

Abstract
The autoimmune enteropathy, coeliac disease (CD), is triggered by ingestion of gluten-containing grains. We recently reported that the chemokine receptor CXCR3 serves as a receptor for specific gliadin peptides that cause zonulin release and subsequent increase in intestinal permeability. To explore the role of CXCR3 in the immune response to gliadin, peripheral blood mononuclear cells from both patients with CD and healthy controls were incubated with either pepsin-trypsin-digested gliadin or 11 α-gliadin synthetic peptides in the presence or absence of a blocking anti-CXCR3 monoclonal antibody. Supernatants were analysed for interleukin-6 (IL-6), IL-8, IL-10, IL-13, IP-10 (CXCL10), tumour necrosis factor-α and interferon-γ. Gliadin broadly induced cytokine production irrespective of the clinical condition. However, IL-8 production occurred only in a subgroup of individuals and cells of the phagocytic lineage were the main source. Induction of IL-8 was reproduced by one of a comprehensive panel of synthetic α-gliadin peptides and was abrogated when CXCR3 was blocked before stimulation with either gliadin or this peptide in the CD group but not in the control group, suggesting that gliadin-induced IL-8 production was CXCR3-dependent gliadin induced IL-8 production only in CD.
Keywords: coeliac disease, CXCR3, gliadin, interleukin-8, peripheral blood mononuclear cells
Introduction
A compromised barrier function, in addition to genetic predisposition and disproportional immune reactivity to gluten, is a prerequisite to the pathogenesis of the autoimmune enteropathy coeliac disease (CD).1 We have observed that gliadin, the immunogenic component of gluten, induces an increase in intestinal permeability that is small and transient in non-CD intestinal tissue, but large and persistent over time in CD intestinal tissue.2 We have recently shown that gliadin-induced tight junction disassembly is caused by binding of gliadin to the chemokine receptor CXCR3 and subsequent zonulin release.3 CXCR3 is expressed more abundantly at the intestinal level (i.e. epithelium and lamina propria) in patients with CD than in non-CD individuals.3
Besides the intestinal epithelium, CXCR3 is widely expressed on immune cells, such as activated T cells,4γδ T cells,5 eosinophils,6 B cells,7 plasma cells8 and plasmacytoid dendritic cells (DC).9 Three chemokines, CXCL9 (Mig), CXCL10 (IP-10) and CXCL11 (I-TAC), bind to different domains of CXCR3 and differentially activate the receptor,10 each inducing distinct biological effects on receptor internalization, chemotaxis and calcium mobilization.10,11 Not surprisingly, we have reported that the CXCR3-dependent effect of gliadin on intestinal epithelial barrier function is not reproduced by CXCL10.3
By breaking down the intestinal barrier, gliadin gains entrance to the lamina propria. Once in the submucosa, its recognition as a foreign antigen will activate the host immune response. Given that gliadin induces a strong cytokine response in both healthy and CD immune cells,12,13 we hypothesized that binding to CXCR3 also mediates the activation of the immune response to gliadin after tight junction disassembly, and that differences may exist in CXCR3-dependent regulation of immunity between non-CD individuals and patients with CD. We show here that gliadin markedly activates immune cells, inducing the production of important pro-inflammatory cytokines in all individuals regardless of the clinical condition. Only some of these effects of gliadin appeared to be mediated by CXCR3 binding. Interestingly, gliadin-induced production of interleukin-8 (IL-8), a chemokine with potent neutrophil chemoattractant and activation properties,14 appeared to be CXCR3-mediated only in patients with CD.
Materials and methods
Donors
Blood was drawn from healthy controls (HC) and from patients with CD in remission. All patients with CD had been previously diagnosed with CD by biopsy and antibody screening, and had been on a gluten-free diet (GFD) for at least 6 months. To ensure compliance to the GFD, all participating patients with CD were screened for the presence of anti-tissue transglutaminase (tTG) and anti-gliadin antibodies (AGA) as previously described.15 These patients with CD in remission are referred to throughout this article as CD-GFD. Independent ethics review board approval was obtained in advance for the protocol, and volunteers gave their informed consent.
Isolation and stimulation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMC) were prepared by density gradient centrifugation. The PBMC (1 × 106/ml) were incubated with pepsin-trypsin-digested (PT-) gliadin (1 mg/ml) or medium alone [RPMI-1640 Dutch Modification; (Sigma, St Louis, MO) complemented with 2 mm l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum (Invitrogen, Carlsbad, CA)] at 37° in a humidified atmosphere of 5% CO2 for 24 hr. In some wells, 10 μg/ml monoclonal anti-human CXCR3 antibody (clone 49801) or its appropriate isotype-matched control (clone 11711; both from R&D Systems, Minneapolis, MN) were added to the cell cultures 30 min before addition of PT-gliadin or medium to the wells.
Different aliquots of PBMC were used to either isolate purified T cells (untouched human T-cell kit; Invitrogen) or deplete monocytes, B cells, myeloid DC or plasmacytoid DC, by capturing the respective cell fractions with monoclonal mouse anti-human CD14 (clone 61D3; eBioscience, San Diego, CA), mouse anti-human CD19 (clone HIB19; BD Biosciences, San Jose, CA), mouse anti-human BDCA1 (clone AD5-8E7; Miltenyi Biotec, Auburn, CA), or mouse anti-human BDCA4 (clone AD5-17F6; Miltenyi Biotec). The PBMC were incubated with FcR blocking reagent (Miltenyi Biotec) before addition of the appropriate antibody. Subsequently, cells were incubated with sheep anti-mouse IgG Dynabeads M-450 and tagged cells were removed with a Dynal magnet (Invitrogen). The remaining cells (1 × 106/ml) were incubated with PT-gliadin or medium alone for 24 hr, after which supernatants were collected for assessment of cytokine levels by Luminex (Cytokine Core Facility, University of Maryland School of Medicine, Baltimore, MD).
Preparation of PT-gliadin and synthetic peptides
Gliadin (Sigma) was dissolved in 0·2 m HCl. Subsequently, the mixture was incubated with pepsin (Sigma) under continuous stirring at room temperature for 18 hr, and, at the end of this incubation, the pH was adjusted to 7·4. The peptic digest was further digested by addition of purified trypsin (Sigma) and left overnight at 37°. The solution was boiled for 30 min, divided into aliquots and lyophilized. The design and synthesis of the α-gliadin peptide library, consisting of 25 overlapping, 20-mer peptides, were described previously.3 To narrow the search for those peptides responsible for CXCR3-dependent IL-8 production, a first set of experiments was designed to test five peptide pools each containing five randomly chosen, different peptides (Table 1). Peptide pools (20 μg peptide/ml) were incubated with PBMC from HC and CD-GFD patients in the presence or absence of a blocking anti-CXCR3 antibody, and supernatants were measured for IL-8 concentrations. Thereafter, the experiments were repeated with individual peptides from the pool(s) closely reproducing PT-gliadin effects (4 μg peptide/ml).
Table 1.
The α-gliadin synthetic peptide library used in this study
| Sequences | Peptide | Pool | CXCL111 |
|---|---|---|---|
| Pepsin-trypsin-digested gliadin | 69 | ||
| MVRVPVPQLQPQNPSQQHPQ | 4012 | 2 | 98 |
| PQNPSQQHPQEQVPLVQQQQ | 4013 | 2 | 112 |
| EQVPLVQQQQFLGQQQSFPP | 4014 | 1 | 90 |
| FLGQQQSFPPQQPYPQPQPF | 4015 | 4 | 96 |
| QQPYPQPQPFPSQQPYLQLQ | 4016 | 5 | 103 |
| PSQQPYLQLQPFPQPQLPYL | 4017 | 1 | 94 |
| PFPQPQLPYLQPQPFRPQQP | 4018 | 4 | 93 |
| QPQPFRPQQPYPQPQPQYSQ | 4019 | 5 | 99 |
| YPQPQPQYSQPQQPISQQQQ | 4020 | 1 | 112 |
| PQQPISQQQQQQQQQQQQQQ | 4021 | 4 | 86 |
| QQQQQQQQQQQQILQQILQQ | 4022 | 5 | 55 |
| QLIPCMDVVLQQHNIAHGRS | 4024 | 5 | 116 |
| QQHNIAHGRSQVLQQSTYQL | 4025 | 2 | 106 |
| QVLQQSTYQLLQELCCQHLW | 4026 | 1 | 36 |
| LQELCCQHLWQIPEQSQCQA | 4027 | 3 | 90 |
| QIPEQSQCQAIHNVVHAIIL | 4028 | 3 | 76 |
| IHNVVHAIILHQQQKQQQQP | 4029 | 1 | 93 |
| HQQQKQQQQPSSQVSFQQPL | 4030 | 4 | 126 |
| SSQVSFQQPLQQYPLGQGSF | 4031 | 3 | 100 |
| QQYPLGQGSFRPSQQNPLAQ | 4032 | 3 | 100 |
| RPSQQNPLAQGSVQPQQLPQ | 4033 | 3 | 108 |
| GSVQPQQLPQFEEIRNLALQ | 4034 | 2 | 95 |
| FEEIRNLALQTLPAMCNVYI | 4035 | 5 | 109 |
| TLPAMCNVYIPPYCTIVPFG | 4036 | 2 | 110 |
| PPYCTIVPFGIFGTNYR | 4037 | 4 | 108 |
Competitive binding assays performed in our previous study,3 using 125I-labelled CXCL11 and competitors (pepsin-trypsin-digested-gliadin or peptides) on CXCR3-expressing membranes from CHO-K1 cell lines. Data are in % binding of CXCL11 to the cell membranes in the presence of competitors; values below 100% indicate displacement of CXCL11 by its competitors. More details on the procedure are described in Ref. 3.
Statistical analysis
Values, given in median (interquartile range), were analysed for statistical significance using two-tailed Mann–Whitney U-test. Where appropriate, a paired Wilcoxon test was performed as indicated. Data on PBMC and subset-depleted parallel cultures were analysed with the Kruskal–Wallis test and the Dunn's multiple comparison test. Differences were considered statistically significant at P < 0·05.
Results
CD patient population
All patients with CD were known to have normal serum IgA levels and had both anti-tTG and AGA IgG and IgA titres within normal limits, with the exception of one patient who had a slightly elevated AGA IgG titre (19·07 EU). Therefore, these patients were all in remission.
PT-gliadin is a potent stimulus for cytokine production by PBMC
Production of interferon-γ (IFN-γ), tumour necrosis factor-α (TNF-α), IL-6, IL-8, IL-10, IL-13 and CXCL10 was assessed in supernatants of PBMC from HC (n = 10) and CD-GFD patients (n = 7) cultured with PT-gliadin or medium alone. The results shown in Fig. 1 indicate that PT-gliadin is a potent inducer of cytokine production in PBMC from both HC and CD-GFD patients. Three cytokines, IL-6, IFN-γ and IL-13, were induced at significantly higher levels in CD-GFD patients compared with HC. Interleukin-6 and IFN-γ were produced at 50 (38·8–106) ng/106 cells and 10·3 (9·1–78) pg/106 cells in CD-GFD patients versus 16·6 (7·1–43·9) ng/106 cells (P < 0·05) and 4·1 (0·1–9·7) pg/106 cells (P < 0·05) in HC, respectively. Interleukin-13 was produced at very low concentrations, but significantly higher in CD-GFD patients than in HC, that is, respectively, 8·8 (6·2–9·9) pg/106 cells versus 0·7 (0·1–2·3) pg/106 cells (P < 0·05). Production of TNF-α, IL-8 and IL-10 tended to be higher in CD-GFD patients compared with HC, but without reaching significance; TNF-α, IL-8 and IL-10 were produced at 2213·4 (1933–3327) pg/106 cells, 109·34 (2·5–199·3) ng/106 cells and 1893·4 (1320–2347) pg/106 cells in CD-GFD patients versus 1255 (1060–2545) pg/106 cells (NS), 2·8 (2·6–79·8) ng/106 cells (NS) and 1440 (768–1768) pg/106 cells in HC (NS), respectively. It is important to note, though, that IL-8 production was induced only in a subgroup of individuals, namely 30% of HC and 57% of CD-GFD patients. CXCL10 was not detected in culture supernatants of PBMC from either HC or CD-GFD patients in response to PT-gliadin (data not shown).
Figure 1.
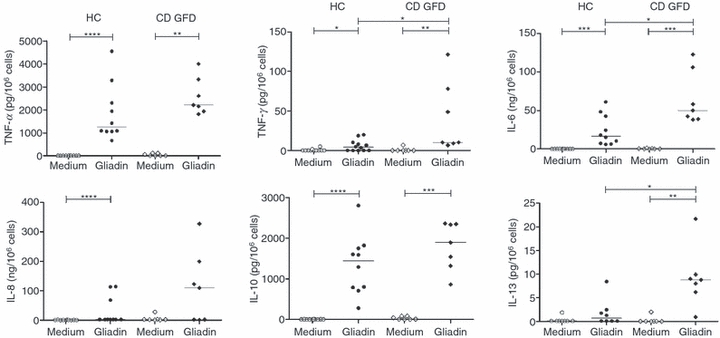
Pepsin-trypsin-digested (PT-) gliadin is a potent stimulus for cytokine production by peripheral blood monoonuclear cells (PBMC). PBMC from healthy controls (HC) and patients with coeliac disease on a gluten-free diet (CD-GFD) were incubated with medium alone or PT-gliadin (1 mg/ml) for 24 hr. Supernatants were assayed for interleukin-6 (IL-6), IL-8, IL-10, tumour necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and IL-13 production as indicated. Each dot represents a single donor. Horizontal lines are drawn at the median value in each group. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Cytokine production is CXCR3-dependent in PBMC from a subgroup of CD-GFD but not HC
We have previously shown that gliadin can increase zonulin release and intestinal permeability via binding to the chemokine receptor, CXCR3.3 To investigate whether CXCR3 is also involved in PT-gliadin-induced cytokine production in PBMC, cells from HC and CD-GFD patients were pre-incubated with a CXCR3-blocking monoclonal antibody or an isotype control for 30 min, followed by stimulation with PT-gliadin for 24 hr. We found that CXCR3 was not involved in PT-gliadin-induced cytokine production in PBMC from HC (Fig. 2a), but, interestingly, appeared to be involved in PT-gliadin-induced IL-8 and IL-6 production in cells from CD-GFD patients (Fig. 2b). Most strikingly, IL-8 production in PBMC from CD-GFD patients, but not HC, was almost completely abrogated upon CXCR3 blockade, corresponding to a reduction by 98·3 ± 0·4% of the levels detected in the presence of control antibody (P < 0·05). After blocking of CXCR3, PBMC from a subgroup of CD-GFD patients produced lower levels of IL-6 in response to PT-gliadin, accounting for an average reduction of IL-6 concentrations by 32·7 ± 12·1% compared with levels detected in PBMC that were not pre-treated with anti-CXCR3 (P < 0·05) (Fig. 2b). The PT-gliadin-induced production of IL-10, IL-13, TNF-α and IFN-γ was not significantly affected by pre-incubation of PBMC from CD-GFD patients or HC with anti-CXCR3 antibody compared with isotype control-treated PBMC (NS) (Fig. 2b).
Figure 2.
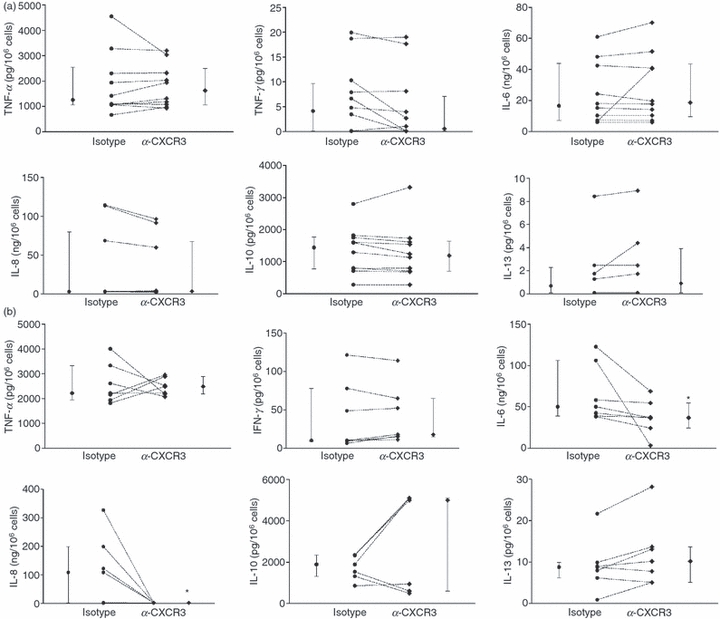
Pepsin-trypsin-digested (PT-) gliadin-induced production of interleukin-8 (IL-8) is CXCR3-dependent in peripheral blood mononuclear cells (PBMC) from patients with coeliac disease fed a gluten-free diet (CD-GFD). The PBMC were pre-incubated for 30 min with a blocking anti-human CXCR3 antibody (10 μg/ml), followed by incubation with medium alone or PT-gliadin (1 mg/ml) for 24 hr. Supernatants were assayed for IL-6, IL-8, IL-10, tumour necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and IL-13 concentrations. (a) Blocking of the chemokine receptor, CXCR3, did not change PT-gliadin-induced cytokine production in PBMC from healthy controls (HC), whereas (b) blocking of CXCR3 affected greatly the PT-gliadin-induced IL-8 and, to a much lesser extent, IL-6 production in PBMC from CD-GFD patients, while IL-10, IL-13, TNF-α and IFN-γ production was unchanged. Each dot represents a single donor. Medians and interquartile ranges for each group of donors are also shown. *P < 0·05 relative to isotype control.
Identification of α-gliadin motif responsible for the production of IL-8 in PBMC
In an effort to identify the α-gliadin domains responsible for IL-8 production and its association with CXCR3 binding, peptides from the synthetic library were divided into five randomly chosen pools (Table 1) and incubated with PBMC from CD-GFD patients or HC subjects for 24 hr. Two of these peptides, QVLQQSTYQLLQELCCQHLW and QQQQQQQQQQQQILQQILQQ, which had been previously found to bind to CXCR3 in an I-TAC/CXCL11 displacement assay,3 were included in pool 1 and pool 5, respectively. Results showed that peptides within pool 5, and to some extent pool 4, triggered CXCR3-dependent IL-8 production in CD-GFD patients. In the attempt to identify the specific peptide(s) within the two active pools triggering an IL-8 response, individual peptides from pool 4 and pool 5 were subjected to further testing. Additionally, given that the effects of individual peptides may be masked in complex polypeptide pools, we also tested the CXCR3-binding peptide included in the unresponsive pool 1. These experiments revealed that peptide 4037, PPYCTIVPFGIFGTNYR from pool 4, corresponding to aa 270–286 of α-gliadin, induced IL-8 production in six of the 17 CD-GFD patients (35%) and in five of 16 HC (31%), with net production amounting to 61 (30·3–93·4) ng/106 cells and 34·3 (30·9–35·5) ng/106 cells, respectively. In analogy to the effect of PT-gliadin, pre-treatment with anti-CXCR3 antibodies decreased IL-8 production induced by the peptide in PBMC from CD-GFD patients (P < 0·05), but it did not affect IL-8 production in PBMC from HC (NS) (Fig. 3; for the effects of the individual peptides tested in these experiments, see supplementary material Fig. S1).
Figure 3.
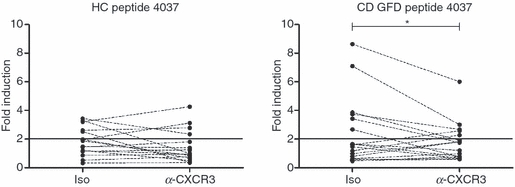
Production of interleukin-8 (IL-8) in response to a comprehensive panel of α-gliadin peptides. Eleven peptides from the α-gliadin synthetic peptide library were separately tested as outlined in the Materials and methods section to identify the sequence(s) responsible for IL-8 induction by peripheral blood mononuclear cells from 16 healthy controls (HC) and 17 patients with coeliac disease fed a gluten-free diet (CD GFD). The effect of peptide 4037, corresponding to aa 270–286 of α-gliadin, is shown. Each dot corresponds to a single donor. *P < 0·05 relative to isotype control (iso) from responders (six out of 17 patients). The IL-8 response to the entire set of tested peptides is shown in Fig. S1.
Cellular source of gliadin-induced IL-8 production
We asked whether the observed differences in CXCR3 involvement in PT-gliadin-induced IL-8 production in PBMC from HC and CD-GFD patients were the result of the interaction with distinct cellular sources of IL-8. To elucidate this point, PBMC from six HC and three CD-GFD patients, previously identified as ‘IL-8 responders’, were subjected to selective depletion of monocytes, B cells or myeloid or plasmacytoid DC, or to T-cell separation as indicated in the Materials and methods section. Statistical analysis showed that in HC, relative to unfractionated PBMC [37·5 (36·3–47·9) ng/106 cells; set as 100%], PT-gliadin-induced IL-8 production was significantly decreased in suspensions depleted of distinct cell subsets (P < 0·01). Dunn's multiple comparison test further identified the suspensions deprived of monocytes or BDCA4+-plasmacytoid DC as those exhibiting a significant decrease in IL-8 production to respective values of 52·4 (28·6–82·6) % (P < 0·05) or 35·3 (24·9–69·8) % (P < 0·05), in contrast to suspensions deprived of B cells [96·6 (83–116) % (NS)] or BDCA1+-myeloid DC [86 (80·1–95·5) % (NS)] (Fig. 4). A similar pattern was observed in PBMC from CD-GFD patients (P < 0·05), in which cultures deprived of monocytes or BDCA4+-plasmacytoid DC produced, respectively, 3·4 (2·7–59·9) % and 29·7 (22·9–60·7) % of levels detected in unfractionated PBMC. As seen in HC, IL-8 production was not affected, relative to unfractionated PBMC [165·2 (126·3–178·5) ng/106 cells; set as 100%], in cultures deprived of B cells [69·9 (67·6–97·2) %, NS] or BDCA1+-myeloid DC [150·1 (46·9–177·9) %, NS]. As a result of the small number of observations, Dunn's multiple comparison test did not give P-values for distinct suspensions from CD-GFD. Purified T cells from HC or CD-GFD did not produce IL-8 in response to PT-gliadin (not shown), which ruled out the contribution of this lineage to IL-8 production, and cytokine production in general, in the context of a non-adaptive response to this molecule. Altogether, these data indicate that cells from the phagocytic lineage, i.e. the CD14+ and BDCA4+ subsets, were required for gliadin-induced IL-8 production in both HC and CD-GFD patients. Production of IL-6, IL-10 and TNF-α was also associated with these two main PBMC subsets (data not shown).
Figure 4.
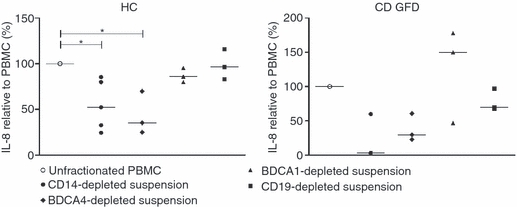
Identification of monocytes and plasmacytoid dendritic cells as cellular sources of gliadin-induced interleukin-8 (IL-8) production. Peripheral blood mononuclear cells (PBMC) from healthy controls (HC) and patients with coeliac disease fed a gluten-free diet (CD-GFD) were subjected to depletion of monocytes (CD14+), B cells (CD19+), myeloid (BDCA1+) or plasmacytoid (BDCA4+) dendritic cells as indicated, or untouched T cells were separated as indicated in the Materials and methods. All fractions were incubated in the presence of medium alone (unstimulated) or pepsin-trypsin-digested (PT-) gliadin (1 mg/ml) for 24 hr. Supernatants were assayed for IL-8. Each dot corresponds to a single donor. Median values are indicated as horizontal lines. Kruskal–Wallis test comparing all suspensions in each group of subjects showed significant differences both in HC (P < 0·01) and CD GFD (P < 0·05). *P < 0·05 at the Dunn's multiple comparison post-test.
Discussion
Gliadin is a complex glycoprotein present in gluten-containing grains, wheat, rye and barley.16 Gliadin exists in three variants, α-, γ- and ω-gliadin, the α-gliadin variant being most prevalent.17α-Gliadin is a multi-functional, complex protein that contains multiple motifs which account for its reported cytotoxic, immunogenic and intestinal permeability effects via, respectively, a 13-mer peptide (aa 31–43),18,19 a 33-mer peptide (aa 57–89),20 and two 20-mer CXCR3-binding peptides [QVLQQSTYQLLQELCCQHLW (aa 120–140) and QQQQQQQQQQQQILQQILQQ (aa 160–180)].3 In this study, we identify another 17-mer peptide, PPYCTIVPFGIFGTNYR (aa 270–286), responsible for CXCR3-dependent production of IL-8 in CD. This observation adds to the multiple functions of α-gliadin mentioned above, and provides a mechanistic explanation for its reported capability to induce a rapid and sustained neutrophil influx in experimental models of gliadin challenge in CD patients in remission,21–23 a phenomenon possibly mediated by the production of a potent neutrophil chemoattractant and activator such as IL-8.
We show here, in line with previous studies,12,13 that gliadin, the environmental trigger for CD, is capable of inducing prominent pro-inflammatory cytokine and chemokine production by immune cells not only in CD-GFD patients but also in HC. Our data showing PT-gliadin-induced production of IL-8, a potent neutrophil chemoattractant and activator, are unexpected. First, we show that PT-gliadin induces production of IL-8 only in a subpopulation of individuals, regardless of clinical condition, and at higher median levels in CD-GFD patients compared with HC, albeit without reaching statistical significance. In contrast, the other cytokines tested were induced in all individuals, and in the case of IFN-γ and IL-6, at significantly higher levels in CD-GFD patients than HC. Second, PT-gliadin-induced IL-8 production appeared to depend on CXCR3 engagement in all responder CD-GFD patients but not in HC. These data suggest that IL-8 production in response to PT-gliadin is regulated differently between responsive CD-GFD patients and HC. Third, we show that cells of the mononuclear phagocyte system, i.e. monocytes and BDCA4+ plasmacytoid DC, were responsible for IL-8 production in response to PT-gliadin both in CD-GFD patients and HC. This finding rules out the idea that the differential involvement of CXCR3 in CD is the result of the activation of distinct cellular sources of IL-8.
CXCR3 is involved in the early mucosal steps of the response to gliadin (i.e. increased intestinal permeability), which set the stage for the subsequent cascade of events, including neutrophil recruitment,22,23 IL-15 production,24 intraepithelial lymphocyte recruitment25 and, finally, activation of the adaptive immune response eventually leading to autoimmunity.26 It is noteworthy that, among HC and CD-GFD patients, those individuals who were ‘IL-8 responders’ showed consistent responses to gliadin upon repeated sampling (data not shown), indicating that IL-8 responsiveness is an intrinsic feature of PBMC from these individuals, and only minimally affected by experimental and environmental variables.
Using a pool-wise approach, we tested 25 synthetic peptides spanning the α-gliadin sequence (Table 1), 11 of which were individually selected for further examination. This allowed us to identify aa 270–286 (peptide 4037) as an immunomodulatory gliadin domain responsible for CXCR3-dependent production of IL-8 in a subgroup of CD-GFD patients, but not in HC. The fact that none of the other tested peptides showed similar effects, including the two CXCR3-binding peptides that we previously identified as zonulin-releasing peptides,3 suggests a distinct specificity of this immunomodulatory effect. However, as complex peptide pools can enhance or mask the activity of individual peptides, we cannot exclude that peptide(s) within the non-responsive pools, hence not selected for further examination, might contribute to IL-8 induction in our model. Intriguingly, the peptide identified as inducing CXCR3-dependent IL-8 production does not appear to bind to CXCR3 according to our previously described CXCR3 binding assay using radiolabelled CXCL11.3 Given that peptide 4037 showed CXCR3-mediated IL-8 production only in a subgroup of patients with CD but not HC, we concluded that the most likely explanation is that the patients carry a structural/conformational change in CXCR3 that allows gliadin peptide 4037, that did not show binding activity to the conventional receptor3, to bind with higher affinity to the modified receptor. Alternatively, it can be speculated that, similar to other CXCR3 ligands, CXCL9 and CXCL10,10 this peptide may use a different domain of CXCR3 for its function, and so would be unable to compete with CXCL11 in a conventional CXCR3 binding assay.
The data presented here suggest that there are substantial differences in the mechanisms of IL-8 production in non-CD individuals and patients with CD, which involve distinct signalling pathways downstream of the chemokine receptor, CXCR3. Interleukin-8 is rapidly released in the early phase of inflammation,27 is produced by a broad variety of cells, including epithelial cells,28,29 monocytes and macrophages,30 T cells,31 and endothelial cells,32 and is tightly regulated at the transcriptional and post-transcriptional levels.33 That IL-8 may play a crucial role in gliadin-induced disease is suggested in a number of studies showing rapid neutrophil recruitment to the gut mucosa of CD-GFD patients exposed to gliadin,22,23 and significantly elevated numbers of mucosal neutrophils even at baseline in CD-GFD patients.21 This might be associated with, and accounted for, by persistently elevated zonulin levels and intestinal permeability, which have indeed been shown to remain elevated in CD-GFD patients.2,34
Specific PT-gliadin peptides can also be presented by HLA molecules on antigen-presenting cells in both patients with CD and HC. This has been shown to be the prevailing mechanism for gliadin-induced production of the pro-inflammatory cytokines, IL-1 and TNF-α, and for CXCR3-independent production of other cytokines in the cascade, such as IL-6 and IL-8. The present findings showing inhibition of IL-8 production after blocking of the CXCR3 receptor in CD-GFD patients but not non-CD individuals, suggest a peculiar mechanism of IL-8 regulation in CD. Possible explanations include (i) structural and functional differences in the CXCR3 receptor in CD-GFD patients relative to HC, at least in selected lineages, (ii) deviant signalling downstream of CXCR3, and (iii) the modulation of CXCR3 activity through a mechanism of receptor dimerization and cross-talk, as previously suggested for distinct (patho-)physiological processes.35 Structural diversities by alternative splicing have been reported for CXCR3, splice variants CXCR3-alt36 and CXCR3-B37,38 being associated with differential responses to CXCR3 ligands. In particular, CXCR3-alt surface expression is impaired compared with full-length CXCR3, and although CXCR3-alt-expressing cells show defective chemotaxis to CXCR3 ligands, proximal G coupling is still conserved. Hence, in this case the structural alterations of CXCR3 are associated with a decreased level of expression and impaired, but not abrogated, physiological function. Splice variant CXCR3-B is structurally different from CXCR3 in the N-terminal region. In this case, the receptors have opposite biological activities, suggesting that they trigger different signal transduction pathways.35 The antibody used for CXCR3 blocking in this and other studies most probably recognizes both forms (R&D Systems, technical service, personal communication), but we cannot exclude possible, yet unreported, differences in binding affinity for either molecular form of the receptor.37 With respect to the possible regulation of CXCR3 activity through receptor dimerization and cross-talk, engagement of CXCR4 by its ligand, CXCL12, has been shown to be essential for plasmacytoid DC to gain CXCR3 responsiveness.39 We can hypothesize that CXCR3-dependent IL-8 production in CD might involve the association of CXCR3 with receptors and signalling pathways that skew the response to specific ligands, in this case gliadin, to a different biological outcome. Whether CXCR3-dependent IL-8 production is associated with CD-linked mutations in the CXCR3 gene sequence, which in turn might affect its dimerization with an as yet unidentified co-receptor(s) and/or cause differential signalling, is under investigation.
In conclusion, our findings demonstrate that in addition to its reported cytotoxic,18,19 immunogenic20 and intestinal permeabilizing activities,3 PT-gliadin is a potent agonist of IL-8 and other cytokine production irrespective of the clinical condition. Unlike other cytokines, IL-8 is only induced by PT-gliadin in a responder subgroup, a pattern that appears to be more frequent among CD-GFD patients than individuals without CD. Furthermore, only in CD-GFD patients, but not in non-CD individuals, is IL-8 induction by PT-gliadin mediated by CXCR3 engagement. By testing a synthetic library spanning the α-gliadin sequence, we have identified a peptide, PPYCTIVPFGIFGTNYR, that fully reproduces this effect. Given the abundant mucosal CXCR3 expression in patients with CD,3 the data presented here may explain the reported increased neutrophil influx in CD and may point towards a substantial role of IL-8 in disease pathogenesis in at least a subgroup of patients. Further investigation is required to elucidate the differential involvement of CXCR3 observed in this study.
Acknowledgments
We are indebted to volunteers, with and without coeliac disease, for their participation in the study. This work was supported by the Center for Celiac Research, University of Maryland School of Medicine, Baltimore, MD.
Disclosures
K.M.L., S.K., F.Ch., D.K., E.L.P. and V.C. have no relationships to declare. A.F. is a stock holder of ALBA Therapeutics.
Supporting information
Additional supporting information may be found in the online version of this article:
Figure S1. Production of IL-8 in response to eleven peptides from the alpha-gliadin synthetic peptide library that were tested to identify the sequence(s) responsible for IL-8 production by PBMC from healthy donors (n = 16) and CD-GFD (n = 17). *P < 0.05 relative to isotype control from responders.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:416–22. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 2.Drago S, El Asmar R, Di Pierro M, et al. Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–19. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 3.Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–04. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–34. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poggi A, Catellani S, Fenoglio D, Borsellino G, Battistini L, Zocchi MR. Adhesion molecules and kinases involved in γδ T cells migratory pathways: implications for viral and autoimmune diseases. Curr Med Chem. 2007;14:3166–70. doi: 10.2174/092986707782793835. [DOI] [PubMed] [Google Scholar]
- 6.Katoh S, Fukushima K, Matsumoto N, Ehara N, Matsumoto K, Yamauchi A, Hirashima M. Accumulation of CXCR3-expressing eosinophils and increased concentration of its ligands (IP10 and Mig) in bronchoalveolar lavage fluid of patients with chronic eosinophilic pneumonia. Int Arch Allergy Immunol. 2005;137:229–35. doi: 10.1159/000086335. [DOI] [PubMed] [Google Scholar]
- 7.Johansson C, Ahlstedt I, Furubacka S, Johnsson E, Agace WW, Quiding-Jarbrink M. Differential expression of chemokine receptors on human IgA+ and IgG+ B cells. Clin Exp Immunol. 2005;141:279–87. doi: 10.1111/j.1365-2249.2005.02843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol. 2003;3:822–9. doi: 10.1038/nri1203. [DOI] [PubMed] [Google Scholar]
- 9.Penna G, Vulcano M, Sozzani S, Adorini L. Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum Immunol. 2002;63:1164–71. doi: 10.1016/s0198-8859(02)00755-3. [DOI] [PubMed] [Google Scholar]
- 10.Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–27. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- 11.Colvin RA, Campanella GS, Manice LA, Luster AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26:5838–49. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palova-Jelinkova L, Rozkova D, Pecharova B, Bartova J, Sediva A, Tlaskalova-Hogenova H, Spisek R, Tuckova L. Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J Immunol. 2005;175:7038–45. doi: 10.4049/jimmunol.175.10.7038. [DOI] [PubMed] [Google Scholar]
- 13.Thomas KE, Sapone A, Fasano A, Vogel SN. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in celiac disease. J Immunol. 2006;176:2512–21. doi: 10.4049/jimmunol.176.4.2512. [DOI] [PubMed] [Google Scholar]
- 14.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84:1045–9. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch Intern Med. 2003;163:286–92. doi: 10.1001/archinte.163.3.286. [DOI] [PubMed] [Google Scholar]
- 16.Fasano A. Celiac disease – how to handle a clinical chameleon. N Engl J Med. 2003;348:2568–70. doi: 10.1056/NEJMe030050. [DOI] [PubMed] [Google Scholar]
- 17.Bernardin JE, Kasarda DD, Mecham DK. Preparation and characterization of α-gliadin. J Biol Chem. 1967;242:445–50. [PubMed] [Google Scholar]
- 18.Maiuri L, Troncone R, Mayer M, Coletta S, Picarelli A, De Vincenzi M, Pavone V, Auricchio S. In vitro activities of A-gliadin-related synthetic peptides: damaging effect on the atrophic coeliac mucosa and activation of mucosal immune response in the treated coeliac mucosa. Scand J Gastroenterol. 1996;31:247–53. doi: 10.3109/00365529609004874. [DOI] [PubMed] [Google Scholar]
- 19.Picarelli A, Di Tola M, Sabbatella L, Anania MC, Di Cello T, Greco R, Silano M, De Vincenzi M. 31–43 amino acid sequence of the α-gliadin induces anti-endomysial antibody production during in vitro challenge. Scand J Gastroenterol. 1999;34:1099–02. doi: 10.1080/003655299750024896. [DOI] [PubMed] [Google Scholar]
- 20.Qiao SW, Bergseng E, Molberg O, Xia J, Fleckenstein B, Khosla C, Sollid LM. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol. 2004;173:1757–62. doi: 10.4049/jimmunol.173.3.1757. [DOI] [PubMed] [Google Scholar]
- 21.Diosdado B, van Bakel H, Strengman E, Franke L, van Oort E, Mulder CJ, Wijmenga C, Wapenaar MC. Neutrophil recruitment and barrier impairment in celiac disease: a genomic study. Clin Gastroenterol Hepatol. 2007;5:574–81. doi: 10.1016/j.cgh.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Kristjansson G, Hogman M, Venge P, Hallgren R. Gut mucosal granulocyte activation precedes nitric oxide production: studies in coeliac patients challenged with gluten and corn. Gut. 2005;54:769–74. doi: 10.1136/gut.2004.057174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kristjansson G, Serra J, Loof L, Venge P, Hallgren R. Kinetics of mucosal granulocyte activation after gluten challenge in coeliac disease. Scand J Gastroenterol. 2005;40:662–9. doi: 10.1080/00365520510015566. [DOI] [PubMed] [Google Scholar]
- 24.Maiuri L, Ciacci C, Auricchio S, Brown V, Quaratino S, Londei M. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology. 2000;119:996–06. doi: 10.1053/gast.2000.18149. [DOI] [PubMed] [Google Scholar]
- 25.Biagi F, Luinetti O, Campanella J, Klersy C, Zambelli C, Villanacci V, Lanzini A, Corazza GR. Intraepithelial lymphocytes in the villous tip: do they indicate potential coeliac disease? J Clin Pathol. 2004;57:835–9. doi: 10.1136/jcp.2003.013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sapone A, Lammers KM, Mazzarella G, Mikhailenko I, Carteni M, Casolaro V, Fasano A. Differential mucosal IL-17 expression in two gliadin-induced disorders: gluten sensitivity and the autoimmune enteropathy celiac disease. Int Arch Allergy Immunol. 2010;152:75–80. doi: 10.1159/000260087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sallusto F, Baggiolini M. Chemokines and leukocyte traffic. Nat Immunol. 2008;9:949–52. doi: 10.1038/ni.f.214. [DOI] [PubMed] [Google Scholar]
- 28.Lammers KM, Jansen J, Bijlsma PB, Ceska M, Tytgat GN, Laboisse CL, van Deventer SJ. Polarised interleukin 8 secretion by HT 29/19A cells. Gut. 1994;35:338–42. doi: 10.1136/gut.35.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elner VM, Strieter RM, Elner SG, Baggiolini M, Lindley I, Kunkel SL. Neutrophil chemotactic factor (IL-8) gene expression by cytokine-treated retinal pigment epithelial cells. Am J Pathol. 1990;136:745–50. [PMC free article] [PubMed] [Google Scholar]
- 30.Strieter RM, Chensue SW, Basha MA, Standiford TJ, Lynch JP, Baggiolini M, Kunkel SL. Human alveolar macrophage gene expression of interleukin-8 by tumor necrosis factor-α, lipopolysaccharide, and interleukin-1 beta. Am J Respir Cell Mol Biol. 1990;2:321–6. doi: 10.1165/ajrcmb/2.4.321. [DOI] [PubMed] [Google Scholar]
- 31.Smyth MJ, Zachariae CO, Norihisa Y, Ortaldo JR, Hishinuma A, Matsushima K. IL-8 gene expression and production in human peripheral blood lymphocyte subsets. J Immunol. 1991;146:3815–23. [PubMed] [Google Scholar]
- 32.Sica A, Matsushima K, van Damme J, Wang JM, Polentarutti N, Dejana E, Colotta F, Mantovani A. IL-1 transcriptionally activates the neutrophil chemotactic factor/IL-8 gene in endothelial cells. Immunology. 1990;69:548–53. [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–55. [PubMed] [Google Scholar]
- 34.Duerksen DR, Wilhelm-Boyles C, Veitch R, Kryszak D, Parry DM. A comparison of antibody testing, permeability testing, and zonulin levels with small-bowel biopsy in celiac disease patients on a gluten-free diet. Dig Dis Sci. 2010;55:1026–31. doi: 10.1007/s10620-009-0813-5. [DOI] [PubMed] [Google Scholar]
- 35.Salanga CL, O’Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci. 2009;66:1370–86. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol. 2004;173:6234–40. doi: 10.4049/jimmunol.173.10.6234. [DOI] [PubMed] [Google Scholar]
- 37.Lasagni L, Francalanci M, Annunziato F, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–49. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrai I, Rombouts K, Lasagni L, et al. Activation of p38(MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol. 2008;40:1764–74. doi: 10.1016/j.biocel.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Briere F, Trinchieri G, Caux C. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med. 2003;198:823–30. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.