

Abstract
The glycosylphosphatidylinositol (GPI)-anchored neutrophil-specific receptor NB1 (CD177) presents the autoantigen proteinase 3 (PR3) on the membrane of a neutrophil subset. PR3-ANCA-activated neutrophils participate in small-vessel vasculitis. Since NB1 lacks an intracellular domain, we characterized components of the NB1 signaling complex that are pivotal for neutrophil activation. PR3-ANCA resulted in degranulation and superoxide production in the mNB1pos/PR3high neutrophils, but not in the mNB1neg/PR3low subset, whereas MPO-ANCA and fMLP caused similar responses. The NB1 signaling complex that was precipitated from plasma membranes contained the transmembrane receptor Mac-1 (CD11b/CD18) as shown by MS/MS analysis and immunoblotting. NB1 co-precipitation was less for CD11a and not detectable for CD11c. NB1 showed direct protein-protein interactions with both CD11b and CD11a by surface plasmon resonance analysis (SPR). However, when these integrins were presented as heterodimeric transmembrane proteins on transfected cells, only CD11b/CD18 (Mac-1)-transfected cells adhered to immobilized NB1 protein. This adhesion was inhibited by mAb against NB1, CD11b, and CD18. NB1, PR3, and Mac-1 were located within lipid rafts. In addition, confocal microscopy showed the strongest NB1 co-localization with CD11b and CD18 on the neutrophil. Stimulation with NB1-activating mAb triggered degranulation and superoxide production in mNB1pos/mPR3high neutrophils, and this effect was reduced using blocking antibodies to CD11b. CD11b blockade also inhibited PR3-ANCA-induced neutrophil activation, even when β2-integrin ligand-dependent signals were omitted. We establish the pivotal role of the NB1-Mac-1 receptor interaction for PR3-ANCA-mediated neutrophil activation.
Keywords: Glycosylphosphatidylinositol Anchors, Integrin, Neutrophil, Signal Transduction, Superoxide Ion
Introduction
Anti-neutrophil cytoplasmic auto-antibodies (ANCA)2 are found in patients with Wegener granulomatosis, microscopic polyangitis, Churg-Strauss syndrome, and necrotizing crescentic glomerulonephritis (1, 2). PR3- and MPO-ANCA binding to their target antigens on the membrane of neutrophils and monocytes leads to cell activation (3, 4). ANCA pathogenicity and the pivotal role of the ANCA-neutrophil interaction were established in several animal models (5–9). In contrast to MPO, membrane-PR3 (mPR3) has a bimodal expression pattern resulting in distinct mPR3low and mPR3high subsets. The percentage of mPR3high neutrophils ranges from 0 to 100%, is genetically determined, and in a large part explained by the HLA region (10–12). Patients with ANCA vasculitis have a higher percentage of mPR3high neutrophils that correlates with disease parameters (13–15). In vitro experiments indicate that mPR3high neutrophils respond with increased PR3-ANCA-mediated superoxide generation and degranulation, whereas other stimuli triggered a similar response in both neutrophil subsets (16). Neutrophil antigen B1 (NB1, CD177) is exclusively expressed in and on mPR3high neutrophils and functions as a presenting receptor for PR3 on the cell membrane on this neutrophil subset (17–19). In a recent report, the percentage of NB1-expressing neutrophils was higher in ANCA vasculitis patients, compared with healthy controls. Furthermore, within the ANCA group, the percentage was higher in those patients who had relapsing disease (20). Together, these data indicate that the mNB1pos/PR3high phenotype is clinically relevant in ANCA vasculitis.
NB1 is a GPI-anchored molecule that lacks an intracellular domain. The link between mPR3 presentation by the non-signaling NB1 receptor and neutrophil activation in response to PR3-ANCA is still missing. We hypothesized that additional components that have not yet been identified must be recruited into a larger NB1 signaling complex. Examples from other GPI-linked receptors implicate candidates such as various integrins (21, 22, 23, 25, 26), gp130 (23), the transmembrane protein tyrosine kinase Ret (24), and the formyl peptide receptor-like 1 (FPRL1) (23) that are often dynamically organized in lipid rafts. We aimed to identify constituents of the PR3-NB1 receptor complex that are functionally important when PR3-ANCA activate neutrophils. Clarification of these initial signaling processes may identify novel treatment targets for ANCA vasculitis.
EXPERIMENTAL PROCEDURES
Materials
TNF-α was obtained from R&D Systems (Wiesbaden-Nordenstedt, Germany). Phorbol-2-myristate-13-acetate (PMA), the bacterial peptide f-met-leu-phe (fMLP), 2.2′-azino-bis (3-ethylbenzthiazoline-6-sulfonicacid), dihydrorhodsamine (DHR), and Histopaque were from Sigma. HBSS, PBS, and Trypan Blue were from PAA (Colbe, Germany) and dextran was purchased from Roth (Karlsruhe, Germany), Percoll was from GE Healthcare (Amsterdam, Netherlands). The PR3 mAb 43-8-3 was from BioGenes (Berlin, Germany), the CLB 12.8 from CLB (Amsterdam, the Netherlands) and the mAb to MPO from Acris (Herford, Germany). The mAb to NB1 (MEM166) was from Serotec (Düsseldorf, Germany) and Biolegend, the polyclonal goat anti-CD11b, anti-CD18, and anti-NB1 Ab from R&D Systems (Wiesbaden-Nordenstedt, Germany), the mAb Cdc42 from BD (BD Biosciences, San Jose, CA), the flotillin-1 Ab and blocking CD11b mAb (clone 2LPM19c) from Santa Cruz Biotechnology (Santa Cruz, CA and Bayport MN, respectively), blocking CD18 mAbs (clone 7E4, MHM23, and IB4, respectively) were from Immunotech (Marseille, France), rabbit mAbs to CD11a, CD11b, and CD11c were obtained from Epitomics (Burlingame, CA), horseradish peroxidase-labeled secondary antibodies were from GE Healthcare and Santa Cruz Biotechnology, the FITC-conjugated Fab-fragments of goat anti-mouse IgG were from DAKO (Hamburg, Germany). Hybridoma producing mAb 7D8 against NB1 was kindly provided by Dr. D. Stroncek (Department of Transfusion Medicine, NIH, Bethesda, MD). Purified recombinant human CD11b/CD18 integrin was from R&D systems (Wiesbaden-Nordenstedt, Germany). NB1 and αIIb-β3 was purified from human neutrophils or platelets as previously described (27, 28). Endotoxin-free reagents and plastic disposables were used in all experiments.
Preparation of Human Neutrophils and Human IgG
Neutrophils from healthy human donors were isolated from heparinized whole blood as described previously (16). Cell viability was >99% by Trypan Blue exclusion. Normal IgG and ANCA-IgG were prepared from normal volunteers and PR3- and MPO-ANCA patients with active disease using a High-Trap-protein-G column in an Äkta-FPLC system (GE Healthcare). We detected a single band on Coomassie-stained gels under non-reducing conditions.
Separation of mNBpos and mNB1neg as Well as mPR3low and mPR3high Neutrophil Subsets by Magnetic Beads
Neutrophil subsets were separated with MACS separation columns (Miltenyi Biotec, Bergisch Gladbach, Germany) as described in the manufacturer's manual. Isolated neutrophils were stained with a mAb to NB1 (MEM166) or PR3 (CLB). MACS rat anti-mouse IgG1 were added, and cells were pipetted onto a MACS LD Column. The flow-through containing the nonlabeled mNB1neg or mPR3low neutrophils was collected. Columns were removed from the magnet and labeled mNB1pos or mPR3high neutrophils were collected. Cells were stained with a FITC-labeled secondary antibody Fab fragment. NB1 and PR3 expression was assessed by flow cytometry using a FACSort (Becton Dickinson, Heidelberg, FRG), and 10,000 events per sample were collected.
Lipid Raft Preparation and Subcellular Fractionation
Lipid raft preparation was done as described by Kowalski et al. (29) with minor modifications. TNF-α-primed neutrophils (2 ng/ml for 15 min) were pelleted, resuspended in MBS (MES-buffered saline, 25 mm MES, pH 6.5, 150 mm NaCl) containing 1% Triton X-100 and protease inhibitors (10 μg/ml quercetin, 10 μg/ml, leupeptin, 0.1 mm aprotinin, 0.2 mm Na3VO4, and 1 mm PMSF), incubated for 20 min on ice and passed five times through a 21-gauge needle. Lysates were spun down (400 × g, 10 min, 4 °C) to remove cell debris and nuclei. 2 ml of supernatant was added to 2 ml of 85% sucrose in MBS (final concentration 42.5% sucrose), transferred into a Beckmann ultracentrifuge tube and overlaid with 6 ml of 35% sucrose in MBS. An additional 2 ml of 5% sucrose in MBS was placed on the top of the tube. Samples were centrifuged, 1-ml fractions were removed from the top of the gradient, and protein concentration was measured.
Subcellular fractionation to obtain pure membrane fractions from freshly isolated neutrophils was performed as previously described with some modifications (30). Briefly, TNF-α primed cells were incubated with diisopropylfluorophosphate (DFP) for 5 min on ice, centrifuged and resuspended in disruption buffer (100 mm KCl, 3 mm NaCl, 1 mm ATPNa, 3.5 mm MgCl, 10 mm PIPES, pH 7.2) containing 0.5 mm PMSF. The cavitate from cells disrupted in a precooled nitrogen bomb (350 psi, 5 min) was collected dropwise into EGTA (final concentration 1.5 mm). Intact cells and nuclei were pelleted (400 × g, 15 min, 4 °C), and supernatant was transferred into a Beckmann ultracentrifuge tube. 18 ml of Percoll, density 1.065 g/ml, were gently layered under the supernatant. Finally, 2 ml of Percoll, density 1.12 g/ml was placed carefully at the bottom of the tube. The prepared one-layer gradient was centrifuged at 37,000 × g for 30 min (Beckman fixed-angle Ti70 rotor), this give rise to two distinct bands containing granules and plasma membranes. 1.5-ml fractions were carefully harvested with a fine needle from the bottom of the ultracentrifuge tube and tested for MPO activity and alkaline phosphatase (AP) activity as described. Fractions with the highest AP activity were pooled. Percoll was removed, and membranes were pelleted by centrifugation (100,000 × g, 45 min, 4 °C). Membranes were resuspended in extraction buffer (1% digitonin in PBS) at room temperature for 30 min.
Mass Spectrometry
Proteins eluted from the affinity column by high pH treatment were converted to peptides using an in-solution digest with endopeptidase LysC and trypsin. Peptides were collected on a C18-column, desalted and prepared for mass spectrometry (31). The peptide mix was separated on an in-house packed 15 cm analytical column (75-μm inner diameter, 3 μm C18-reverse phase beads, Dr. Maisch, Reprosil-AQ Pur), with a 155-min gradient from 5% to 40% acetonitrile in 0.5% acetic acid using an Eksigent nanoLC HPLC system. The effluent was directly electrosprayed into the mass spectrometer. For data collection an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with an electrospray ion source (Proxeon Biosystems) was used. For the data analysis, the MaxQuant and the Mascot software package (Matrix Science) were used (32).
Adhesion of CD11a/CD18 and CD11b/CD18 Stably Transfected Cells to Immobilized NB1 Protein
Cell adhesion to transfected cells was tested as described previously with minor modifications (33). Briefly, microtiter plates were coated with 100 μl of BSA, ICAM-1 (R&D System, Wiesbaden, Nordenstedt, Germany) or purified NB1 protein (500 ng/well) in bicarbonate buffer pH 9.6 and blocked with 3% BSA for 1 h at room temperature. CD11b/CD18, αIIb-β3 or CD11a/CD18 (generous gift from Dr. T. Chavakis, Dresden, Germany) transfected cells were incubated with 5 μl of 2′7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF, Molecular Probes, Leiden, The Netherlands) for 30 min in the dark. Cells were washed twice with HBSS buffer, and 50-μl cell suspensions (2 × 106/ml) were plated onto the precoated wells at 37 °C for 30 min. For inhibition studies, cells were incubated with mAbs (20 μg/ml). Thereafter, fluorescence intensity in each well was measured on fluorescence microplate reader Flx-800 (Biotek, Neufahrn, Germany) before and after washing twice. The total amount of bound cells was calculated.
Surface Plasmon Resonance Analysis (SPR)
SPR analysis was performed on a ProteOn XPR36 system (Bio-Rad, Herculas, CA) as described (33). Purified recombinant CD11b, CD11a, or αIIb-β3 protein (60 pmol in 10 mm sodium acetate buffer, pH 4.5) was immobilized on a GLM sensor chip by amine coupling using standard procedures. Purified NB1 protein or BSA (0.1 μm in PBS) was injected as analyte over the chip at a flow rate of 20 μl/min in a total volume of 250 μl at 25 °C. The sensorgrams were evaluated using the ProteOn evaluation software package. Protein-protein interaction was recorded as relative response unit (RU) of NB1 minus BSA for 800 s.
Immunoprecipitation and Western Blot Analysis
Cell lysis, SDS-PAGE, and Western blot were performed as described previously (18). For immunoprecipitation studies mAbs against NB1 were covalently bound to tosylactivated Dynabeads M-280 (Invitrogen, Carlsbad, CA) according to the manufacturer's directions, and digitonin membrane extracts were incubated with anti-NB1-beads (5 μg of coupled mAbs) overnight at 4 °C. Precipitates were washed with TBS-Tween buffer and subjected to PAGE and Western blotting. The blots were developed with the indicated antibody and visualized by an enhanced chemiluminescence detection system (Thermo Scientific, Rockford, IL).
Measurement of Respiratory Burst
Superoxide was measured using the assay of SOD-inhibitable reduction of ferricytochrome c as described by Pick and Mizel (34). Neutrophils were pretreated with 5 μg/ml cytochalasin B for 15 min. Cells were treated with 10 μg/ml blocking mAb to CD11b, CD18. or isotype control for 30 min (as indicated), primed with 2 ng/ml TNF-α for 15 min before stimulating antibodies were added. The final concentrations were 5 μg/ml for the mAbs and 150 μg/ml for purified IgG preparations. Experiments were done in duplicate. Samples were incubated in 96-well plates at 37 °C for up to 60 min, and the absorption of samples with and without 300 units/ml SOD was scanned repetitively at 550 nm using a Microplate Autoreader (Molecular Devices, Munich, Germany). 45 min results are reported. When indicated, ultra-low attachment plates were used (Corning, NY) instead of polystyrene plates. The final ferricytochrome c concentration was 50 μmol/liter, and the final cell concentration was 0.75 × 106/ml. To reduce cell-cell interactions, the experiments on ultra-low adhesion plates employed 0.5 × 106/ml neutrophils.
For the DHR assay prewarmed neutrophils (1 × 107/ml HBSS) were loaded with DHR (1 μm) for 10 min at 37 °C. After treatment with blocking antibodies to CD11b, CD18 or isotype control for 30 min, cells were primed with 2 ng/ml TNF-α cells and 5 × 105 cells were incubated with the stimuli in a total assay volume of 100 μl in polypropylene tubes. Antibodies were added and the reactions were stopped after another 30 min by adding ice-cold PBS/1% bovine serum albumin. Samples were analyzed using a FACScan (Becton Dickinson, Heidelberg, Germany). Data were collected from 10,000 cells per sample. The shift of green fluorescence in the FL-1 mode was determined, and the mean fluorescence intensity (MFI, representing the amount of generated hydrogen peroxide) is reported.
Degranulation Assay
Neutrophils (5 × 105) were preincubated with 5 μg/ml cytochalasin B and for 30 min with blocking mAb to CD11b, CD18, or isotype control, respectively. Cells were primed in a 96-well microtiter plate for 15 min with 2 ng/ml TNF-α and stimulated with ANCA or human control IgG for up to 60 min. Cell-free supernatants were collected by centrifugation and β-glucuronidase activity was assessed by the cleavage of phenolphtaleinglucuronic acid (Sigma-Aldrich). The reaction was stopped by adding 1 ml of a 0.02 mol/liter solution of glycine buffer, pH 10.4. Absorbance values were measured at 405 nm. Nonstimulated neutrophils served as baseline, whereas the total neutrophil β-glucuronidase content was obtained by incubation 5 × 105 neutrophils with 1% Triton X-100.
Assessment of Neutrophil Adhesion
96-well polystyrene or ultra-low attachment plates were used. Neutrophils (5 × 105) in 100 μl of HBSS were treated with 2 ng/ml TNF-α. Adherent cells were estimated using the MPO assay. Briefly, adherent cells were lysed in 100 μl of 0.5% Triton X-100. Substrate (100 μl; 2.2′-azino-bis (3-ethylbenzthiazoline-6-sulfonic acid) was added, and absorbance was read at 450 nm. Absorbance of the experimental sample was compared with a standard curve that showed an excellent correlation between absorbance and cell number.
Confocal Microscopy
Neutrophils were fixed in 0.5% paraformaldehyde and blocked with 5% BSA in PBS containing human Fc block. Cells were incubated with polyclonal goat anti-NB1 together with mAbs to CD11a, CD11b and CD18, respectively. After washing samples were incubated with the corresponding Alexa488 and Alexa555 conjugated secondary Abs. Cells were washed and cytospun onto a slide. Fluorescence images were acquired at a laser scanning confocal microscope Leica TCS SP5 using a 63× NA1.4 PL APO oil immersion objective. A 405 nm diode laser, a 488 nm argon laser and a 561 nm DPSS laser were used to excite DAPI, Alexa488, and Alexa555 with bandpass filters 415–460, 495–550 and 570–640 nm, respectively, to detect fluorescence emission, respectively in sequential image acquisition mode. Each cell was imaged by 5 to 7 confocal z-sections without over saturation of individual pixels. Single stained samples and samples without primary antibodies were used to check for cross talk of fluorophores and background signal. Pixel shift between green and red channels was determined with tetraspeck beads under identical conditions. Colocalization analysis (Pearson's Correlation coefficient) of the pixel-shift corrected images was performed with the Leica LASAF 2.1.1 software by applying an ROI around the cell.
CD11a and CD11b Expression on the Neutrophil Membrane by Flow Cytometry
Aliqouts of 103 washed neutrophils were incubated with PBS containing 20 μg/ml FITC-labeled mAb against CD11a, CD11b, or mouse IgG (BD Pharmingen) for 30 min at 4 °C. After washings with PBS, fluorescence-labeled neutrophils were analyzed on a FACS Calibur flow cytometer (BD Biosciences, Heidelberg, Germany).
Statistical Analysis
Results are given as mean ± S.E. Comparisons between multiple groups were done using one or two way ANOVA. Specific differences between multiple groups were then determined by post-hoc tests. Differences were considered significant if p < 0.05.
RESULTS
PR3-ANCA-induced Degranulation, Respiratory Burst, and Membrane PR3/NB1 Expression
We first studied the significance of the membrane NB1/PR3 phenotype for ANCA-induced degranulation and respiratory burst. Cells were sorted for mNB1 yielding very pure NB1neg and NB1pos subsets as shown in Fig. 1A. We verified that the NB1neg subset is characterized by low mPR3 expression (mPR3low) and the NB1pos subset by high mPR3 (mPR3high) expression (Fig. 1A). Using the β-glucoronidase assay, we observed that PR3-ANCA triggered significantly more degranulation in the NB1pos/mPR3high neutrophils compared with the NB1neg/mPR3low subset (Fig. 1B). In contrast, MPO-ANCA and fMLP resulted in similar degranulation, irrespectively of the membrane NB1/PR3 phenotype. Parallel assessment of ferricytochrome c reduction (Fig. 1C) and DHR oxidation (Fig. 1D) in neutrophils of the same preparation indicates that extracellular superoxide generation in response to PR3-ANCA, but not to MPO-ANCA was significantly higher in NB1pos/mPR3high neutrophils compared with the NB1neg/mPR3low cells. In contrast to the ferricytochrome c assay that measures extracellular superoxide, both neutrophil subsets showed similar intracellular hydrogen peroxide generation to PR3- and MPO-ANCA as assessed using the DHR assay. This first part of our study underscores the significance of the mPR3/NB1 phenotype for important PR3-ANCA induced neutrophil functions, namely degranulation and superoxide production. We focused our next experiments on the role of NB1 in PR3-ANCA-induced activation.
FIGURE 1.

Degranulation, NADPH oxidase-dependent ferricytochrome c reduction and intracellular DHR oxidation in NB1neg and NBpos neutrophils. Neutrophils from mNB1 and mPR3 bimodal donors (unsorted cells) were separated by magnetic cell sorting using an anti-NB1 mab. Sorting resulted in two distinct subsets that are NB1neg and NB1pos as well as mPR3low and mPR3high. Typical flow cytometry examples are depicted (panel A). The mNB1neg/mPR3low and mNB1pos/mPR3high subsets were then primed with TNF-α and subsequently stimulated with IgG from 2 different patients with PR3-ANCA or MPO-ANCA, or from normal (control). Incubation with fMLP was done for comparison. Degranulation is shown in panel B (n = 3). Ferricytochrome c reduction is shown in panel C (n = 6). Bars for ferricytochrome c measurements on the left depict superoxide generation at 45 min with a typical example for the time course up to 60 min on the right. DHR oxidation is shown in panel D (n = 6). Bars for DHR oxidation on the left depict data after 30 min stimulation with typical histograms on the right. * indicates significant activation of mNB1pos/mPR3high neutrophils when compared with the mNB1neg/mPR3low cells (p < 0.05).
Plasma Membrane Preparations and Extraction of Membrane Proteins
Because PR3-presenting mNB1 lacks an intracellular domain, we aimed to identify additional members of the NB1 membrane complex that allow for PR3-ANCA-mediated cell activation. The amount of PR3 and NB1 that is membrane expressed is rather small compared with the granule compartment. Because our study focused on NB1 membrane partners, we wanted to exclude, to a large extend, non-membrane NB1 sources from our analysis. Accordingly, we prepared pure membrane fractions and selected those fractions that were positive for the membrane marker alkaline phosphatase and negative for the granule marker MPO as shown in Fig. 2A. The fractions were then subjected to digitonin solubilization because digitonin extracts membrane molecules and minimizes disruption of complexes that have formed in situ. This approach diminishes the possibility that artificial complexes are generated in vitro. We reasoned that the resulting extract contained mNB1 together with its potential binding partners. The presence of NB1 and PR3 in the membrane extracts was confirmed by Western blotting as shown in Fig. 2B.
FIGURE 2.

NB1 is co-immunoprecipitated with Mac-1 from digitonin extracts of plasma membrane preparations. Neutrophil plasma membranes were prepared and assayed for the membrane marker alkaline phosphatase and the granule marker MPO (panel A). Membrane fractions were then solubilized with digitonin. The presence of NB1 and PR3 in the membrane extracts was confirmed by Western blotting (panel B). NB1 was precipitated from neutrophil membranes using an affinity matrix. The eluted material was analyzed using tandem mass spectrometry analysis (MS/MS). CD11b and CD18 were identified with typical MS characteristics shown in panels C and D. Immunoprecipitated NB1 complexes were probed for CD11a, CD11b, CD11c, CD18, and NB1 using specific antibodies and Western blot analysis (panel E). Instead of an anti-NB1 antibody (αNB1), an isotype control (Iso), and beads not coupled to an antibody (empty) were used as negative controls. Digitonin extracts were used as positive control.
Characterization of the mNB1-PR3 Complex by Immunoprecipitation, Mass Spectrometry, and Western Blot Analysis
We next immunoprecipitated NB1 from digitonin-extracted membranes. After elution from the matrix, the sample was subjected to tryptic digestion and analyzed by tandem mass spectrometry analysis (MS/MS). Several molecules were identified that are listed in the supplemental Table S1. Among these molecules, we detected the CD11b and CD18 chain of the β2-integrin Mac-1 with typical MS characteristics shown in Fig. 2, C and D. In accordance with these findings, Western blotting analysis with antibodies against CD11b and CD18 subunits showed significant amounts of CD11b and CD18 co-precipitated with mNB1-PR3 (Fig. 2E). In contrast, CD11a was barely detected, even after exposure time was prolonged to 30 min, and CD11c was not detected at all.
NB1 Interacts with the Extracellular Mac-1 Domain and Both Molecules Co-localize on the Neutrophil Membrane
Next, we assayed direct protein interactions by surface plasmon resonance (SPR) between NB1 and CD11a and CD11b by the SPR technique. As shown in Fig. 3A, purified NB1 protein interacts with immobilized CD11b as well as CD11a on a sensor chip with similar binding property. On the cell surface, however, both proteins are expressed as heterodimer together with the CD18 subunit. To mimic this situation, cell lines expressing CD11b/CD18 or CD11a/CD18 were tested for their capability to adhere on immobilized NB1 protein. CD11b/CD18 co-transfected cells adhered strongly to NB1 protein when compared with a cell line expressing the αIIbβ3 integrin (Fig. 3B). Importantly, preincubation with inhibitory mAb (MEM166), but not with non-inhibitory mAb against NB1 (7D8) blocked cell adhesion. Additionally, incubation with mAbs against CD11b (2LPM19c and ICRF4) and CD18 caused inhibition of cell adhesion to NB1. In contrast, no adhesion of CD11a/CD18-transfected cells on NB1 protein was observed. In the control experiment, CD11a/CD18 cells bound immobilized ICAM-1 and this binding was prevented by the CD11a inhibitory antibody (Fig. 3C). Taken together, these data indicate that CD11b/CD18 represents the major counter receptor for NB1 cis interaction on neutrophils. Confocal microscopy analysis using overlay images, revealed a strong co-localization of NB1 with CD11b and CD18 on the neutrophil membrane of the same cell. The degree of co-localization can best be appreciated by overlay images and in the scattergrams by the amount of co-localizing pixels in region C. In contrast to CD11b, NB1 co-localization with CD11a was clearly inferior (Fig. 4A). Incubation of TNF-α-primed neutrophils with human PR3-ANCA, compared with human control IgG did not further increase the degree of co-localization between NB1 and CD11b and CD18, respectively. The Pearson's colocalization coefficient for CD11b was 0.84 ± 0.06 for the human control IgG and 0.84 ± 0.05 for PR3-ANCA. The values for CD18 were 0.82 ± 0.04 and 0.82 ± 0.05, respectively.
FIGURE 3.

Characterization of NB1 interactions with CD11b/CD18 and CD11a/CD18. Panel A, protein-protein interaction was analyzed by SPR. Recombinant CD11b, CD11a, or αIIb-β3 was immobilized on a sensor chip. NB1 protein at a concentration of 0.5 μm was injected wit a flow rate 20 μl/min at 25°. Protein-protein interaction was recorded as relative response unit (RU) for 300 s. Panels B and C, a cell adhesion assay was performed. Panel B, CD11b/CD18 (Mac-1)-transfected (gray columns) and αIIb-β3-transfected cells (white columns) were added to 96-well plates coated with purified NB1 or BSA for 30 min as indicated. Cell adhesion was measured in the presence of two different anti-NB1 mAbs (αNB1, 7D8, MEM166), two different anti-CD11b (αCD11b, 2LPM19c, ICRF44), anti-CD11a (αCD11b, HI111), and anti-CD18 mAb (αCD18, IB4). Note the significant inhibitory capacity of the MEM166 mAb and the mAbs against CD11b and CD18. Panel C, CD11a/CD18 (LFA-1)-transfected (gray columns) and mock-transfected cells (white columns) were added to 96-well plates coated with purified BSA, NB1, or ICAM-1 protein (10 μg/ml) for 30 min as indicated. After washing twice, no significant adhesion of LFA-1-transfected cells to coated NB1 was observed when compared with coated ICAM-1 (positive control). In addition, anti-LFA-1 mAbs significantly blocked cell adhesion to immobilized ICAM-1. n = 4, * indicates p < 0.05.
FIGURE 4.

NB1 strongly co-localizes with CD11b and CD18 on the neutrophil membrane, and CD11b shows higher expression on the neutrophil membrane compared with CD11a. Panel A, confocal microscopy was performed. Neutrophils were stained for NB1 (red) or for CD11a, CD11b, and CD18, respectively (green). DAPI staining indicates the nuclei in blue. Overlay images demonstrate co-localization of green and red-stained molecules by a shift toward yellow color. Scattergrams in the right panel of each row show the amount of co-localizing pixels in region C after threshold and background correction. The data show a high amount of co-localizing pixels for NB1/CD11b and NB1/CD18. In contrast, only a few co-localizing pixels are found for NB1 and CD11a. 14 to 19 cells were analyzed in each of two independent experiments. Panel B, cell surface expression of CD11a and CD11b was assessed by flow cytometry. The data demonstrate that ∼5-fold more CD11b molecules are expressed on the neutrophil membrane compared with CD11a.
Direct comparison of the amounts of CD11a and CD11b on the neutrophil surface by flow cytometry indicates that ∼5-fold more CD11b molecules are expressed on the neutrophil membrane compared with CD11a (Fig. 4B). Based on these data, we thought CD11b/CD18 as an important molecule within NB1 signaling complex.
NB1, PR3, and Mac-1 Exist in Lipid Rafts
Lipid rafts function as signaling platforms in cell membranes. We asked whether or not NB1, PR3, and Mac-1 are present in lipid rafts of TNF-α-primed neutrophils. Membrane fractionation was performed followed by Western blot analysis using an anti-flotilin-1 antibody for lipid rafts and anti-Cdc42 mAb for non-raft structures. Fig. 5 shows the highest flotillin-1 amount, and therefore the highest lipid raft concentration, in fraction 4. NB1, PR3, CD11b, and CD18 were also observed in fraction 4, indicating that, at least some of these molecules exist in lipid raft domains that promote the interaction of receptor complexes.
FIGURE 5.
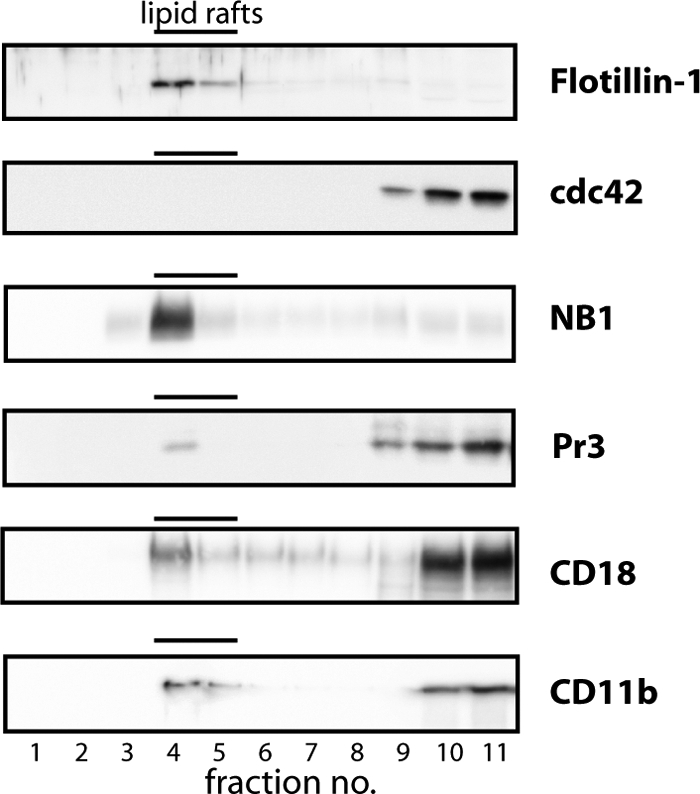
NB1 exists together with Mac-1 and PR3 in lipid rafts. Neutrophil plasma membranes were lyzed and subjected to sucrose density gradient centrifugation. By Western blot analysis, the resulting fractions were probed with specific antibodies to the indicated molecules. Plasma membrane fraction 4 shows the highest amount of the lipid raft marker flotillin-1 in the absence of the non-lipid raft marker Cdc42. Both NB1 and PR3 were also detected in these lipid rafts. One of two similar experiments is depicted.
CD11b/CD18 Blockade Abrogates Anti-NB1 Antibody-induced Degranulation and Superoxide Production
The next set of experiments investigated the functional role of CD11b/CD18 within the NB1 membrane complex. We envisioned that one way to approach this issue was to stimulate the neutrophils with an activating anti-NB1 antibody in the presence or absence of blocking anti-CD11b or anti-CD18 antibodies. By cell sorting using an anti-PR3 mAb (CLB), neutrophils were separated into a NB1pos/mPR3high and NB1neg/mPR3low subset (Fig. 6A). Cells were then primed with TNF-α and stimulated with an anti-NB1 mAb (MEM166), fMLP, or PMA as indicated. The mAb to NB1 resulted in a strong degranulation (Fig. 6B) and induced a weaker, but significant superoxide generation (Fig. 6C). Similar to the PR3-ANCA response, anti-NB1 antibody-induced degranulation and superoxide generation occurred only in the NB1pos/mPR3high subset. No difference was observed when fMLP or PMA were used for stimulation.
FIGURE 6.

NB1 mediates degranulation and superoxide generation in a Mac-1-dependent fashion. Neutrophils from mNB1/mPR3 bimodal donors were separated by magnetic cell sorting using an anti-PR3 mAb. Sorting resulted in two distinct subsets that are either mNB1neg and mNB1pos or mPR3low and mPR3high. A typical flow cytometry example is depicted (panel A). The mNB1neg/mPR3low and mNB1pos mPR3high subsets were then primed with TNF-α and subsequently stimulated with anti-NB1 mAb MEM166 (αNB1) or an isotype control (control). Incubation with fMLP and PMA was done for comparison. The anti-NB1 mAb induced degranulation (panel B, n = 4), ferricytochrome c reduction (panel C, n = 8) only in mNB1pos/mPR3high neutrophils. Blockade of CD11b and CD18 was performed in unsorted cells. Cells were incubated with the blocking antibodies for 30 min, followed by TNF-α priming and anti-NB1 mAb incubation. CD11b and CD18 blockade prevented NB1-mediated degranulation (panel D, n = 3) and superoxide generation (panel E, n = 6). * indicates significant inhibition (p < 0.05).
We then explored whether or not the anti-NB1-mediated responses were decreased with Mac-1 blockade. Because our data had established that only NB1pos/mPR3high neutrophils responded to anti-NB1 stimulation, we performed these experiments without prior cell sorting. We observed that degranulation and superoxide production were significantly inhibited when cells were preincubated with blocking antibodies to CD11b or CD18 prior to the anti-NB1 stimulation (Fig. 6, D and E). These data provide direct evidence for a functional role of CD11b/CD18 in mNB1-mediated neutrophil activation.
CD11b and CD18 Blockade Abrogate Anti-PR3 Antibody-mediated Degranulation and Superoxide Production in a Ligand-independent Fashion
We next studied the functional consequences of blocking Mac-1 on NB1-dependent neutrophil activation in response to mAb to PR3 and PR3-ANCA from patients using the ferricytochrome c assay. mAbs to PR3 triggered degranulation and superoxide generation in TNF-α-primed neutrophils, and this response was abrogated by preincubation with blocking Abs to CD11b and CD18 (Fig. 7, A and B). An isotype control was without effect. To ensure that this mechanism is also at work when human PR3-ANCA preparations were used, we repeated the superoxide assay with 2 different PR3-ANCA IgG preparations. Fig. 7C illustrates that blocking either CD11b or CD18 (7E4) abrogated PR3-ANCA-induced superoxide generation. The use of an additional blocking CD18 Ab recognizing a different epitope (MHM23) gave similar results. Superoxide generation increased from 2.3 ± 0.9 nmol O2−/7.5 × 105 cells with control IgG to 19.5 ± 1.9 nmol O2−/7.5 × 105 cells with PR3-ANCA. The blocking CD18 antibody reduced PR3-ANCA-induced superoxide to 3.2 ± 0.5 nmol (n = 3 independent experiments using 2 different PR3-ANCA preparations). These data indicate that the NB1-dependent neutrophil activation by PR3-ANCA utilizes Mac-1.
FIGURE 7.

Mac-1 blockade abrogates degranulation and superoxide generation induced by anti-PR3 antibodies in a ligand-independent fashion. Neutrophils were preincubated for 30 min with blocking mabs to CD11b, CD18, and isotype control, respectively. After TNF-α priming, cells were treated with mabs to PR3 (αPR3) or isotype control (panels A, B, and E) or with IgG from 2 different patients with PR3-ANCA or human controls (panels C and F). Degranulation (panel A, n = 3) and ferricytochrome c reduction (panels B and C, n = 6) were measured. Blocking mAb to CD11b, CD18, but not an isotype control significantly reduced activation of TNF-α-primed neutrophils in response to mabs to PR3 and PR3-ANCA from patients. Neutrophil adhesion on a plastic and an ultra-low adhesion surface in parallel (panel D, n = 7). 5 × 105 neutrophils were stimulated with 2 ng/ml TNF-α. After 45 min., wells were washed and adherent cells were estimated by the MPO activity assay. Neutrophils on ultra-low attachment plates were preincubated with blocking mabs to CD11b, CD18, and isotype control, respectively. After TNF-α priming, cells were treated with mabs to PR3 (panel E, n = 7) or with IgG from 2 different patients with PR3-ANCA patients (panel F, n = 5). Appropriate isotype control antibodies (isotype) and IgG preparations from normal (control) were used for comparison. Adhesion and ferricytochrome c reduction was measured in parallel. * indicates p < 0.05.
Blocking CD11b or CD18 resulted in decreased superoxide production in response to anti-PR3 mAbs and to PR3-ANCA. Given the fact that NB1 and Mac-1 interact and co-localize, these findings are consistent with the hypothesis that NB1 and CD11b/18 have inter-receptor interactions that transduce the PR3-ANCA-mediated activation. Alternatively, the β2-integrin could be activated by inside out signaling and consequently provide a second, ligand-dependent outside-in signal. To gain further insight, we performed the superoxide assay in 96-well plates with an ultra-low hydrogel attachment surface and, in addition, agitated the cells during the assay. Cell adhesion assays established that, under these culture conditions, neutrophil adhesion in response to TNF-α was indeed abrogated (Fig. 7D). We then repeated the ferrricytochrome c assay under these strict non-adherent conditions and observed that CD18 blockade still abrogated superoxide production in response to anti-PR3 mAb and to human PR3-ANCA (Fig. 7, E and F). Thus, these results strongly implicate NB1-Mac-1 receptor interaction as an important signaling mechanism in anti-PR3 antibody-induced neutrophil activation, whereas co-stimulation via ligand-mediated outside-in signals was not necessary.
DISCUSSION
Our data enhance our earlier findings, that PR3 is presented on the membrane of a neutrophil subset by the GPI-anchored NB1 receptor (18). NB1 lacks a transmembrane domain that is necessary for intracellular signal transduction and subsequent neutrophil activation by PR3-ANCA. We show here that PR3-ANCA stimulates degranulation and superoxide production in the mNB1/mPR3-expressing subset only and that the β2-integrin Mac-1 is part of a larger PR3-NB1 signaling complex. The CD11b and CD18 chain are functionally important for neutrophil activation via the NB1 signaling complex and are at work when degranulation and superoxide generation in mNB1pos/mPR3high neutrophils are stimulated by PR3-ANCA. The data also suggest that NB1 and Mac-1 cooperate by receptor-receptor interaction rather than ligand-dependent co-stimulation.
PR3-ANCA and MPO-ANCA activate cytokine-primed neutrophils. PR3, but not MPO shows a bimodal membrane expression pattern on neutrophils. High mPR3 expression is only found on those neutrophils that co-express the GPI-linked NB1 receptor (17, 18, 20). We performed previously ectopic expression studies showing that NB1 was a sufficient receptor for PR3 (18, 35). Korkmaz et al. (19) elegantly showed that a hydrophobic patch of the PR3 molecule mediates membrane binding via NB1 receptors. No data so far explain how PR3-ANCA-mediated neutrophil activation via the GPI-linked non-signaling NB1 receptor can occur. This issue may be even of broader interest, since NB1 is also implicated in other antibody-mediated diseases, such as neonatal neutropenia and transfusion-related acute lung injury (TRALI) and functions as a counter-receptor for PECAM-1 (CD31) (27, 36–38). We performed parallel assessment of extracellular superoxide and intracellular oxidants in ANCA-stimulated neutrophils. Our data clearly confirm that the mNB1/mPR3 phenotype matters for degranulation and for the NADPH oxidase-mediated extracellular superoxide generation to PR3-ANCA. Interestingly, these head-to-head comparisons with respect to the respiratory burst indicate that both the mNB1/mPR3low and mNB1/mPR3high phenotype responded to PR3-ANCA with similar intracellular oxidant production. Thus, there is only an apparent controversy between our data and a recent study by Hu et al. (20). These investigators separated neutrophils in mNB1/mPR3low and mNB1/mPR3high subset followed by anti-PR3 mAb stimulation. Respiratory burst was measured by DHR oxidation. Results were variable, but suggested that similar amounts of intracellular oxidants were generated by both cell subsets. We believe that our parallel experiments settle a potentially time- and labor-consuming conflict. It is conceivable that the mNB1/mPR3 phenotype causes differences in ANCA internalization that in turn result in differences between extra- and intracellular oxidant production. Future studies are needed to address this issue.
GPI-linked receptors utilize transmembrane proteins to initiate cell signaling and activation. We detected several signaling molecules that were complexed with NB1 in carefully digitonin-extracted membranes. We focused on the transmembrane integrin chains CD11b and CD18 that form the β2-integrin Mac-1 because they were previously shown to cooperate with GPI-anchored receptors in neutrophils. For example Mac-1 cooperates with the GPI-linked receptors for uPA, CD14, FcgIII and GPI-80 in adhesion, phagocytosis, chemotactic movement, and respiratory burst (21, 22, 25, 26, 39). Previously, David et al. (40) described co-localization of mPR3 and CD11b/CD18 in neutrophil plasma membrane preparations. Our data extend this report by showing that the mPR3-presenting NB1 receptor and Mac-1 co-immunoprecipitate, interact physically and co-localize on the neutrophil membrane and that all 3 molecules, namely PR3, NB1, and Mac-1 exist in lipid rafts. Lipid rafts provide a highly interactive platform allowing the dynamic regulation of signaling proteins.
Three α-subunits (CD11a, CD11b, CD11c) are expressed together with the CD18 subunit on the neutrophil cell surface. Analysis of direct protein-protein interaction showed that NB1 could also react with CD11a. However, this reaction was not observed on the cell level; CD11a/CD18-transfected cells did not interact with immobilized NB1 protein. Together with the fact that CD11b, compared with CD11a, exist in higher surface density, our results implicate that CD11b/CD18 acts as the major co-receptor for NB1 in neutrophils. However, based on our MS/MS results, it is very likely that more NB1-associated molecules might exist in an even larger membrane-associated signaling complex. The functional role of these molecules needs to be explored in future studies.
Using different Mac-1 blocking antibodies to either CD11b or CD18, and even different epitopes to the latter, we demonstrate that both β2-integrin transmembrane chains are functional important for PR3-ANCA-induced degranulation and superoxide generation. We reasoned that Mac-1 could participate in at least two ways in PR3-ANCA-initiated activation. One could envision receptor-receptor interaction between Mac-1 and NB1 and alternatively, a ligand-dependent co-stimulatory outside-in Mac-1 signal. Our data using strict non-adherent conditions strongly suggest that NB1-Mac-1 receptor interactions occur and that no β2-integrin outside-in signaling is needed. Furthermore, we show by different approaches that the extracellular NB1 and Mac-1 domains interact with each other and that both molecules co-localize on the neutrophil plasma membrane. Several models of inter-receptor interactions are conceivable, including receptor coordination, compartmentalization, lateral transactivation, and modulation of receptor expression (25, 41). Future experiments will identify the specific mechanisms that are at work.
Our results could become highly clinically relevant. We identified Mac-1 as a new member of a larger NB1 membrane complex. We provide evidence that Mac-1 is functionally important for NB1-mediated neutrophil activation by PR3-ANCA. We suggest that targeting the PR3-NB1-Mac-1 signaling complex might be a novel strategy to block PR3-ANCA-mediated neutrophil activation.
Supplementary Material
Acknowledgment
We thank Monika Burg-Roderfeld for excellent assistance with the SPR experiments.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG: KE 576/7-1), and a grant from the Experimental and Clinical Research Center (ECRC) Berlin-Buch and from the Transregional Collaborative Research Center (SFB/TRR84) supported this study.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
- ANCA
- anti-neutrophil cytoplasmic auto-antibodies
- GPI
- glycosylphosphatidylinositol
- fMLP
- f-met-leu-phe
- PMA
- phorbol-2-myristate-13-acetate
- DHR
- 2.2′-azino-bis (3-ethylbenzthiazoline-6-sulfonicacid), dihydrorhodsamine
- DFP
- diisopropylfluorophosphate.
REFERENCES
- 1. van der Woude F. J., Rasmussen N., Lobatto S., Wiik A., Permin H., van Es L. A., van der Giessen M., van der Hem G. K., Hauu The T. (1985) Lancet i, 425–429 [DOI] [PubMed] [Google Scholar]
- 2. Falk R. J., Jennette J. C. (1988) N. Engl. J. Med. 318, 1651–1657 [DOI] [PubMed] [Google Scholar]
- 3. Falk R. J., Terrell R. S., Charles L. A., Jennette J. C. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 4115–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savage C. O., Pottinger B. E., Gaskin G., Pusey C. D., Pearson J. D. (1992) Am. J. Pathol. 141, 335–342 [PMC free article] [PubMed] [Google Scholar]
- 5. Xiao H., Heeringa P., Hu P., Liu Z., Zhao M., Aratani Y., Maeda N., Falk R. J., Jennette J. C. (2002) J. Clin. Invest. 110, 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfister H., Ollert M., Froehlich L. F., Quantanilla-Martinez L., Colby T. V., Specks U., Jenne D. (2004) Blood 104, 1411–1418 [DOI] [PubMed] [Google Scholar]
- 7. Little M. A., Smyth C. L., Yadav R., Ambrose L., Cook H. T., Nourshargh S., Pusey C. D. (2005) Blood 106, 2050–2058 [DOI] [PubMed] [Google Scholar]
- 8. Xiao H., Heeringa P., Liu Z., Huugen D., Hu P., Maeda N., Falk R. J., Jennette J. C. (2005) Am. J. Pathol. 167, 39–45 [DOI] [PubMed] [Google Scholar]
- 9. Schreiber A., Xiao H., Falk R. J., Jennette J. C. (2006) J. Am. Soc. Nephrol. 17, 3355–3364 [DOI] [PubMed] [Google Scholar]
- 10. Halbwachs-Mecarelli L., Bessou G., Lesavre P., Lopez S., Witko-Sarsat V. (1995) FEBS Lett. 374, 29–33 [DOI] [PubMed] [Google Scholar]
- 11. Schreiber A., Busjahn A., Luft F. C., Kettritz R. (2003) J. Am. Soc. Nephrol. 14, 68–75 [DOI] [PubMed] [Google Scholar]
- 12. von Vietinghoff S., Busjahn A., Schönemann C., Massenkeil G., Otto B., Luft F. C., Kettritz R. (2006) J. Am. Soc. Nephrol. 17, 3185–3191 [DOI] [PubMed] [Google Scholar]
- 13. Witko-Sarsat V., Lesavre P., Lopez S., Bessou G., Hieblot C., Prum B., Noël L. H., Guillevin L., Ravaud P., Sermet-Gaudelus I., Timsit J., Grünfeld J. P., Halbwachs-Mecarelli L. (1999) J. Am. Soc. Nephrol. 10, 1224–1233 [DOI] [PubMed] [Google Scholar]
- 14. Rarok A. A., Stegeman C. A., Limburg P. C., Kallenberg C. G. (2002) J. Am. Soc. Nephrol. 13, 2232–2238 [DOI] [PubMed] [Google Scholar]
- 15. Schreiber A., Otto B., Ju X., Zenke M., Goebel U., Luft F. C., Kettritz R. (2005) J. Am. Soc. Nephrol. 16, 2216–2224 [DOI] [PubMed] [Google Scholar]
- 16. Schreiber A., Luft F. C., Kettritz R. (2004) Kidney Int. 65, 2172–2183 [DOI] [PubMed] [Google Scholar]
- 17. Bauer S., Abdgawad M., Gunnarsson L., Segelmark M., Tapper H., Hellmark T. (2007) J. Leukoc. Biol. 81, 458–464 [DOI] [PubMed] [Google Scholar]
- 18. von Vietinghoff S., Tunnemann G., Eulenberg C., Wellner M., Cristina, Cardoso M., Luft F. C., Kettritz R. (2007) Blood 109, 4487–4493 [DOI] [PubMed] [Google Scholar]
- 19. Korkmaz B., Kuhl A., Bayat B., Santoso S., Jenne D. E. (2008) J. Biol. Chem. 283, 35976–35982 [DOI] [PubMed] [Google Scholar]
- 20. Hu N., Westra J., Huitema M. G., Bijl M., Brouwer E., Stegeman C. A., Heeringa P., Limburg P. C., Kallenberg C. G. (2009) Arthritis Rheum. 60, 1548–1557 [DOI] [PubMed] [Google Scholar]
- 21. Bohuslav J., Horejsi V., Hansmann C., Stöckl J., Weidle U. H., Majdic O., Bartke I., Knapp W., Stockinger H. (1995) J. Exp. Med. 181, 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe T., Sendo F. (2002) Biochem. Biophys. Res. Commun. 294, 692–694 [DOI] [PubMed] [Google Scholar]
- 23. Blasi F., Carmeliet P. (2002) Nat. Rev. Mol. Cell Biol. 3, 932–943 [DOI] [PubMed] [Google Scholar]
- 24. Klein R. D., Sherman D., Ho W. H., Stone D., Bennett G. L., Moffat B., Vandlen R., Simmons L., Gu Q., Hongo J. A., Devaux B., Poulsen K., Armanini M., Nozaki C., Asai N., Goddard A., Phillips H., Henderson C. E., Takahashi M., Rosenthal A. (1997) Nature 387, 717–721 [DOI] [PubMed] [Google Scholar]
- 25. Petty H. R., Todd R. F., 3rd (1996) Immunol. Today 17, 209–212 [DOI] [PubMed] [Google Scholar]
- 26. Stöckl J., Majdic O., Pickl W. F., Rosenkranz A., Prager E., Gschwantler E., Knapp W. (1995) J. Immunol. 154, 5452–5463 [PubMed] [Google Scholar]
- 27. Sachs U. J., Andrei-Selmer C. L., Maniar A., Weiss T., Paddock C., Orlova V. V., Choi E. Y., Newman P. J., Preissner K. T., Chavakis T., Santoso S. (2007) J. Biol. Chem. 282, 23603–23612 [DOI] [PubMed] [Google Scholar]
- 28. Socher I., Andrei-Selmer C., Bein G., Kroll H., Santoso S. (2009) Transfusion 49, 943–952 [DOI] [PubMed] [Google Scholar]
- 29. Kowalski M. P., Dubouix-Bourandy A., Bajmoczi M., Golan D. E., Zaidi T., Coutinho-Sledge Y. S., Gygi M. P., Gygi S. P., Wiemer E. A., Pier G. B. (2007) Science 317, 130–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kjeldsen L., Sengelov H., Borregaard N. (1999) J. Immunol. Methods 232, 131–143 [DOI] [PubMed] [Google Scholar]
- 31. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Fröhlich F., Walther T. C., Mann M. (2008) Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 32. Cox J., Mann M. (2008) Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 33. Santoso S., Sachs U. J., Kroll H., Linder M., Ruf A., Preissner K. T., Chavakis T. (2002) J. Exp. Med. 196, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pick E., Mizel D. (1981) J. Immunol. Methods 46, 211–226 [DOI] [PubMed] [Google Scholar]
- 35. von Vietinghoff S., Eulenberg C., Wellner M., Luft F. C., Kettritz R. (2008) Clin Exp Immunol. 152, 508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pocock C. F., Lucas G. F., Giles C., Vassiliou G., Cwynarski K., Rezvani K., Apperley J. F., Goldman J. M. (2001) Br. J. Haematol. 113, 483–485 [DOI] [PubMed] [Google Scholar]
- 37. Sachs U. J., Hattar K., Weissmann N., Bohle R. M., Weiss T., Sibelius U., Bux J. (2006) Blood 107, 1217–1219 [DOI] [PubMed] [Google Scholar]
- 38. Bayat B., Werth S., Sachs U. J., Newman D. K., Newman P. J., Santoso S. (2010) J. Immunol. 184, 3889–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peyron P., Bordier C., N′Diaye E. N., Maridonneau-Parini I. (2000) J. Immunol. 165, 5186–5191 [DOI] [PubMed] [Google Scholar]
- 40. David A., Kacher Y., Specks U., Aviram I. (2003) J. Leukoc. Biol. 74, 551–557 [DOI] [PubMed] [Google Scholar]
- 41. Miranti C. K., Brugge J. S. (2002) Nat Cell Biol. 4, E83–E90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.