 200 million people) of the world population.1,2 One remarkable feature of HCV infection is that the majority (>80%) of infected individuals progress to chronic hepatitis, which is associated with the development of liver cirrhosis and hepatocellular carcinoma, a leading cause for liver transplantation.1,2 The propensity of HCV to persist in the vast majority of infected individuals suggests that the virus has evolved one or more mechanisms to evade host immunity.
Virus-specific cytotoxic T lymphocytes (CTL) are critical for the elimination of the virus-infected cells. Strong virus-specific CTL responses have been demonstrated in those individuals whose HCV infection spontaneously resolves.3 In chronically HCV-infected subjects, however, virus-specific CTL responses are significantly diminished both in the liver and in peripheral blood.4,5 This apparent suppression of CTL responses suggests that HCV may interfere with CD4+ helper T-cell activation and differentiation, so allowing the establishment of chronic infection. Interleukin-12 (IL-12), a key pro-inflammatory cytokine produced by activated monocyte/macrophages (M/Mφ), provides a pivotal link between innate and adaptive immunity by regulating the T helper type 1 (Th1) response. In chronically HCV-infected subjects, IL-12 production and Th1 responses are significantly suppressed.6,7 In addition to HCV, several other viruses, including cytomegalovirus, HIV and poxvirus, also impair Th1 immunity by inhibiting IL-12 production.8–10 This suppression of Th1 by dysregulating IL-12 production may represent a common shared mechanism for host immune evasion; how IL-12 production by M/Mφ is suppressed during viral infections remains largely unknown.
Recently, it has been shown that the virus-induced negative cell modulator programmed death 1 (PD-1) contributes significantly to immunodysregulation.11–13 PD-1 is an inhibitory receptor predominantly expressed on activated lymphocytes as well as M/Mφ. Its ligand, PDL-1, is expressed on both haematopoietic and parenchymal cells.14 It has been proposed that PDL-1 expressed on hepatocytes and endothelial cells plays a role in immune regulation by interaction with PD-1 expressed on immune cells; PDL-1 expressed on distinct immune cells is also able to interact and regulate other immune cells expressing PD-1. The engagement of PD-1/PDL-1 induces immune-receptor tyrosine phosphorylation and delivers negative signals to receptor signalling pathways, such as janus kinase (JAK)/signal transducer and activator of transcription (STAT), so preventing cell activation. It has been reported that CD4+ and CD8+ T-cell responses, including virus-specific interferon-γ (IFN-γ) production, are severely suppressed in chronically HCV-infected subjects through the PD-1 inhibitory pathway.15
Because IL-12 is a key cytokine for regulating IFN-γ and T-cell responses during viral infection, in this study we sought to determine whether the PD-1 inhibitory pathway is involved in regulating IL-12 production by M/Mφ during chronic HCV infection. To address this possibility, we examined the expressions of PD-1, its ligand PDL-1, as well as IL-12, in human blood-derived M/Mφ stimulated with toll-like receptor 4 (TLR4) ligand – lipopolysaccharide (LPS) and TLR7/8 ligand – R848, which are known to synergistically induce IL-12 production.16,17 We employed M/Mφ derived from a cohort of subjects with chronic HCV infection, subjects with resolved HCV infection, and healthy subjects. We found that the PD-1 pathway is involved in negative regulation of M/Mφ IL-12 production by limiting STAT-1 phosphorylation during chronic HCV infection.
Materials and methods
Subjects
An institutional review board-approved protocol at East Tennessee State University and James H. Quillen VA Medical Center (ETSU/VA IRB) has contributed to a longstanding database for the storage of blood samples from HCV-infected individuals for the purpose of viral immunological studies. Samples were subjected to density gradient separation of peripheral blood mononuclear cells (PBMC), divided into aliquots and cryopreserved in liquid nitrogen storage as described previously.18 All subjects gave written consent for this study. The subjects recruited comprised three groups of populations with overall characteristics as delineated in Table 1. The first group included 25 chronically HCV-infected subjects before treatment with pegylated interferon and ribavirin (IFN/RBV). HCV genotype and viral load testing were performed by Lexington VA Medical Center. The second group comprised 15 HCV-resolved individuals including four subjects with spontaneously resolved HCV, and 11 subjects with HCV cleared by IFN/RBV treatment with sustained virological response (SVR); so all subjects were HCV RNA negative but HCV antibody positive. The samples from SVR patients were collected at 6 months after IFN/RBV treatment, which should provide enough wash-out time to define that any changes were the result of clearance of HCV rather than any regulatory effect of IFN/RBV on the immune system. The third group comprised 14 healthy subjects negative for HBV, HCV and HIV. The majority of the study subjects were male and Caucasian. The mean age of the HCV-infected or HCV-resolved individuals was higher but comparable to that of healthy subjects.
Table 1.
Demographic features of the subjects recruited in the study
| ID | Age | Sex | Race (Caucasian) | ALT/AST (U/l) | HCV genotype | Viral load (IU/ml) |
|---|---|---|---|---|---|---|
| P01 | 59 | M | Yes | 37/50 | 1a | 4 790 000 |
| P02 | 38 | M | Yes | 56/72 | 1a | 38 200 000 |
| P03 | 54 | M | Yes | 89/73 | 1a | 1 890 000 |
| P04 | 55 | M | Yes | 85/47 | 3a | 50 000 000 |
| P05 | 33 | M | Yes | 103/62 | 2b | 40 100 000 |
| P06 | 62 | M | Yes | 108/99 | 1a | 97 965 |
| P07 | 49 | M | Yes | 43/62 | 3a | 11 641 000 |
| P08 | 58 | M | Yes | 72/73 | 1b | 530 590 |
| P09 | 39 | M | Yes | 22/25 | 1a | 992 000 |
| P10 | 59 | M | No | 74/48 | 1a | 2 110 000 |
| P11 | 52 | M | Yes | 64/28 | 3a | 10 000 000 |
| P12 | 55 | M | Yes | 43/47 | 1a | 3 640 000 |
| P13 | 53 | M | Yes | 52/47 | 1a | 2 180 000 |
| P14 | 60 | M | No | 57/150 | 3a | 26 800 000 |
| P15 | 56 | F | No | 22/25 | 1b | 5 780 000 |
| P16 | 64 | M | Yes | 177/103 | 2b | 50 000 000 |
| P17 | 31 | M | No | 69/46 | 1b | 23 000 |
| P18 | 69 | M | Yes | 112/78 | 1a | 2 210 000 |
| P19 | 66 | M | No | 54/43 | 1a | 1 100 000 |
| P20 | 68 | M | No | 66/53 | 1a | 790 000 |
| P21 | 56 | M | Yes | 74/57 | 1a | 1 360 233 |
| P22 | 51 | M | Yes | 33/23 | 1b | 13 321 000 |
| P23 | 46 | M | No | 34/56 | 3a | 2 560 000 |
| P24 | 51 | M | Yes | 77/61 | 1a | 188 897 |
| P25 | 55 | M | Yes | 43/45 | 1b | 1 132 878 |
| Group summary | No. of subjects | Mean age (range), years | % of males | % of genotype 1 | HCV-RNA (IU/ml) |
|---|---|---|---|---|---|
| HCV-infected patients | 25 | 54 (31–69) | 96 | 72 | 23 000–50 000 000 |
| HCV-resolved individuals | 15 | 51 (37–66) | 100 | NA | NA |
| Healthy subjects | 14 | 44 (27–58) | 85 | NA | NA |
ALT/AST, alanine aminotransferase/aspartate aminotransferase; NA, not applicable.
Cell isolation and culture
Human PBMC were isolated from the peripheral blood of study subjects by density gradient centrifugation with lympholyte-H (Cedarlane Laboratories, Ontario, Canada). The PBMC were viably frozen and thawed as described previously.18,19 A human monocytic cell line, THP-1 cells, was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in RPMI-1640 (ATCC, Manassas, VA), containing 10% fetal bovine serum, 100 μg/ml penicillin-streptomycin, and 0·05 mmβ-mercaptoethanol (Sigma-Aldrich, St Louis, MO) at 37° with 5% CO2 atmosphere.
Flow cytometry
Intracellular IL-12 (p40/p70) production in activated M/Mφ was determined by flow cytometric analysis using the MACS Inside Stain Kit (Miltenyi Biotec Inc., Auburn, CA) per the manufacturer's instruction. Briefly, 1 × 106 purified PBMC were stimulated with 5 μg/ml LPS and 5 μg/ml R848 for 72 hr, with 2 μg/ml secretion inhibitor brefeldin A for 6 hr before harvesting the cells for intracellular immunofluorescent staining. The stimulated PBMC were washed twice with 200 μl FACS medium (pH 7·2 PBS buffer containing 2 mm EDTA and 0·5% BSA) by centrifugation at 300 g for 5 min, then stained for cell surface markers for M/Mφ using 10 μl FITC-conjugated anti-human CD14 antibody (clone: TÜK4; Miltenyi Biotec Inc.) in 90 μl FACS medium in a 96-well plate for 1 hr at room temperature. The cells were washed as described above; resuspended with 200 μl Inside Fix for 20 min, followed by washing with 200 μl Inside Perm; then cells were incubated in 90 μl Inside Perm with 10 μl allophycocyanin-conjugated (APC-) anti-human-IL-12 antibody (clone: C-8.6; Miltenyi Biotec Inc.) at room temperature for 1 hr. The cells were then washed three times in FACS medium and stored at 4° in the dark before analysis by FACSCalibur flow cytometry (BD, Franklin Lakes, NJ) and cellquest software. The primary isotype controls were used to determine the level of background staining and 20 000 events were collected after gating on M/Mφ populations.
To determine the expression level of PD-1, PDL-1, IL-6, IL-10, tumour necrosis factor-α (TNF-α), phospho-STAT-1, TLR4, and TLR7 on the surface or inside M/M-ϕ, 1 × 106 isolated PBMC were activated with LPS/R848, followed by FACS staining as described above. The source and clone of each antibody used are as followings: APC-anti-PD-1 (clone: M1H4; eBioscience, San Diego, CA) APC-anti-PDL-1 (clone: M1H1; eBioscience), phycoerythrin-conjugated (PE-) anti-IL-6 (clone: MQ2-13A5; eBioscience), PE-anti-IL-10 (clone: B-T10; Miltenyi Biotec Inc), APC-anti-TNF-α (clone: MAb11; eBioscience), PE-anti-phospho-STAT-1 (clone: K51-856; BD Biosciences, San Jose, CA), PE-anti-TLR4 (clone: 285219; R&D Systems Inc., Minneapolis, MN) and PE-anti-TLR7 (clone: 533707; R&D Systems Inc.). Isotype-matched control antibodies (R&D Systems Inc.) were used to determine background levels of staining.
For blocking assays, PBMC isolated from six HCV-infected subjects were incubated with 10 μg/ml anti-PDL-1 (anti-human CD274 or B7-H1 clone: M1H1; eBioscience) or IgG isotype control antibody in RPMI-1640 with 10% FCS overnight before stimulation with LPS and R848 for 72 hr, and expressions of the PD-1, IL-12 and phospho-STAT-1 in CD14+ M/Mφ were carried out as described above. The concentration of blocking antibody was chosen based on literature and our previous reports.18,20 To determine the direct effect of blocking the PD-1 pathway on monocytes versus other cell types, the THP-1 monocytic cell line was pre-incubated with or without anti-PDL-1 or control IgG antibody overnight, then stimulated with LPS/R848 in the presence or absence of HCV core protein for 72 hr, followed by FACS analysis of IL-12 expression.
Statistical analysis
Study results are summarized for each group and results are expressed as mean ± SD. Comparison between two groups is performed using multiple comparison testing – least significant difference or Tukey's procedure depending on the analysis of variance F-test by spss 18 software (IBM, Somers, NY). Paired t-tests were used to compare data before and after IFN/RBV therapy or anti-PDL-1 versus control IgG treatment. A Pearson correlation program was used to determine the correlation between PD-1 or PDL-1 expression and IL-12 production in M/Mφ from the study subjects. P-values < 0·05 were considered significant, and **< 0·01 were considered very significant.
Results
IL-12 production by CD14+ M/MØ is inhibited in subjects with chronic HCV infection
We have previously demonstrated that HCV core protein inhibits T-cell activation and M/Mφ IL-12 production through interaction with a complement receptor-gC1qR in vitro.21,22 To confirm IL-12 inhibition during clinical HCV infection, we isolated PBMC from 25 HCV-infected subjects, four HCV spontaneously resolved and 11 treatment resolved subjects, and 14 healthy subjects, stimulated with TLR ligand LPS and R848 ex vivo, and analysed by flow cytometric analysis for intracellular IL-12 production by CD14+ M/Mφ. Percentage of IL-12+ cells in CD14+ M/Mφ was calculated and plotted in Fig. 1. Interleukin-12 production was significantly suppressed in subjects with chronically HCV-infected subjects when compared with the HCV-resolved or healthy subjects. No difference was identified between HCV-resolved individuals and healthy subjects.
Figure 1.
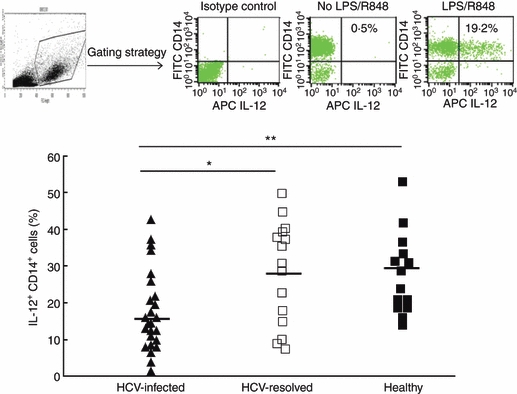
Interleukin-12 (IL-12) production is suppressed in CD14+ monocyte/macrophage (M/Mφ) during chronic hepatitis C virus (HCV) infection. Peripheral blood mononuclear cells from HCV-infected (▲, n = 25), HCV-resolved (□, n= 15), and healthy subjects (▪, n = 14) were stimulated with lipopolysaccharide (LPS) and R848 ex vivo, followed by cell surface staining for CD14 and intracellular staining for IL-12. The top panel shows the gating strategy for identifying M/Mφ subsets and representative flow cytometric isotype control (left) and dot plot (right) measuring IL-12 production in gated CD14+ M/Mφ with or without LPS/R848 stimulation. The percentage of IL-12-producing cells was calculated in CD14+ cell populations from the study subjects and plotted in the lower panel. Each symbol represents an individual subject, and the horizontal line demonstrates the mean of IL-12+ CD14+ M/Mφ. The P value is shown as *< 0·05, **< 0·01 above the group of study subjects.
PD-1 expression is up-regulated on CD14+ M/Mφ during HCV infection
To determine whether the PD-1 inhibitory pathway is involved in the regulation of IL-12 production in M/Mφ, we examined PD-1 and PDL-1 expression on the surface of M/Mφ stimulated with LPS and R848 as described above, followed by flow cytometric analysis. Percentages of PD-1+ or PDL-1+ cells on CD14+ M/Mφ were calculated and plotted in Fig. 2. In contrast to the down-regulation of IL-12 production in HCV infection, PD-1 expression on M/Mφ from chronically HCV-infected subjects was detected significantly higher than those from healthy subjects. A significant difference was also observed when comparing the PD-1 expression on M/Mφ in HCV-infected versus HCV-resolved subjects, whereas no significant difference was found between HCV-resolved and healthy subjects. Upon grouping all virus-negative subjects (HCV-resolved + healthy subjects), statistical significance of PD-1 expression was maintained. Although we also found relatively higher levels of PDL-1 expression on M/Mφ from HCV-infected subjects than either HCV-resolved individuals or healthy subjects, the differences were not statistically significant (Fig. 2, right panel).
Figure 2.
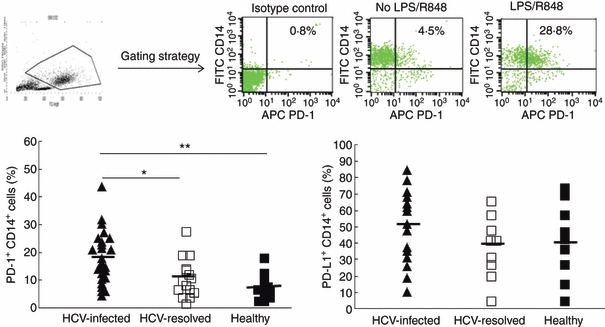
Programmed death 1 (PD-1)/PD-1 ligand (PDL-1) expression is up-regulated on CD14+ monocyte/macrophages (M/Mφ) during chronic hepatitis C virus (HCV) infection. Peripheral blood mononuclear cells from HCV-infected (▲), HCV-resolved (□), and healthy subjects (▪) were stimulated with lipopolysaccharide (LPS) and R848 ex vivo, followed by cell surface double staining for CD14, PD-1, and PDL-1. The top panel shows the gating strategy for identifying M/Mφ subsets and representative flow cytometric isotype control (left) and dot plot (right) measuring PD-1 expression in gated CD14+ M/Mφ with or without Toll-like receptor (TLR) stimulation. The percentage of PD-1+ (left panel) or PDL-1+ (right panel) cells was calculated in CD14+ cell populations from the study subjects and plotted in the lower panel. Each symbol represents an individual subject, and the horizontal line demonstrates the mean of PD-1+ or PDL-1+ CD14+ M/Mφ. The P value is denoted as *< 0·05, **< 0·01 above the group of study subjects.
PD-1 expression is inversely associated with IL-12 production by M/Mφ
Because we had observed what appeared to be an inverse correlation between PD-1 and IL-12 expressions in all of our subject populations, we next sought to determine this association in M/Mφ. We analysed expression levels in 20 subjects (HCV-positive and HCV-negative) by Pearson correlation using spss 18 software. As shown in Fig. 3(a), PD-1 was inversely associated with IL-12 production in M/Mφ; PDL-1 expression (Fig. 3b), was not statistically significantly associated with the degree of IL-12 expression. To assess whether the presence of HCV might be contributing to PD-1 up-regulation, we also longitudinally studied PD-1 expressions and IL-12 production in M/Mφ in several HCV-infected subjects before treatment and following a successful antiviral treatment. We observed a significant decrease in the up-regulated PD-1 (Fig. 3c) expressions and, correspondingly, an increase in IL-12 production (Fig. 3d), in M/Mφ from those achieving SVR.
Figure 3.
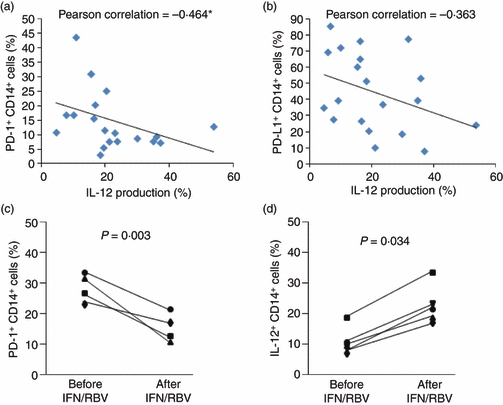
Programmed death 1 (PD-1) expression is inversely associated with interleukin-12 (IL-12) production by monocyte/macrophage (M/Mφ). M/Mφ from hepatitis C virus (HCV) -positive and -negative subjects were stimulated with lipopolysaccharide (LPS)/R848 as above followed by staining and flow cytometric analysis. The relationship between PD-1 (a) or PD-1 ligand (PDL-1) (b) expression and IL-12 production in M/Mφ was analysed by Pearson correlation using SPSS 18 software. The association of PD-1 expression and IL-12 production in M/Mφ was also longitudinally studied in HCV-infected subjects before and after antiviral treatment with interferon/ribavirin (IFN/RBV) with sustained virological response (c, n= 4, and d, n = 5). Paired Student's t-test was used to calculate P value here.
IL-12 is selectively inhibited in M/Mφ with impaired STAT-1 phosphorylation
To determine whether IL-12 is selectively inhibited during HCV infection, we simultaneously analysed the expressions of IL-12, IL-6, IL-10 and TNF-α in CD14+ M/Mφ from five HCV-infected individuals and five healthy subjects by flow cytometry. As shown in Fig. 4, only IL-12 expression (Fig. 4a), but not IL-6 (Fig. 4b), IL-10 (Fig. 4c) or TNF-α (Fig. 4d), was found to be significantly suppressed during chronic HCV infection in M/Mφ in response to LPS/R848 stimulation. To determine whether M/Mφ from HCV-infected individuals fail to respond to these TLR ligands ex vivo because they lack the ability to sense pathogens, we also analysed the expression of TLR4 and TLR7 in M/Mφ from HCV-infected versus healthy subjects. TLR4 was found to be expressed more highly on M/Mφ from HCV-infected versus uninfected subjects (Fig. 5a), whereas TLR7 was detected at high but not statistically different levels in both HCV-infected and healthy subjects (Fig. 5b). Given the pivotal role of the intracellular JAK-STAT pathway in TLR signalling, we then examined the activation status of TLR downstream signalling by analysing STAT-1 phosphorylation in M/Mφ from HCV-infected, HCV-resolved, and healthy subjects by flow cytometry. As shown in Fig. 5(c), STAT-1 phosphorylation was inhibited in M/Mφ from chronically HCV-infected patients when compared with HCV-resolved individuals or healthy subjects.
Figure 4.
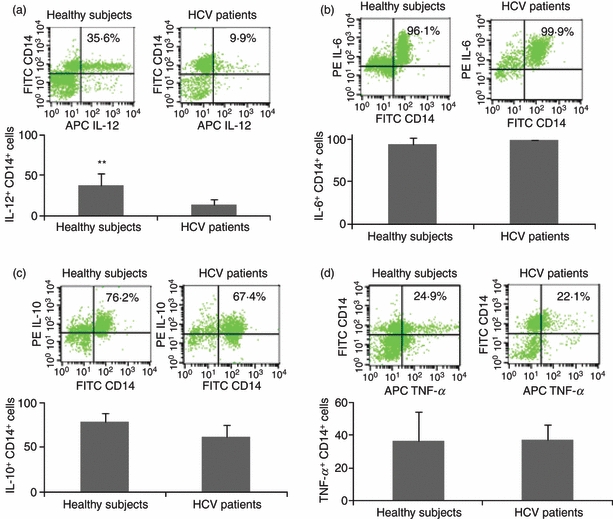
Interleukin-12 is selectively inhibited in monocyte/macrophage (M/Mφ) during hepatitis C virus (HCV) infection. M/Mφ from five HCV-infected and five healthy subjects were stimulated with lipopolysaccharide (LPS)/R848 as above followed by staining and flow cytometric analysis for the expressions of IL-12 (a), IL-6 (b), IL-10 (c), and tumour necrosis factor-α (TNF-α) (d). Representative dot plots with percentage of positive cells are shown above and summary data with bar figures are shown below. **P < 0·01.
Figure 5.
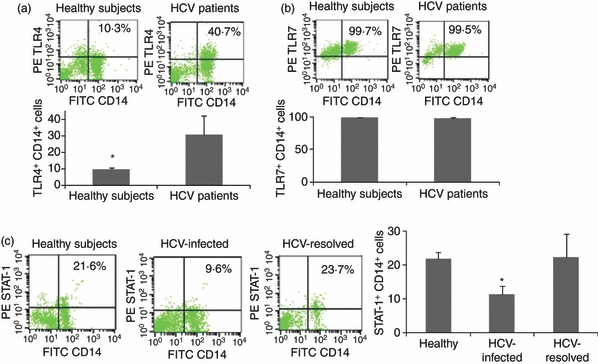
Toll-like receptor (TLR) and phospho-signal transducer and activator of transcription 1 (STAT-1) expressions in monocyte/macrophage (M/Mφ) during hepatitis C virus (HCV) infection. M/Mφ from five HCV-infected and five HCV-uninfected subjects were stimulated with lipopolysaccharide (LPS)/R848 as above followed by staining and flow cytometric analysis for the expressions of TLR4 (a), TLR7 (b), and phospho-STAT-1 (c). Representative dot plots with percentage of positive cells as well as summary data with bar figures are shown. *P < 0·05.
Blocking the PD-1 inhibitory pathway improves IL-12 production and STAT-1 activation
To further address the role of the PD-1 inhibitory pathway in regulating IL-12 production in M/Mφ during HCV infection, we pre-incubated PBMC from HCV-infected subjects with 10 μg/ml of anti-PDL-1 or a control IgG antibody overnight, then stimulated with LPS and R848 ex vivo for 72 hr, followed by flow cytometric analysis for PD-1 expression and IL-12 production in the treated M/Mφ. Similar to the data reporting the functional restoration of HCV-specific CD8+ T cells by PD-1 blockade,20 we observed the recovery of PD-1 expression (Fig. 6a) and improved IL-12 production (Fig. 6b) after ex vivo blocking PD-1/PDL-1 engagement on M/Mφ when compared with cells treated with the control antibody. Correspondingly, blocking the PD-1 pathway significantly restored the impaired STAT-1 phosphorylation in M/Mφ from HCV infection (Fig. 6c). The improvedIL-12 production in M/Mφ could be secondary to cytokines secreted from other immune cells in the culture system rather than blockade of PD-1 signalling on M/Mφ. To observe the direct effect of PD-1 blocking on monocytes, we therefore incubated PDL-1 antibody or control IgG and HCV core, which we have shown to decrease IL-12 expression in monocytes,21 with the LPS/R848-treated monocytic cell line, THP-1, followed by FACS analysis of IL-12 expression. As shown in Fig. 6(d), compared with LPS/R848 stimulation alone, HCV core protein significantly inhibits IL-12 expression, and this core-mediated IL-12 inhibition could be restored by blocking PD-1 pathway.
Figure 6.
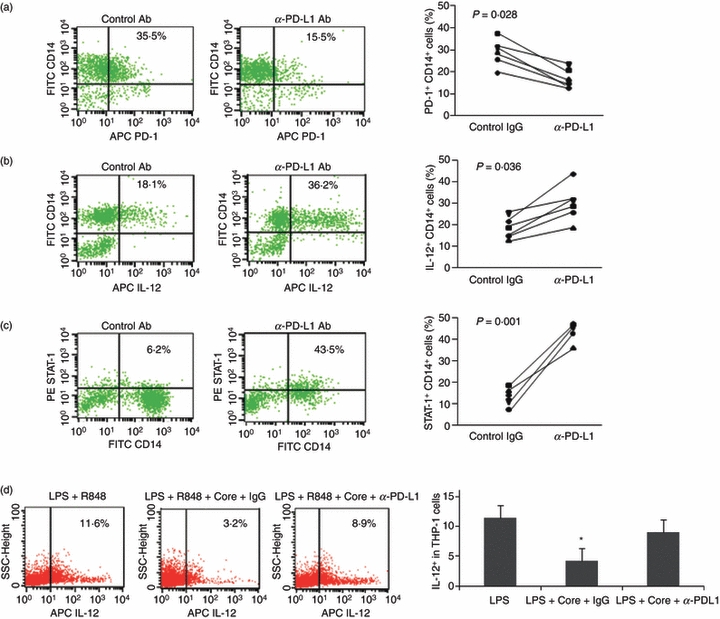
Blocking the programmed death 1 (PD-1) inhibitory pathway reduces PD-1 expression and improves interleukin-12 (IL-12) production and signal transducer and activator of transcription 1 (STAT-1) activation. Peripheral blood mononuclear cells from hepatitis C virus (HCV) -infected individuals were pre-incubated with anti-PD-1 ligand (PDL-1) or a control antibody overnight; then stimulated with lipopolysaccharide (LPS) and R848 ex vivo for 72 hr, followed by flow cytometric analysis for the expressions of PD-1 (a), IL-12 (b), and phospho-STAT-1 (c). Left panel shows representative flow cytometric dot plots measuring PD-1 expression, IL-12 production, and STAT-1 phosphorylation in cells incubated with anti-PDL-1 blocking antibody or the control IgG. Right panel shows the combined data from all subjects tested comparing the effect of anti-PDL-1 to that of the IgG control. Each symbol represents an individual with chronic HCV infection. P-value is shown to compare PD-1 expression, IL-12 production, and STAT-1 phosphorylation in monocyte/macrophage treated with anti-PDL-1 versus control antibody. (d) To determine a direct effect of PD-1 blocking on monocytes, THP-1 cells were pre-incubated with or without PDL-1 or control IgG overnight, then stimulated with LPS/R848 in the presence or absence of HCV core protein, followed by FACS analysis of IL-12 expression. Representative dot plot and summary data from three repeated experiments are shown. *P < 0·05.
Discussion
Inhibition of IL-12 leading to impaired Th1 polarization and CTL responses represents a common mechanism for host immune evasion by several viral infections, including hepatitis C. The underlying mechanism(s) for IL-12 inhibition during viral infections, however, remains elusive. Whereas extensive studies report that the PD-1 inhibitory pathway plays a pivotal role in suppressing T-cell responses,11–15,18,20 little is known about its function in regulating M/Mφ production of IL-12, a key pro-inflammatory cytokine linking innate and adaptive immunity during viral infection. In this report, we found that PD-1 is significantly up-regulated on peripheral M/Mφ, and this up-regulation is inversely correlated with IL-12 production by these cells in chronically HCV-infected subjects when compared with HCV-resolved individuals or healthy subjects. Interestingly, the poor response of M/Mφ from HCV-infected individuals to TLR ligands appears not because they lack the ability to sense pathogen, but probably because of an impaired phosphorylation of intracellular JAK/STAT pathway. Importantly, successful HCV treatment by IFN/RBV, as well as blocking PD-1/PDL-1 engagement ex vivo, could significantly decrease PD-1 expression and improve IL-12 production and STAT-1 phosphorylation in TLR-stimulated M/Mφ, suggesting that the PD-1 inhibitory pathway is involved in the regulation of IL-12 production by limiting STAT-1 phosphorylation during HCV infection.
Interleukin-12 is a 70 000–75 000 molecular weight (MW) heterodimeric cytokine composed of a 40 000 MW heavy chain and a 35 000 MW light chain. It is produced mainly by activated M/Mφ as well as myeloid dendritic cells, primarily in response to stimulation by bacterial or viral antigens via TLR expressed on the cell surface. Microbial gene products also stimulate up-regulation of co-stimulatory or inhibitory receptors on M/Mφ or myeloid dendritic cells and modulate the secretion of IL-12. The M/Mφ-produced IL-12 induces various effects on both natural killer and T cells, playing an essential role in the transition between innate and adaptive immune responses and particularly crucial for CD4+ T-cell differentiation and the production of Th1 cytokines such as IFN-γ.23 We have previously demonstrated that HCV core protein inhibits T-cell activation and M/Mφ IL-12 production through interaction with a complement receptor, gC1qR, in vitro.18,21,22 In this translational study, we have further demonstrated that human M/Mφ-derived IL-12 production is inhibited in chronically HCV-infected subjects, and this inhibition is inversely associated with the up-regulation of PD-1 expression on these cells, suggesting that the PD-1 inhibitory pathway is involved in IL-12 suppression during HCV infection.
PD-1 is an inducible molecule belonging to the immunoglobulin superfamily. It is mainly expressed on activated T and B lymphocytes, and plays a pivotal role in the negative regulation of adaptive immune responses during chronic viral infections.11–15 Although ample evidence supports roles for PD-1 in negative regulation of adaptive immune responses, little is known about its expression and function in innate cells and immunity. Recently, it has been demonstrated that in addition to T and B lymphocytes, PD-1 can be induced on M/Mφ and dendritic cells by bacterial or viral infection or TLR engagements,14,24 and PD-1/PDL-1 interaction negatively regulates LPS-mediated IL-12 production in murine macrophages.25 It was also reported that PD-1 is induced by TLR ligands on monocytes and is responsible for IL-10 production.26 In this report, we further show that PD-1 is induced on human blood-derived M/Mφ and is involved in inhibiting IL-12 production during HCV infection. Our findings reveal a novel aspect of negative regulator in innate immune regulation in viral infection. Interestingly, PD-1 expression is decreased upon treatment with blocking PDL-1 antibody. Although we could not exclude the possibility that neutralizing anti-PDL-1 antibody prevented PD-1 detection by FACS, we also detected PD-1 protein by Western blot in purified monocytes, in the presence of anti-PD-1 or anti-PDL-1 versus control antibody, and we found that PD-1 is decreased upon treatment with these blocking antibodies (data not shown).
Expression of PDL-1 on M/Mφ during chronic HCV infection was not statistically significantly higher than the control subjects, perhaps because PDL-1 is expressed highly and scattered on immune cells, and because only relatively few subjects were recruited in this study. Interaction with PDL-1 is, however, required for PD-1-mediated suppression of IL-12 production. While PDL-1 has been shown to be up-regulated in various tissues and cells in response to viral infection, it may not be the rate-limiting molecule in the dysregulation of M/Mφ cytokine responses. That is, in addition to the PDL-1/PD-1 engagement that may occur between M/Mφ, it is feasible that the ample PDL-1 available on other cells/tissues, including hepatocytes and endothelial cells, interacts with up-regulated PD-1 on M/Mφ to signal IL-12 inhibition during chronic HCV infection.
The molecular mechanism underlying PD-1-mediated IL-12 inhibition is yet to be clarified. It has been proposed that PD-1 may suppress T-cell responses via the immunoreceptor tyrosine inhibitory motif and/or the immunoreceptor tyrosine switch motif in its cytoplasmic tail, delivering a negative signal to the JAK/STAT pathway.27 In B cells, cross-linking of PD-1 along with the B cell receptor leads to tyrosine phosphorylation of the PD-1 cytoplasmic domain, recruitment of SHP-2 phosphatase, and reduced phosphorylation of B-cell receptor proximal kinases.28 Our experiments suggest that pursuing the downstream mechanisms involved in suppression of IL-12 in M/Mφ would be salient. It is possible that PD-1, induced by HCV gene products, can directly inhibit M/Mφ production of IL-12 by down-regulation of TLR signalling as a negative feedback mechanism to diminish innate immune responses and reduce pathological damage. Supporting this concept are our preliminary data indicating that, upon successful treatment with IFN/RBV or ex vivo blocking PD-1/PDL-1 engagement, the inhibited phosphorylation of STAT-1 is reversed. Further investigation of the underlying mechanism(s) of PD-1 up-regulation and its cross-talk with other signalling pathways, including suppressor of cytokine signalling 1 (SOCS-1), and T-cell immunoglobulin and mucin domain-containing protein 3 (Tim-3), are ongoing in our laboratory.
Antiviral CD4+ T-cell responses are crucial for the resolution of viral infections by providing help for CD8+ T-cell priming and B-cell antibody generation. However, HCV-specific T and B cells are not detected until 1–3 months after infection,29,30 suggesting that there is a delay in the generation of adaptive immunity during HCV infection compared with other persistent viruses such as HIV. These data support the concept that the early stages of innate immune responses against HCV may be defective. Indeed, multiple independent studies have shown that M/Mφ or DC isolated from subjects chronically infected with HCV display a reduced capacity to induce natural killer cell or T-cell activation, and M/Mφ or myeloid dendritic cell-produced IL-12 has been shown to be significantly suppressed during HCV infection.6,7,31–34 Based on this and our previous studies, we propose a model in which immunosuppressive antigens secreted from HCV-infected hepatocytes would bind to their receptors displayed on immune cells, lead to up-regulation of negative immunoregulators, such as PD-1/PDL-1, Tim-3/Gal-9, A20, and SOCS-1, and result in a cytokine environment (deficient IL-12/TNF-α/IFN-γ) that is permissive for suppression of innate, and in turn adaptive, immune responses in the liver, thus facilitating the establishment of persistent infection.
This study is translational in nature so it is limited by the possibility of viral and host heterogeneity that may be present. For example, the majority of our HCV population is male, Caucasian and infected with HCV genotype 1. Expression levels of PD-1 appear to vary within different ethnic groups.20 Despite this, the effects we have observed in M/Mφ at least support a relationship between PD-1 and IL-12, and it is possible that while the PD-1 levels may vary within groups, the inverse correlation with IL-12 remains. Our future studies will attempt to parse out these different subgroups. Additionally, the mechanisms of IL-12 down-regulation remain elusive. It remains unclear whether M/Mφ from HCV-infected individuals fail to respond to TLR stimulation because they lack the ability to sense pathogens and whether this effect is specific for IL-12. From our preliminary studies, it appears that TLR4, but not TLR7, is up-regulated; and IL-12 is selectively inhibited in M/Mφ isolated from HCV-infected individuals and ex vivo stimulated with LPS/R848 when compared with those from healthy subjects. This result is in line with the studies showing that no significant differences were observed in response to TLR ligands of monocytes from HCV-infected patients compared with healthy subjects as assessed by their similar levels of secretion of IL-6, IL-1β, TNF-α, IL-10, IFN-inducible protein-10 and macrophage inflammatory protein-1a35; and HCV core treatment specifically inhibits the expression of IL-12, but not IL-10, TNF-α, transforming growth factor-β in LPS- or poly(I:C)-stimulated monocyte-derived dendritic cells.7 However, the data are not in agreement with other studies reporting that the expressions of TLRs 2, 4, 7 or 8 in CD14+ cells of PBMC were increased in HCV-infected patients compared with controls, irrespective of HCV genotype or histological stage of disease, and circulating levels of IL-12, IL-6, TNF-α were also increased in peripheral PBMC in HCV-infected patients.36–39 These disparities are possibly because of the differences in the stimulation methods, the cell differential status, and the time-points chosen to evaluate these molecules’ expressions in human M/Mφ under different conditions.
To our knowledge, there have been few studies focusing on PD-1/PDL-1 expression on human blood-derived M/Mφ and the regulation of IL-12 production during HCV infection. Our study has demonstrated that up-regulation of PD-1 correlates with IL-12 inhibition in M/Mφ and that HCV treatment with IFN/RBV or blocking PD-1/PDL-1 engagement ex vivo may reduce PD-1 expression and improve IL-12 production as well as STAT-1 phosphorylation in subjects with HCV infection. These results provide new evidence that the PD-1 inhibitory pathway may mediate functional impairment of early immune responses during HCV infection, further supporting the notion that PDL-1 blockade represents a novel therapeutic approach to this common viral infection.
Acknowledgments
This work was supported by an National Institutes of Health National Institute of Allergy and Infectious Diseases grant to ZQY/JPM (1R15A1084057-01). L. Ni, a joint PhD student, Division of Physiology, Department of Human Function, School of Preclinical Medicine, Beijing University of Chinese Medicine, Beijing, China, was supported in part by the China Scholarship Council (CSC 2008655005). C.L. Zhang, a visiting scholar, holds a grant for viral hepatitis research from Guangzhou Municipal Health Bureau, China. This publication is the result of work supported with resources and the use of facilities at the James H. Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
Disclosures
The authors have no conflicts of interest, including relevant financial interests, to disclose.
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–52. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 3.Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rehermann B, Chang KM, McHutchison JG, Kokka R, Houghton M, Chisari FV. Quantitative analysis of the peripheral blood cytotoxic T lymphocyte response in patients with chronic hepatitis C virus infection. J Clin Invest. 1996;98:1432–40. doi: 10.1172/JCI118931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedemeyer H, He XS, Nascimbeni M, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol. 2002;169:3447–58. doi: 10.4049/jimmunol.169.6.3447. [DOI] [PubMed] [Google Scholar]
- 6.Gelderblom HC, Nijhuis LE, de Jong EC, et al. Monocyte-derived dendritic cells from chronic HCV patients are not infected but show an immature phenotype and aberrant cytokine profile. Liver Int. 2007;27:944–53. doi: 10.1111/j.1478-3231.2007.01507.x. [DOI] [PubMed] [Google Scholar]
- 7.Waggoner SN, Hall CH, Hahn YS. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J Leukoc Biol. 2007;82:1407–19. doi: 10.1189/jlb.0507268. [DOI] [PubMed] [Google Scholar]
- 8.Buisson S, Benlahrech A, Gazzard B, Gotch F, Kelleher P, Patterson S. Monocyte-derived dendritic cells from HIV type 1-infected individuals show reduced ability to stimulate T cells and have altered production of interleukin (IL)-12 and IL-10. J Infect Dis. 2009;199:1862–71. doi: 10.1086/599122. [DOI] [PubMed] [Google Scholar]
- 9.Renneson J, Dutta B, Goriely S, et al. IL-12 and type I IFN response of neonatal myeloid DC to human CMV infection. Eur J Immunol. 2009;39:2789–99. doi: 10.1002/eji.200939414. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Chaudhri G, Jackson RJ, Karupiah G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J Immunol. 2009;183:3324–31. doi: 10.4049/jimmunol.0803985. [DOI] [PubMed] [Google Scholar]
- 11.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 12.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–58. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamoto N, Kaplan DE, Coleclough J, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. 2008;134:1927–37. doi: 10.1053/j.gastro.2008.02.033. 37 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong HY, Lee YJ, Seo SK, et al. Blocking of monocyte-associated B7-H1 (CD274) enhances HCV-specific T cell immunity in chronic hepatitis C infection. J Leukoc Biol. 2008;83:755–64. doi: 10.1189/jlb.0307168. [DOI] [PubMed] [Google Scholar]
- 15.Rutebemberwa A, Ray SC, Astemborski J, et al. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J Immunol. 2008;181:8215–25. doi: 10.4049/jimmunol.181.12.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bohnenkamp HR, Papazisis KT, Burchell JM, Taylor-Papadimitriou J. Synergism of Toll-like receptor-induced interleukin-12p70 secretion by monocyte-derived dendritic cells is mediated through p38 MAPK and lowers the threshold of T-helper cell type 1 responses. Cell Immunol. 2007;247:72–84. doi: 10.1016/j.cellimm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Wenink MH, Santegoets KC, Broen JC, et al. TLR2 promotes Th2/Th17 responses via TLR4 and TLR7/8 by abrogating the type I IFN amplification loop. J Immunol. 2009;183:6960–70. doi: 10.4049/jimmunol.0900713. [DOI] [PubMed] [Google Scholar]
- 18.Yao ZQ, King E, Prayther D, Yin D, Moorman J. T cell dysfunction by hepatitis C virus core protein involves PD-1/PDL-1 signaling. Viral Immunol. 2007;20:276–87. doi: 10.1089/vim.2006.0096. [DOI] [PubMed] [Google Scholar]
- 19.Yao ZQ, Prayther D, Trabue C, Dong ZP, Moorman J. Differential regulation of SOCS-1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology. 2008;125:197–207. doi: 10.1111/j.1365-2567.2008.02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golden-Mason L, Klarquist J, Wahed AS, Rosen HR. Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol. 2008;180:3637–41. doi: 10.4049/jimmunol.180.6.3637. [DOI] [PubMed] [Google Scholar]
- 21.Eisen-Vandervelde AL, Waggoner SN, Yao ZQ, Cale EM, Hahn CS, Hahn YS. Hepatitis C virus core selectively suppresses interleukin-12 synthesis in human macrophages by interfering with AP-1 activation. J Biol Chem. 2004;279:43479–86. doi: 10.1074/jbc.M407640200. [DOI] [PubMed] [Google Scholar]
- 22.Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. 2000;106:1239–49. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 24.Yao S, Wang S, Zhu Y, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113:5811–8. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho HY, Choi EK, Lee SW, Jung KO, Seo SK, Choi IW, Park SG, Choi I. Programmed death-1 receptor negatively regulates LPS-mediated IL-12 production and differentiation of murine macrophage RAW264.7 cells. Immunol Lett. 2009;127:39–47. doi: 10.1016/j.imlet.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Said EA, Dupuy FP, Trautmann L, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010;16:452–9. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheppard KA, Fitz LJ, Lee JM, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 28.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. 2001;98:13866–71. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, Ray SC. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology (Baltimore, MD) 2005;42:104–12. doi: 10.1002/hep.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotta S, Brazzoli M, Piccioli D, Valiante NM, Wack A. Hepatitis C virions subvert natural killer cell activation to generate a cytokine environment permissive for infection. J Hepatol. 2009;52:183–90. doi: 10.1016/j.jhep.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Shiina M, Rehermann B. Cell culture-produced hepatitis C virus impairs plasmacytoid dendritic cell function. Hepatology (Baltimore, MD) 2008;47:385–95. doi: 10.1002/hep.21996. [DOI] [PubMed] [Google Scholar]
- 33.Tu Z, Pierce RH, Kurtis J, Kuroki Y, Crispe IN, Orloff MS. Hepatitis C virus core protein subverts the antiviral activities of human Kupffer cells. Gastroenterology. 2009;138:305–14. doi: 10.1053/j.gastro.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Zhang Z, Zhang S, Fu J, Yao J, Jiao Y, Wu H, Wang FS. B7-H1 up-regulation impairs myeloid DC and correlates with disease progression in chronic HIV-1 infection. Eur J Immunol. 2008;38:3226–36. doi: 10.1002/eji.200838285. [DOI] [PubMed] [Google Scholar]
- 35.Perrin-Cocon L, Agaugué S, Diaz O, Vanbervliet B, Dollet S, Guironnet-Paquet A, André P, Lotteau V. Th1 disabled function in response to TLR4 stimulation of monocyte-derived DC from patients chronically-infected by hepatitis C virus. PLoS ONE. 2008;3:e2260. doi: 10.1371/journal.pone.0002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolganiuc A, Oak S, Kodys K, et al. Hepatitis C core and nonstructural 3 proteins trigger toll like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–24. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 37.Riordan SM, Skinner NA, Kurtovic J, Locarnini S, McIver CJ, Williams R, Visvanathan K. Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm Res. 2006;55:279–85. doi: 10.1007/s00011-006-0082-0. [DOI] [PubMed] [Google Scholar]
- 38.Shehata MA, Abou El-Enein A, El-Sharnouby GA. Significance of toll-like receptors 2 and 4 mRNA expression in chronic hepatitis C virus infection. Egypt J Immunol. 2006;13:141–52. [PubMed] [Google Scholar]
- 39.Sato K, Ishikawa T, Okumura A, et al. Expression of Toll-like receptors in chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2007;22:1627–32. doi: 10.1111/j.1440-1746.2006.04783.x. [DOI] [PubMed] [Google Scholar]