

Abstract
Tumor suppressor RASSF1A (RAS association domain family 1, isoform A) is known to play an important role in regulation of mitosis; however, little is known about how RASSF1A is regulated during the mitotic phase of the cell cycle. In the present study, we have identified Cullin-4A (CUL4A) as a novel E3 ligase for RASSF1A. Our results demonstrate that DNA damage-binding protein 1 (DDB1) functions as a substrate adaptor that directly interacts with RASSF1A and bridges RASSF1A to the CUL4A E3 ligase complex. Depletion of DDB1 also diminishes intracellular interactions between RASSF1A and CUL4A. Our results also show that RASSF1A interacts with DDB1 via a region containing amino acids 165–200, and deletion of this region abolishes RASSF1A and DDB1 interactions. We have found that CUL4A depletion results in increased levels of RASSF1A protein due to increased half-life; whereas overexpression of CUL4A and DDB1 markedly enhances RASSF1A protein ubiquitination resulting in reduced RASSF1A levels. We further show that CUL4A-mediated RASSF1A degradation occurs during mitosis, and depletion of CUL4A markedly reverses mitotic-phase-stimulated RASSF1A degradation. We also note that overexpression of CUL4A antagonizes the ability of RASSF1A to induce M-phase cell cycle arrest. Thus, our present study demonstrates that the CUL4A·DDB1 E3 complex is important for regulation of RASSF1A during mitosis, and it may contribute to inactivation of RASSF1A and promoting cell cycle progression.
Keywords: Cell Cycle, E3 Ubiquitin Ligase, Mitosis, Tumor Suppressor, Ubiquitination, Cul4A, DDB1, RASSF1A
Introduction
RASSF1A is a recently identified important tumor suppressor (1, 2). It is the major transcript of the seven alternatively spliced variants of the RASSF1 gene, including isoforms A–G (1, 2). Loss or altered expression of RASSF1A by either homozygous or heterozygous deletions of the RASSF1 gene (1) or by hypermethylation of the RASSF1 gene promoter (2–7) has been associated with the pathogenesis of a variety of malignancies. Restoration of RASSF1A expression has been shown to suppress tumorigenic growth, both in vitro and in vivo (2, 3, 8–10). Although the in-depth molecular mechanisms by which RASSF1A functions as a tumor suppressor remain to be elucidated, recent evidence indicates that cell cycle regulation is an important aspect of its function. A number of recent studies including ours have shown that RASSF1A induces cell cycle arrest in both G1/S- and mitotic (M)-phases (8, 11–16). A number of mechanisms have been identified for its action on cell cycle control, for example, RASSF1A-mediated G1/S cell cycle arrest has been associated with the inhibition of cyclin D1 accumulating in the nucleus (15) and M-phase of cell cycle regulation is linked to the ability of RASSF1A to modulate the activity of anaphase-promoting complex (16) and microtubule dynamics (8, 11–14).
Despite a decade-long investigation establishing RASSF1A as an important tumor suppressor that plays a crucial role in cell growth control and apoptosis, little is known about its regulation at the protein level. Recently, we identified the first kinase, the mitotic kinase Aurora-A, for RASSF1A; Aurora-A phosphorylates and modulates the ability of RASSF1A to associate with microtubule during mitosis (17). Since then, a number of RASSF1A kinases have also been identified, including cyclin-dependent kinase-4 (18), PKC (19), MST1 (macrophage stimulating-1) (20), and Aurora-B (21), which have diverse effects on RASSF1A function. Clearly, more studies are needed to further investigate the regulation of this important tumor suppressor protein.
CUL4A E3 ligase is a member of the cullin E3 ligase family (22). CUL4A serves as a scaffold protein to form a CUL4A-DDB1-RING complex and regulates several cellular pathways by targeting a variety of proteins for ubiquitination and degradation. The C terminus of CUL4A interacts with RING protein, which in turn recruits the E2 (22, 23). The N terminus of CUL4A interacts with the substrate adaptor DDB1, which either directly interacts with a substrate or indirectly recruits a substrate through a secondary adaptor. The CUL4A E3 ligase brings the E2 and substrate in close proximity where ubiquitin could be transferred from the E2 to the substrate (22, 23). Some of the known CUL4A substrates include DDB2 (24), Cdt1 (25, 26), HOXA9 (22, 23), and c-Jun (27), and they have a diverse cellular functions such as DNA repair and replication, cell differentiation, and transcription regulation (23). Studies have also shown that the CUL4A gene is amplified or overexpressed in a subset of breast cancers and hepatocellular carcinomas (28, 29). Recent reports suggest that CUL4A is implicated in ubiquitin-mediated degradation of several cell cycle regulators, such as the CDK inhibitor p27 (30–32). These studies suggest that overexpression of CUL4A may contribute to dysregulation of cell cycle control in human cancers.
In this study, we have identified CUL4A as a novel RASSF1A E3 ubiquitin ligase. Our studies show that RASSF1A is ubiquitinated and degraded via proteasome-dependent mechanism by the CUL4A·DDB1 E3 ligase complex. We have also provided evidence that CUL4A-mediated RASSF1A ubiquitination occurs during mitotic phase of the cell cycle, and CUL4A suppresses the ability of RASSF1A to induce M-phase arrest. Thus, our studies suggest that CUL4A is important in regulation RASSF1A during M-phase of the cell cycle.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Recombinant RASSF1A proteins with an S-tag were expressed and purified from a pET32b vector expression system according to the instructions by the manufacturer (Novagen, Madison, WI). Mammalian HA- and S-tagged RASSF1A vector (pSRα-HA-S-F1A) was generated by inserting a DNA fragment that contains RASSF1A cDNA downstream to an HA-S-tag sequence into the pSRα vector. It is of note that both HA and S tags are in the same vector. GFP- and Myc-tagged RASSF1A mammalian expression vectors have been described in our previous studies (8). The RASSF1A deletion variants were generated with the QuikChange XL system (Stratagene, La Jolla, CA) using the corresponding full-length RASSF1A vectors as templates. CUL4A and DDB1 expression vectors were kindly provided by Dr. Tomoki Chiba (Tokyo Metropolitan Institute of Medical Science) and Dr. Strubin (University Medical Center, Geneva, Switzerland), respectively. The FLAG-tagged ubiquitin expression vector was described previously (33). DDB1 recombinant proteins were a kind gift from Dr. Ning Zhang (University of Washington).
Antibodies and Reagents
The following antibodies were used: DDB1 (clone 8, BD Transduction Laboratories, Lexington, KY), β-actin (clone AC-15, Sigma-Aldrich), CUL4A (catalog no. A300-739A, Bethyl Laboratories, Inc. Montgomery, TX), FLAG (M2 antibody, Sigma-Aldrich), GFP (clones 7.1 and 8.3, Roche Applied Science), HA (3F10, Roche Applied Science), Myc (antibody 9E10, Santa Cruz Biotechnology, Santa Cruz, CA). Two different sources of RASSF1A antibodies were used; one from eBioscience, Inc. (San Diego, CA; used for immunoprecipitation), and another was generated from our previous studies (8) and used for Western blotting. Reagents, including nocodazole, thymidine, polybrene, and puromycin were purchased from Sigma-Aldrich, Inc.
Cell Culture Conditions
Human cell lines MCF-7 (breast cancer), HeLa (cervical cancer), and HEK293T were regularly maintained in DMEM with supplementation of 10% fetal bovine serum. NIH mouse fibroblast 3T3 cells were regularly maintained in DMEM with supplementation of 10% bovine serum. All transfections were performed using Lipofectamine 2000 reagent (Invitrogen) per the manufacturer's protocol.
Cell Synchronization
To synchronize cells in mitotic phase, we used thymidine and nocodazole double-block approach as described previously (17). Briefly, Hela cells grown at 50% confluency were first synchronized by incubation with 1 mm thymidine for 19 h and then released in fresh medium for 7 h before 0.1 μg/ml nocodazole was added, and 10 h later, cells were either harvested as mitotic cells or released from the block in fresh medium.
Lentivirus-mediated shRNA Silencing
Scramble shRNA construct (Addgene plasmid 1864) (34) were purchased from Addgene, Inc. (Cambridge, MA). All other shRNA constructs were from Open Biosystems, Inc. (Huntsville, AL). The 21-bp nucleotides targeting human CUL4A used in this study was as follows: 5′-GCAGAACTGATCGCAAAGCAT-3′. The 21-bp nucleotides targeting human DDB1 used in this study was as follows: 5′-CGTGTACTCTATGGTGGAATT-3′. Virus production and infection were performed per Addgene's protocol.
Immunoprecipitations and S-tag Protein Pull Down
Immunoprecipitations were performed as we have described previously (17). For S-tag protein pull down, purified recombinant S-tagged proteins were first immobilized on S-protein beads and then incubated with either whole cell lysates or with purified recombinant DDB1 protein overnight followed by centrifugation and extensive wash to collect the pulldown products. For S-tag protein pull down performed in the mammalian system, HEK293T cells transfected with S-tagged protein expression vectors were lysed in lysis buffer (20 mm HEPES (pH 7.4), 2 mm EGTA, 50 mm β-glycerophosphate, 1 mm DTT, 1 mm sodium orthovanadate, 5 mm sodium fluoride, 1% Triton X-100, 10% glycerol, 10 μg/ml leupeptin, 400 μm PMSF, 10 μg/ml aprotinin, 0.2 μg/ml okadaic acid). Cell lysates were incubated with S-protein-agarose beads overnight; after centrifugation, the bead-bound products were washed extensively and subjected to immunoblot analyses.
Mass Spectrometry Analysis
S-tag pulldown products were separated by SDS-PAGE gel and stained with Bio-Rad silver staining reagent (catalog no. 161-0445). Bands cut from the gel were further digested with Trypsin (Promega, Madison, WI) followed by MALDI-TOF analysis performed by Dana Farber Cancer Center Molecular Biology Core Facilities Service (Boston, MA).
Immunostaining and DAPI Nucleus Staining
Immunostaining was performed as described previously (35). Briefly, cells grown on chamber slides were fixed and labeled with anti-FLAG-tag antibody (Sigma-Aldrich) followed by rhodamine-labeled goat anti-mouse secondary antibodies (Pierce). DAPI nucleus staining was performed as described previously (35).
In Vivo Ubiquitination Assay
HEK293T cells were co-transfected with ubiquitin and other expression vectors. Twenty-four hours after transfection, cells were treated with proteasome inhibitor MG132 (5 μm) overnight and then lysed in lysis buffer. Whole cell lysate was then treated with 1% SDS at 95 °C for 10 min (to disrupt protein interaction) and diluted 10X with the lysis buffer followed by S-tag protein pull down as described previously.
RT-PCR Analysis
RT-PCR analyses were performed as described previously (35).
RESULTS
RASSF1A Interacts with DDB1
We have previously shown that RASSF1A affects M-phase of cell cycle progression by modulating the microtubule dynamic and Aurora-A phosphorylates RASSF1A to influence its ability to associate with microtubules and regulate cell cycle (8, 17). In our continuing effort to investigate the function and regulation of RASSF1A, we performed RASSF1A protein pulldown assay in combination with mass spectrometry analysis to identify novel RASSF1A-interacting proteins. This approach led to the identification of DDB1 as one of the potential RASSF1A interaction partners. Interestingly, we found that DDB1 was reproducibly identified from the RASSF1A pulldown products using the MCF-7 and HEK293T cell lysates as bait (data not shown). Furthermore, β- and α-tubulins, two previously reported RASSF1A-assocaited proteins (8), were also found in the RASSF1A pulldown products.
To validate RASSF1A-DDB1 interactions in cells, we first examined the protein interactions by co-immunoprecipitation assay using the exogenous Myc-tagged RASSF1A. As shown in Fig. 1A, the Myc-tagged RASSF1A pulled down endogenous DDB1 from HEK293T cell lysates (lane 2), whereas Myc tag alone did not (lane 5). Furthermore, DDB1 protein was only detected from the anti-Myc immunoprecipitants (for Myc-tag RASSF1A) (lane 2) but not from anti-IgG immunoprecipitants (lane 3). These results indicate that exogenous RASSF1A interacts with endogenous DDB1. We further investigated the protein interaction of endogenous RASSF1A and DDB1. As shown in Fig. 1B, the endogenous RASSF1A also specifically interacts with endogenous DDB1. These results thus indicate that DDB1 is a novel RASSF1A-interacting protein.
FIGURE 1.
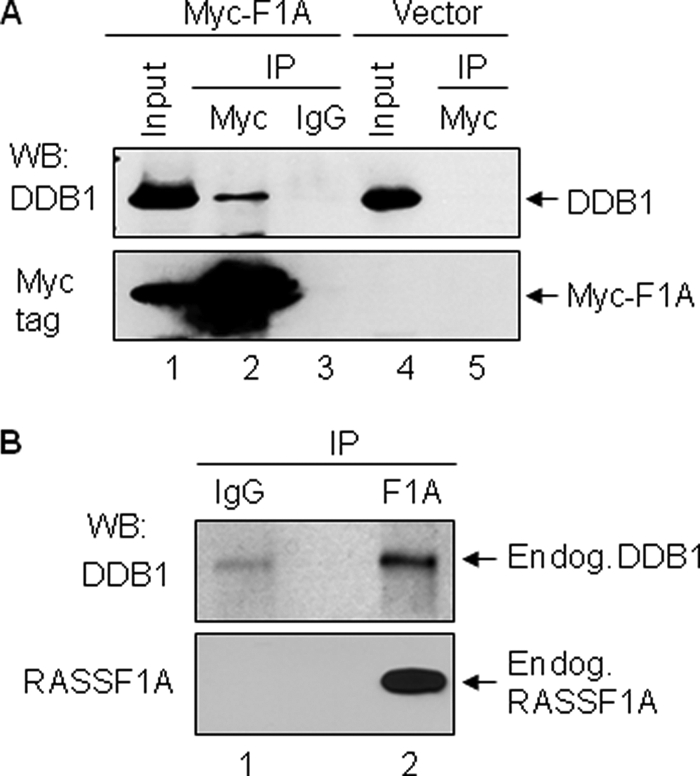
RASSF1A interacts with DDB1 in cells. A, exogenously expressed RASSF1A interacts with endogenous DDB1. HEK293T cells were transfected with either Myc-tagged RASSF1A expression construct (Myc-F1A) or the control vectors. Lysates from the transfectants were subjected to immunoprecipitation (IP) with either Myc tag antibodies or the control IgG as indicated. The immunoprecipitation products and input lysates were analyzed by Western blotting (WB) with antibodies specific to DDB1 and Myc tag. B, endogenous RASSF1A (Endog. RASSF1A) interacts with endogenous DDB1 (Endog.DBB1). HeLa cell lysates were immunoprecipitated with either RASSF1A antibodies (F1A) or the control IgG. Immunoprecipitation products were analyzed by Western blotting with DDB1 and RASSF1A antibodies.
RASSF1A Directly Interacts with DDB1 via Residues 165–200
Next, we investigated the DDB1 interaction domain on RASSF1A. For this purpose, we first generated a set of S-tagged RASSF1A constructs that express recombinant full-length or deletion variants of RASSF1A proteins (Fig. 2A). For each RASSF1A variant, a region of ∼30–60 amino acids harboring a known functional region of the protein was deleted (Fig. 2A). The expression of the recombinant full-length RASSF1A and its deletion variants was confirmed as shown in Fig. 2B (top panel). With the purified recombinant RASSF1A variants, protein pulldown assay was performed to dissect the DDB1-interacting region on RASSF1A. As shown in Fig. 2B, only the recombinant RASSF1AΔ1–50 and RASSF1AΔ165–200 variants did not interact with endogenous DDB1 protein (lanes 2 and 5), whereas all other RASSF1A variants did. In addition, we also used in-cell approach to verify the DDB1 interaction region of RASSF1A. To that end, HEK293T cells were independently transfected with GFP-tagged full-length RASSF1A or Δ165–200 deletion variant RASSF1A or GFP alone expression vectors, and co-immunoprecipitation assays were performed. The RASSF1A Δ1–50 deletion variant could not be similarly used for this approach because the expression levels of this variant were extremely low in cells, which could have been due to enhanced protein instability caused by the deletion of this region. As shown in Fig. 2C, only the full-length RASSF1A was able to co-immunoprecipitate with DDB1 (left panel, lane 4), whereas GFP alone or RASSF1A deletion variant missing residues 165–200 did not show interaction with DDB1 (Fig. 2C, lanes 1 and 2). In addition, we also studied protein interactions of recombinant DDB1 and RASSF1A via protein pulldown assay. Results presented in Fig. 2D indicate that the purified full-length RASSF1A but not the RASSF1A deletion variant (Δ165–200) was able to pull down the recombinant DDB1, indicating (i) a direct interaction between RASSF1A and DDB1 and (ii) residues lying between 165 to 200 of RASSF1A are critical for RASSF1A-DDB1 interactions.
FIGURE 2.

RASSF1A interacts directly with DDB1 via residues 165–200. A, a schematic illustration of recombinant full-length and deletion variants of RASSF1A proteins. The numbers next to each deletion construct indicate the position of the deleted amino acids. The previously identified functional domains are also shown. DAG, diacylglycerol; PEST; proline (P), glutamic acid (E), serine (S), and threonine (T) domain; RA, Ras-association domain. B, recombinant RASSF1A-deleted amino acids 1–50 and 165–200 fail to interact with DDB1 in vitro. Upper panel, Coomassie Blue staining showing the expression of purified recombinant S-tagged full-length (FL) and deletion variants of RASSF1A. Lower panel, detection of RASSF1A-DDB1 interaction in vitro. The recombinant S-tagged RASSF1A proteins (full-length and deletion variants) were incubated with HEK293T cell lysates and then subjected to S-tag protein pulldown. The pulldown products were analyzed by WB using DDB1 antibodies. C, RASSF1A with a 165–200-amino acid deletion fails to interact with DDB1 in cells. HEK293T cells were transfected with either GFP vector-only (lane 1) or GFP-tagged deleted variant of RASSF1A (GFP-F1A-Δ165–200, lanes 2 and 3) or GFP-tagged full-length RASSF1A (GFP-F1A-WT, lanes 4 and 5). Lysates from transfectants were subjected to immunoprecipitation with either DDB1 antibodies or the control IgG as indicated. The immunoprecipitation products (left panel) or lysate input (right panel) were analyzed by WB using GFP or DDB1 antibodies. D, recombinant RASSF1A and DDB1 interact directly in vitro and deletion of amino acids 165–200 of RASSF1A abolishes this interaction. Purified recombinant S-tagged full-length RASSF1A (S-F1A-FL) or deletion variant RASSF1A (S-F1A-Δ165–200) or control S-tag-only peptide (S-tag only) were first incubated with the purified recombinant DDB1 and then subjected to S-tag protein pulldown. The S pulldown products were analyzed by WB using DDB1 antibodies to detect the DDB1-RASSF1A interactions (left panel) or by Coomassie Blue staining to confirm the S-pulldown efficiency (right panel).
RASSF1A Forms a Complex with Ubiquitin E3 Ligase CUL4A via Interaction with DDB1
It has been demonstrated in a number of studies that DDB1 serves as substrate adaptor recruiting CUL4A substrates into the CUL4A·DDB1 E3 complex and facilitating CUL4A-mediated ubiquitination. Next, we sought to investigate whether RASSF1A forms a complex with CUL4A. To this end, HEK293T cells were transfected with HA-S-tag vector alone or HA-S-tag RASSF1A, and cell lysates from the transfectants were then used for S-protein pulldown assay to investigate RASSF1A-CUL4A interactions. As shown in Fig. 3A, HA-S-tag RASSF1A simultaneously pulled down CUL4A and DDB1 (lane 2, upper and middle panel), whereas S-tag alone did not (lane 1). The efficiency of HA-S-tag RASSF1A pull down was verified by anti-HA tag Western blot analysis (lane 2, bottom panel). These results indicate that RASSF1A, DDB1, and CUL4A possibly co-exist in a single complex. To investigate whether RASSF1A interacts with CUL4A endogenously, we performed immunoprecipitations using RASSF1A-specific antibodies or the control IgG followed by anti-CUL4A immunoblotting. Results shown in Fig. 3B indicate that CUL4A was only detected from RASSF1A immunoprecipitants but not from that of control IgG. These results indicate that endogenous RASSF1A also interacts with endogenous CUL4A. To further investigate architecture of the CUL4A·DDB1·RASSF1A complex, we employed a lentivirus shRNA-mediated DDB1 knockdown (KD)3 approach. We reasoned that if RASSF1A indirectly interacts with CUL4A via DDB1, then removal of DDB1 would disrupt the interactions between CUL4A and RASSF1A. Fig. 3C shows that infection of DDB1 shRNA lentivirus in HEK293T cells significantly reduced the expression of DDB1 protein (left top panel). Importantly, our data indicate that RASSF1A and CUL4A interaction was significantly reduced in DDB1 KD cells (right top panel). Our results together suggest that DDB1 serves as an adaptor to mediate the interaction between RASSF1A and CUL4A, although the possibility whether DDB1 is the only adaptor of RASSF1A for CUL4A remains to be further investigated.
FIGURE 3.

CUL4A interacts with RASSF1A via DDB1. A, exogenously expressed RASSF1A pulls down the endogenous CUL4A and DDB1 in cells. Cell lysates from HEK293T cells expressing HA-S-tagged RASSF1A (HA-S-F1A) or HA-S-tag vector alone were subjected to S-tag protein pulldown. The S-tag pulldown products were simultaneously analyzed by WB with CUL4A, DDB1, and HA antibodies. B, endogenous RASSF1A interacts with endogenous CUL4A. HeLa cell lysates were immunoprecipitated (IP) with RASSF1A specific antibodies or with the control IgG. The immunoprecipitation products were analyzed by WB with the indicated antibodies. C, RASSF1A and CUL4A interaction was significantly diminished in DDB1 KD cells. HEK293T cells were first infected with lentivirus-mediated scramble shRNA (Scr. RNAi) or DDB1 shRNA (DDB1 RNAi) and then transfected with S-tagged RASSF1A expression vector. Cell lysates were then either analyzed by WB for the expression of indicated proteins (left panel) or subjected to S-protein pulldown followed by WB with CUL4A or DDB1 antibodies (right panel).
CUL4A Modulates RASSF1A Protein Stability and Half-life
Because RASSF1A interacts with CUL4A·DDB1 E3 complex, we next examined whether CUL4A was capable of affecting RASSF1A protein levels. For this purpose, HEK293T cells were transiently co-transfected with RASSF1A and increasing amount of CUL4A expression vector; the steady-state levels of RASSF1A protein were then analyzed by Western blot analysis. As shown in Fig. 4A, increasing expression of CUL4A caused a gradual reduction of RASSF1A protein levels (top panel, lanes 1–3). The CUL4A-mediated RASSF1A protein reduction could be reversed by treatment with proteasome inhibitor MG132 (Fig. 4A, top panel, lane 4), suggesting that CUL4A-mediated RASSF1A protein reduction involves a proteasome-dependent mechanism. Furthermore, overexpression of CUL4A also resulted in the reduction of endogenous RASSF1A protein (Fig. 4B, top panel). Using lentivirus shRNA-mediated CUL4A knockdown approach, we further examined the effect of CUL4A on RASSF1A protein levels and as shown in Fig. 4C, CUL4A KD significantly enhanced RASSF1A protein levels (Fig. 4C, left top panel, compare lanes 1 and 2). We also investigated the effect of CUL4A KD on RASSF1A mRNA levels and noted that CUL4A KD did not increase RASSF1A mRNA level (Fig. 4C, right panel). These results thus indicate that CUL4A does not reduce the level of RASSF1A mRNA but rather affects RASSF1A at the protein level. Interestingly, we also found that CUL4A KD significantly enhanced half-life of endogenous RASSF1A protein. As shown in Fig. 4D, CUL4A KD resulted in increased RASSF1A protein half-life; from 1.28 h in the scramble cells to 2.92 h in the CUL4A KD cells. Thus, together these results demonstrate that CUL4A plays an important role in regulation of the steady-state levels of RASSF1A protein.
FIGURE 4.

CUL4A negatively regulates RASSF1A protein level and half-life. A, CUL4A-mediated RASSF1A protein reduction is reversed by proteasome inhibitor MG132. HEK293T cells co-transfected with GFP-tagged RASSF1A vector and increasing amounts of FLAG-tagged CUL4A (FLAG-CUL4A) were mock-treated with dimethyl sulfoxide (lanes 1–3) or treated with MG132 (30 μm, lane 4) for 2.5 h. Cell lysates were simultaneously analyzed by WB for GFP-tagged RASSF1A and FLAG-tagged CUL4A on the same membrane. B, expression of CUL4A causes reduction of endogenous RASSF1A protein. HeLa cells (expressing endogenous RASSF1A) were transfected with either FLAG-tagged CUL4A vector (FLAG-CUL4A) or vector-only (Vector) and then subjected to WB for analyzing the expression of RASSF1A and CUL4A proteins. Endog, endogenous; Exog, exogenous. C, CUL4A knockdown increases the levels of endogenous RASSF1A protein but not RASSF1A mRNA. HeLa cells infected with scramble shRNA (Scr. RNAi) or CUL4A shRNA (CUL4A RNAi) lentivirus were split into two-halves; one-half was used for analyzing the expression of RASSF1A and CUL4A proteins (left panel), and the other half was used for analyzing RSSF1A mRNA by RT-PCR (right panel). β-Actin was used as an internal control in RT-PCR assay. RASSF1A expression vector (lane 1) was included as a positive control for PCR assay. D, CUL4A knockdown remarkably increases RASSF1A protein stability and half-life. HeLa cells infected with scramble shRNA or CUL4A shRNA lentivirus was treated with cycloheximide (CHX; 10 μg/ml) for the indicated times. Whole cell lysates were subjected to WB with RASSF1A antibodies. Relative band intensity of the RASSF1A signals was quantified using ImageJ software. The half-life of the RASSF1A protein was calculated based on the linear function of the four time points. For all WB analyses, β-actin was used to serve as a loading control.
DDB1 Facilitates CUL4A-mediated Ubiquitination of RASSF1A
We further performed in-cell ubiquitination assays to determine whether CUL4A is indeed a RASSF1A E3 ligase. To do this, we transfected HEK293T cells with HA-S-tagged RASSF1A expression vector or control HA-S-vector along with Myc-tagged CUL4A and FLAG-tagged ubiquitin. Cell lysates from the transfectants were then subjected to S-tag protein pulldown followed by in-cell ubiquitination assay. As shown in Fig. 5A, HA-S-tagged RASSF1A protein pulled down by S-protein beads exhibited smearing patterns in cells expressing exogenous CUL4A and ubiquitin (Fig. 5A, lane 2). Such protein pattern was not observed in cells expressing HA-S-tag alone (Fig. 5A, lane 1). To confirm that such protein smearing patterns was due to the ubiquitination of RASSF1A protein, on the duplicated membrane (with the same cell lysates), we also evaluated the specific ubiquitination signal by probing the membrane with anti-FLAG tag antibodies (for FLAG-tagged ubiquitin). Results shown in Fig. 5B (lane 2) demonstrate that RASSF1A protein was indeed ubiquitinated in cells expressing the exogenous CUL4A. In addition, our data also indicate that ubiquitination of RASSF1A was significantly enhanced in cells also co-expressing DDB1 protein (Fig. 5C, compare lanes 3 and 4). Thus, our results indicate that CUL4A is a novel E3 ligase of RASSF1A and CUL4A-mediated RASSF1A ubiquitination is facilitated by DDB1.
FIGURE 5.

In vivo ubiquitination assay demonstrates that RASSF1A is ubiquitinated by CUL4A-DDB1 in cells. A and B, HEK293T cells were co-transfected with FLAG-tagged ubiquitin (FLAG-Ub) and Myc-tagged CUL4A (Myc-CUL4A) in combination with either HA-S-tagged RASSF1A (HA-S-F1A) or HA-S-tag only. All cells were treated with MG132 (5 μm) for 16 h prior to be lysed. Cell lysate were first treated with 1% SDS prior to S-tag pulldown. The S-pulldown products were analyzed by WB with either HA-tag antibodies (for HA-S-tagged RASSF1A) (A) or with FLAG tag antibodies (for FLAG-tagged ubiquitin; B). C, HEK293T cells were transfected with the indicated expression vectors or the corresponding empty vectors as indicated by -. Cells were treated as in A and the S-tag pulldown were performed, and their products were analyzed with HA antibodies for HA-tagged RASSF1A. Shorter (left panel) or longer (right panel) exposure of the same membrane are shown.
CUL4A Modulates RASSF1A during Mitotic Phase of Cell Cycle
CUL4A·DDB1 E3 ligase has been reported to regulate their prospective substrates during DNA damage response or cell cycle progression. We next sought to determine the physiological condition that facilitates CUL4A-mediated RASSF1A ubiquitination in cells. To do that, we examined RASSF1A protein expression patterns during DNA damage response or cell cycle progression and determined the effect of CUL4A on RASSF1A during these processes. For this purpose, the lentivirus shRNA-mediated RNAi approach was employed, and RASSF1A protein expression was monitored in cells expressing the CUL4A RNAi or the control scramble RNAi. Fig. 6A shows RASSF1A expression patterns after cells exposed to UV at various times and as is shown, although CUL4A KD strongly enhanced RASSF1A protein levels in general, there is no significant difference in RASSF1A expression patterns. In both CUL4A KD and control scramble RNAi cells, RASSF1A protein expression was gradually decreased (0–12 h) and then diminished (18–24 h) after UV exposure. These results indicate that RASSF1A down-regulation after UV treatment appears to be a CUL4A-independent event because it occurred similarly in both the scramble and CUL4A KD cells and CUL4A depletion had no effect to reverse this RASSF1A protein modulation. Next, we examined the possible involvement of CUL4A in regulation of RASSF1A during cell cycle progression. To this end, cells were first arrested and synchronized at the early mitotic phase by treatment of nocodazole, and then were released to enter cell cycle upon the removal of nocodazole. RASSF1A expression patterns were then determined at various times that represent different stages of the cells cycle (35). As shown in Fig. 6B, in the scramble RNAi cells, a strong RASSF1A protein reduction was observed at 0.5, 1.5, and 12 h following nocodazole release (upper panel). And such reduction in RASSF1A levels at the 1.5 h time point was reversed by the treatment of proteasome inhibitor MG132 in these cells (data not shown). In addition, we also performed DAPI nuclear staining on the matching samples to determine cell cycle status after nocodazole release and noted that most of the cells were accumulated at mitotic phase (metaphase, anaphase, telophase, and cytokinesis) after nocodazole was removed for 0.5–2 h (data not shown). Interestingly, as shown in Fig. 6B in the CUL4A KD cells, no RASSF1A protein reduction was seen at 0.5- and 1.5-h time points (lower panels). However, RASSF1A protein reduction was retained at 12-h post-nocodazole treatment (Fig. 6B, lower panels). These results suggest that the depletion of CUL4A alleviated RASSF1A reduction mediated by CUL4A in the mitotic phase (0.5 and 1.5 h after nocodazole removal). By contrast, CUL4A depletion did not negate RASSF1A reduction at 12-h post nocodazole treatment. Of note and as established in previous studies, at this time point, Hela cells are expected to be enriched at G1- and S-phases after nocodazole treatment (35). Thus, our results suggest that CUL4A modulation of RASSF1A does not occur during G1-S-phases but takes place during M-phase of the cell cycle. To further investigate the functional significance of CUL4A-mediated RASSF1A degradation during mitosis, we examined the effect of CUL4A on RASSF1A-mediated M-phase cell cycle arrest. HEK293T cells were transfected with RASSF1A expression vector along with CUL4A expression construct or the control vector; cell cycle profiles of the transfected cells were then determined by DAPI nuclear staining. As shown in Fig. 6C, M-phase cells were significantly increased when RASSF1A was overexpressed alone without CUL4A; however, when CUL4A was co-expressed with RASSF1A, the M-phase arrest by RASSF1A was clearly inhibited. Thus, our data suggest that Cul4A-mediated RASSF1A protein degradation plays an important role in modulating RASSF1A function during mitosis.
FIGURE 6.

CUL4A regulates RASSF1A during mitosis. A, UV-induced RASSF1A protein reduction is not affected by CUL4A depletion. HeLa cells infected with scramble shRNA or CUL4A shRNA lentivirus were exposed to UV (20 J/m2) and then lysed at the indicated times. All cell lysates were analyzed by WB using antibodies specific to RASSF1A and CUL4A. B, CUL4A depletion reverses CUL4A-mediated RASSF1A protein degradation during M-phase of the cell cycle. Briefly, Hela cells infected with scramble shRNA or CUL4A shRNA lentivirus were first synchronized by thymidine and nocodazole (NZ) double-block then released from the nocodazole-induced mitotic arrest as described under “Experimental Procedures.” Cell lysates harvested at different times were analyzed by WB for the expression of RASSF1A. β-actin serves as a loading control. M, G1, and S; different cell cycle phases. C, overexpression of CUL4A antagonized the ability of RASSF1A to induce cell cycle arrest in the M-phase. HEK293T cells were transfected with GFP-RASSF1A or GFP-only vectors along with FLAG-tagged CUL4A or the control vectors as indicated. Seventy-two hours later, cells were fixed and stained with anti-FLAG antibodies (red) to detect expression of FLAG-tagged CUL4A; the cells were also stained with DAPI nuclear dye (blue) to determine the mitotic status of the cells based on their nuclear morphology. Cells expressing GFP or GFP-RASSF1A with or without FLAG tag CUL4A are counted. Error bars indicate the S.E. of the results from three independent experiments.
DISCUSSION
In this article, we have identified the CUL4A·DDB1 complex as a novel E3 ligase for tumor suppressor RASSF1A. Our studies demonstrate that DDB1 functions as a substrate adapter; it directly interacts with RASSF1A and bridges RASSF1A to the CUL4A E3 ligase complex. Depletion of DDB1 significantly diminishes RASSF1A-CUL4A interactions in cells (Fig. 3C). However, presently, we cannot rule out the possibility that RASSF1A may also work with other adaptor to associate with CUL4A. Our studies show that RASSF1A interacts with DDB1 via a region containing amino acids 165–200; deletion of this region abolishes RASSF1A-DDB1 interactions. Our data also show that CUL4A depletion significantly enhances RASSF1A protein levels and half-life, whereas overexpression of CUL4A and DDB1 markedly enhances RASSF1A protein ubiquitination and reduces the levels of RASSF1A protein. Thus, our studies clearly established that CUL4A-DDB1 is a novel E3 ligase for the RASSF1A tumor suppressor.
A number of previous studies including ours have demonstrated that RASSF1A plays important roles in regulation of the cell cycle. It has been shown that overexpression of RASSF1A arrests cell cycle at both G1- and M-phases (8). RASSF1A appears to regulate cell cycle via a number of mechanisms. For example, RASSF1A inhibits the accumulation of cyclin D1 and arrests cell cycle progression in the G1-phase (15). RASSF1A also regulates M-phase cell cycle progression via a number of different ways; it inactivates anaphase-promoting complex/CDC20 complex and arrests cells at prometaphase (16), and it also stabilizes microtubule and regulates microtubule dynamics and mitotic spindle formation (8, 11–14). Thus, it is evident that RASSF1A is an important player in cell cycle regulation and inactivation of RASSF1A by protein modification could significantly affect its function on cell cycle regulation. Our previous studies have demonstrated that RASSF1A is phosphorylated by mitotic kinase Aurora-A, and its phosphorylation by Aurora-A abolishes RASSF1A-microtubule association and disrupts negative control of RASSF1A on cell cycle progress during mitosis. Interestingly, like the p53 tumor suppressor, RASSF1A appears to be regulated at the protein level by multiple mechanisms. Song et al. (18) have reported recently that RASSF1A protein is ubiquitinated and degraded at the G1-S transition involving the CUL1·SKP2 E3 complex and depletion of SKP2 induces a delay in G1-S progression. Interestingly, in our present study, we have also observed that RASSF1A protein degradation during G1-S phase is not affected by the depletion of CUL4A (Fig. 6B). Thus, it is likely that modulation at G1-S transition is due to CUL1·SKP2 and CUL4A may not play a regulatory role. The novel findings of our study is that RASSF1A is also a substrate target of CUL4A·DDB1 E3 ligase complex. Our results indicate that CUL4A depletion significantly enhances RASSF1A protein stability and levels; RASSF1A is degraded during M-phase of the cell cycle and depletion of CUL4A reverses M-phase RASSF1A degradation. These results suggest that the M-phase-promoted RASSF1A protein degradation is mediated by CUL4A. Our data show that the levels of RASSF1A fluctuated during different times in mitosis (Fig. 6). This could suggest that RASSF1A degradation might occur during particular subphase(s) of the mitosis. Further studies are needed to address this possibility. Interestingly, it has previously been shown that depletion of CUL4A arrests cell cycle in mitosis (36). Based on our studies, one of the possible substrates for CUL4A that affects mitotic progression could be RASSF1A because CUL4A depletion enhances RASSF1A expression (Fig. 4C), and overexpression of CUL4A antagonized the ability of RASSF1A to induce M-phase arrest (Fig. 6C). Together, these studies suggest that CUL4A-mediated RASSF1A degradation is likely to play an important role in regulation of cell cycle progression in M-phase.
Based on our present study and that reported by Song et al. (18), RASSF1A appears to be sequentially regulated by two Cullin family E3 ligases including CUL1 and CUL4A during cell cycle progression with CUL1 playing a role during G1-S-phase transition and CUL4A during mitosis. Thus, both CUL1 and CUL4A share the same substrate RASSF1A but access it at different stages in the cell cycle. A number of previous studies have also shown that CUL1 and CUL4A E3 ligases share some of the same substrates, for example, Cdt1 (37), p27 (30–32, 38), and p21 (39–41). The substrate specificity of these Cullin E3 ligases are largely determined by their N-terminal available regions, which interact with specific adaptor proteins, and in the case of CUL1, also the F-box substrate receptor proteins (22). Presently, it is not clear that what specific signal(s) or factor(s) determine the interaction of RASSF1A to a specific adaptor during different stages of the cell cycle that bring RASSF1A into either the CUL1 or the CUL4A E3 complexes. Further studies are needed to delineate these regulatory processes.
As mentioned above, studies have shown that increased levels of RASSF1A arrest cell cycle in mitosis via a number of different cellular mechanisms (8, 11–16), and thus, it could be expected that reduction of RASSF1A protein may be necessary for normal mitotic cell cycle progression. Previous studies have also shown that RASSF1A is a microtubule associated protein (MAP) and microtubule stabilizer (8, 11–14). During mitosis, microtubule is highly dynamic, undergoes rapid reorganization, and forms microtubule spindles, which facilitates the segregation of replicated chromosomes (42). It is shown that during mitosis, the activities of MAPs are tightly controlled to assist the proper assembling and shrinkage of the microtubule spindles (42). In addition to its phosphorylation by mitotic kinase Aurora-A that leads to its disassociation from microtubules, and as indicated in the present study, RASSF1A is also regulated by ubiquitination by CUL4A that triggers its degradation. Both of these protein regulatory mechanisms appear to work simultaneously to control the activity of RASSF1A during M-phase of the cell cycle.
In summary, our current studies have identified CUL4A as a novel RASSF1A E3 ligase. Our studies indicate that DDB1 serves as an adaptor to bridge CUL4A and RASSF1A interactions and to facilitate CUL4A-mediated RASSF1A ubiquitination. We also provide evidence that RASSF1A is degraded by CUL4A during mitosis and increased levels of CUL4A antagonized the ability of RASSF1A to induce cell cycle arrest in the M-phase. These studies provide valuable new information about the regulation of RASSF1A, a key tumor suppressor particularly in context to its role in cell cycle regulation.
Acknowledgments
We thank Dr. Tomoki Chiba (Tokyo Metropolitan Institute of Medical Science), Dr. Strubin (University Medical Center, Geneva, Switzerland) for kindly providing the CUL4A and DDB1 expression vectors, respectively, and Dr. Ning Zhang (University of Washington) for kindly providing the DDB1 recombinant protein.
This work was supported in part by National Institutes of Health Grants CA113868 and CA128096 (to Y. H.).
- KD
- knockdown
- WB
- Western blot.
REFERENCES
- 1. Lerman M. I., Minna J. D. (2000) Cancer Res. 60, 6116–6133 [PubMed] [Google Scholar]
- 2. Dammann R., Li C., Yoon J. H., Chin P. L., Bates S., Pfeifer G. P. (2000) Nat. Genet. 25, 315–319 [DOI] [PubMed] [Google Scholar]
- 3. Burbee D. G., Forgacs E., Zöchbauer-Müller S., Shivakumar L., Fong K., Gao B., Randle D., Kondo M., Virmani A., Bader S., Sekido Y., Latif F., Milchgrub S., Toyooka S., Gazdar A. F., Lerman M. I., Zabarovsky E., White M., Minna J. D. (2001) J. Natl. Cancer Inst. 93, 691–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agathanggelou A., Honorio S., Macartney D. P., Martinez A., Dallol A., Rader J., Fullwood P., Chauhan A., Walker R., Shaw J. A., Hosoe S., Lerman M. I., Minna J. D., Maher E. R., Latif F. (2001) Oncogene 20, 1509–1518 [DOI] [PubMed] [Google Scholar]
- 5. Tomizawa Y., Kohno T., Kondo H., Otsuka A., Nishioka M., Niki T., Yamada T., Maeshima A., Yoshimura K., Saito R., Minna J. D., Yokota J. (2002) Clin. Cancer Res. 8, 2362–2368 [PubMed] [Google Scholar]
- 6. Chan M. W., Chan L. W., Tang N. L., Lo K. W., Tong J. H., Chan A. W., Cheung H. Y., Wong W. S., Chan P. S., Lai F. M., To K. F. (2003) Int. J. Cancer 104, 611–616 [DOI] [PubMed] [Google Scholar]
- 7. Yu M. Y., Tong J. H., Chan P. K., Lee T. L., Chan M. W., Chan A. W., Lo K. W., To K. F. (2003) Int. J. Cancer 105, 204–209 [DOI] [PubMed] [Google Scholar]
- 8. Rong R., Jin W., Zhang J., Sheikh M. S., Huang Y. (2004) Oncogene 23, 8216–8230 [DOI] [PubMed] [Google Scholar]
- 9. Dreijerink K., Braga E., Kuzmin I., Geil L., Duh F. M., Angeloni D., Zbar B., Lerman M. I., Stanbridge E. J., Minna J. D., Protopopov A., Li J., Kashuba V., Klein G., Zabarovsky E. R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 7504–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuzmin I., Gillespie J. W., Protopopov A., Geil L., Dreijerink K., Yang Y., Vocke C. D., Duh F. M., Zabarovsky E., Minna J. D., Rhim J. S., Emmert-Buck M. R., Linehan W. M., Lerman M. I. (2002) Cancer Res. 62, 3498–3502 [PubMed] [Google Scholar]
- 11. Liu L., Tommasi S., Lee D. H., Dammann R., Pfeifer G. P. (2003) Oncogene 22, 8125–8136 [DOI] [PubMed] [Google Scholar]
- 12. Dallol A., Agathanggelou A., Fenton S. L., Ahmed-Choudhury J., Hesson L., Vos M. D., Clark G. J., Downward J., Maher E. R., Latif F. (2004) Cancer Res. 64, 4112–4116 [DOI] [PubMed] [Google Scholar]
- 13. Vos M. D., Martinez A., Elam C., Dallol A., Taylor B. J., Latif F., Clark G. J. (2004) Cancer Res. 64, 4244–4250 [DOI] [PubMed] [Google Scholar]
- 14. van der Weyden L., Tachibana K. K., Gonzalez M. A., Adams D. J., Ng B. L., Petty R., Venkitaraman A. R., Arends M. J., Bradley A. (2005) Mol. Cell. Biol. 25, 8356–8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shivakumar L., Minna J., Sakamaki T., Pestell R., White M. A. (2002) Mol. Cell. Biol. 22, 4309–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song M. S., Song S. J., Ayad N. G., Chang J. S., Lee J. H., Hong H. K., Lee H., Choi N., Kim J., Kim H., Kim J. W., Choi E. J., Kirschner M. W., Lim D. S. (2004) Nat. Cell Biol. 6, 129–137 [DOI] [PubMed] [Google Scholar]
- 17. Rong R., Jiang L. Y., Sheikh M. S., Huang Y. (2007) Oncogene 26, 7700–7708 [DOI] [PubMed] [Google Scholar]
- 18. Song M. S., Song S. J., Kim S. J., Nakayama K., Nakayama K. I., Lim D. S. (2008) Oncogene 27, 3176–3185 [DOI] [PubMed] [Google Scholar]
- 19. Verma S. K., Ganesan T. S., Parker P. J. (2008) FEBS Lett. 582, 2270–2276 [DOI] [PubMed] [Google Scholar]
- 20. Vichalkovski A., Gresko E., Cornils H., Hergovich A., Schmitz D., Hemmings B. A. (2008) Curr. Biol. 18, 1889–1895 [DOI] [PubMed] [Google Scholar]
- 21. Song S. J., Kim S. J., Song M. S., Lim D. S. (2009) Cancer Res. 69, 8540–8544 [DOI] [PubMed] [Google Scholar]
- 22. Petroski M. D., Deshaies R. J. (2005) Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 23. Jackson S., Xiong Y. (2009) Trends Biochem. Sci. 34, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsuda N., Azuma K., Saijo M., Iemura S., Hioki Y., Natsume T., Chiba T., Tanaka K., Tanaka K. (2005) DNA Repair 4, 537–545 [DOI] [PubMed] [Google Scholar]
- 25. Kim Y., Kipreos E. T. (2007) Mol. Cell. Biol. 27, 1394–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhong W., Feng H., Santiago F. E., Kipreos E. T. (2003) Nature 423, 885–889 [DOI] [PubMed] [Google Scholar]
- 27. Wertz I. E., O'Rourke K. M., Zhang Z., Dornan D., Arnott D., Deshaies R. J., Dixit V. M. (2004) Science 303, 1371–1374 [DOI] [PubMed] [Google Scholar]
- 28. Chen L. C., Manjeshwar S., Lu Y., Moore D., Ljung B. M., Kuo W. L., Dairkee S. H., Wernick M., Collins C., Smith H. S. (1998) Cancer Res. 58, 3677–3683 [PubMed] [Google Scholar]
- 29. Yasui K., Arii S., Zhao C., Imoto I., Ueda M., Nagai H., Emi M., Inazawa J. (2002) Hepatology 35, 1476–1484 [DOI] [PubMed] [Google Scholar]
- 30. Bondar T., Kalinina A., Khair L., Kopanja D., Nag A., Bagchi S., Raychaudhuri P. (2006) Mol. Cell. Biol. 26, 2531–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Higa L. A., Yang X., Zheng J., Banks D., Wu M., Ghosh P., Sun H., Zhang H. (2006) Cell Cycle 5, 71–77 [DOI] [PubMed] [Google Scholar]
- 32. Li B., Jia N., Kapur R., Chun K. T. (2006) Blood 107, 4291–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki H., Chiba T., Kobayashi M., Takeuchi M., Suzuki T., Ichiyama A., Ikenoue T., Omata M., Furuichi K., Tanaka K. (1999) Biochem. Biophys. Res. Commun. 256, 127–132 [DOI] [PubMed] [Google Scholar]
- 34. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 35. Whitfield M. L., Sherlock G., Saldanha A. J., Murray J. I., Ball C. A., Alexander K. E., Matese J. C., Perou C. M., Hurt M. M., Brown P. O., Botstein D. (2002) Mol. Biol. Cell 13, 1977–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kopanja D., Stoyanova T., Okur M. N., Huang E., Bagchi S., Raychaudhuri P. (2009) Oncogene 28, 2456–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishitani H., Sugimoto N., Roukos V., Nakanishi Y., Saijo M., Obuse C., Tsurimoto T., Nakayama K. I., Nakayama K., Fujita M., Lygerou Z., Nishimoto T. (2006) EMBO J. 25, 1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakayama K., Nagahama H., Minamishima Y. A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N., Kitagawa M., Nakayama K., Hatakeyama S. (2000) EMBO J. 19, 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang W., Nacusi L., Sheaff R. J., Liu X. (2005) Biochemistry 44, 14553–14564 [DOI] [PubMed] [Google Scholar]
- 40. Furstenthal L., Swanson C., Kaiser B. K., Eldridge A. G., Jackson P. K. (2001) Nat Cell Biol. 3, 715–722 [DOI] [PubMed] [Google Scholar]
- 41. Nishitani H., Shiomi Y., Iida H., Michishita M., Takami T., Tsurimoto T. (2008) J. Biol. Chem. 283, 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wittmann T., Hyman A., Desai A. (2001) Nat. Cell Biol. 3, E28–34 [DOI] [PubMed] [Google Scholar]