

Abstract
ATP-binding cassette (ABC) systems translocate a wide range of solutes across cellular membranes. The thermophilic Gram-negative eubacterium Thermus thermophilus, a model organism for structural genomics and systems biology, discloses ∼46 ABC proteins, which are largely uncharacterized. Here, we functionally analyzed the first two and only ABC half-transporters of the hyperthermophilic bacterium, TmrA and TmrB. The ABC system mediates uptake of the drug Hoechst 33342 in inside-out oriented vesicles that is inhibited by verapamil. TmrA and TmrB form a stable heterodimeric complex hydrolyzing ATP with a Km of 0.9 mm and kcat of 9 s−1 at 68 °C. Two nucleotides can be trapped in the heterodimeric ABC complex either by vanadate or by mutation inhibiting ATP hydrolysis. Nucleotide trapping requires permissive temperatures, at which a conformational ATP switch is possible. We further demonstrate that the canonic glutamate 523 of TmrA is essential for rapid conversion of the ATP/ATP-bound complex into its ADP/ATP state, whereas the corresponding aspartate in TmrB (Asp-500) has only a regulatory role. Notably, exchange of this single noncanonic residue into a catalytic glutamate cannot rescue the function of the E523Q/D500E complex, implicating a built-in asymmetry of the complex. However, slow ATP hydrolysis in the newly generated canonic site (D500E) strictly depends on the formation of a posthydrolysis state in the consensus site, indicating an allosteric coupling of both active sites.
Keywords: ABC Transporter, ATPases, Membrane Proteins, Multidrug Transporters, Transport Drugs
Introduction
Movement of solutes across cellular membranes is an essential biological process mediated by a number of transport proteins. One of the largest protein families involved in translocation processes are the ATP-binding cassette (ABC)2 transporters, which are integral membrane proteins present in all three phyla of life. Members of this superfamily energize the transmembrane movement of a broad range of solutes, such as lipids, sugars, ions, amino acids, peptides, proteins, or noxious compounds and therefore play a crucial role in various cellular processes (1, 2). In bacteria, they act either as import systems that mediate uptake of, for example, nutrients or as export systems that are mainly involved in cellular detoxification and self-defense.
ABC transporters share a common architecture comprising two transmembrane domains (TMDs) that assemble the translocation pathway and two nucleotide-binding domains (NBDs), which convert the chemical energy of ATP binding/hydrolysis in conformational changes of the TMDs. ABC importers typically consist of four subunits. An additional binding protein captures solutes and hands them over to the outward facing TMDs. In contrast, ABC exporters comprise all four domains (two NBDs and two TMDs) in one polypeptide (full-length transporter) or in two half-transporters, each consisting of one TMD and one NBD, which together assemble to a homodimeric or heterodimeric transport complex (3, 4).
Various bacterial and eukaryotic ABC export systems have been identified that mediate a multidrug resistance phenotype (5, 6). These multidrug resistance proteins are able to export a wide range of chemically unrelated compounds and therefore play a fundamental role in cellular resistance against noxious compounds, like antibiotics or anticancer drugs. Because multidrug ABC proteins cause severe problems in public health, they are a prime target in clinical research (7). In humans, multidrug resistance is caused by several ABC proteins, such as the full-length transporter P-gp (ABCB1), MRP1 (ABCC1), or homooligomeric BCRP (ABCG2) (8–10). In bacteria, most multidrug transport proteins are secondary transporters. However, several ABC-type multidrug transporters have been identified, in particular in Gram-positive bacteria, including LmrA from Lactococcus lactis, Sav1866 from Staphylococcus aureus, or BmrA from Bacillus subtilis, all of which assemble as homodimeric ABC complexes (11–13). Additionally, a small number of heterodimeric efflux pumps, e.g. LmrCD from L. lactis, YheI/H from B. subtilis, or SmdAB from Serratia marcescens, are known in prokaryotes mediating resistance against antimicrobial agents (14–16). The assembly of two functionally different ABC subunits is a well known feature of eukaryotic transporters like TAP1/2 (ABCB2/3), CFTR, or MRP1 (17–19). Typically, the NBDs of these ABC systems are not functionally equivalent and interchangeable, as was shown for P-gp (20), suggesting a functional asymmetry regarding nucleotide binding and hydrolysis (18, 21–23). Similar observations were made for the bacterial multidrug ABC transporter LmrCD (24). However, the mechanism of heterodimeric ABC transporter-mediated multidrug efflux in prokaryotes, in particular in Gram-negative bacteria, is poorly understood.
To obtain insight in the catalytic cycle of asymmetric ABC export complexes, we identified and characterized the solely heterodimeric ABC half-transporter encoded in the genome of the thermophilic eubacterium Thermus thermophilus HB27. Because of its homology to various multidrug transport proteins, such as LmrCD or P-gp, we named the subunits T. thermophilus multidrug resistance protein A and B (TmrA and TmrB). Both ABC proteins were expressed in Escherichia coli. TmrAB-mediated Hoechst 33342 uptake was shown in inside-out oriented vesicles (IOV), implicating a role of the ABC transporter in the phenomenon of multidrug resistance. To obtain mechanistic data of the ABC export system from a hyperthermophilic Gram-negative eubacterium, the TmrAB subunits were co-purified as a stable heterodimeric ABC transport complex. Biochemical analysis of ATP binding and hydrolysis revealed a functional nonequivalence of the NBDs demonstrating the asymmetric function of TmrAB subunits in the ATP hydrolysis cycle.
EXPERIMENTAL PROCEDURES
Materials
Radiolabeled compounds and DDM were purchased from Hartmann Analytics and Carl Roth, respectively. Enzymes for cloning were ordered from Fermentas. The concentration of total membrane or solubilized proteins was determined using the Bradford assay (Pierce) and micro BCA assay (Pierce), respectively.
Expression of TmrAB
The tmrA/B genes (TTC0976 and TTC0977) were amplified from genomic DNA of T. thermophilus HB27 by PCR using the oligonucleotides 5′-GGAATTCCATATGACCGGTCGGTCA-3′ and 5′-GGAATTCCATATGACCGGTCGGTCAG-3′. The PCR product was cloned into the pET22b+ vector (Novagen) as an NdeI/HindIII fragment, resulting in pET-TmrAB. To introduce a TEV-cleavable His10 tag at the C terminus of TmrA, the primer pair 5′-GGCCGCTCAGTGATGGTGATGGTGATGGTGATGGTGATGGCCCTGAAAGTATAGGTTTTCACCACCACCACCA-3′ and 5′-AGCTTGGTGGTGGTGGTGAAAACCTATACTTTCAGGGCCATCACCATCACCATCACCATCACCATCACTGAGC-3′ was hybridized and ligated into pET-TmrAB by using the HindIII and NotI restriction sites. The resulting expression vector is called pET-TmrAB-His10. Single as well as double mutants were obtained by using LCR with the following mutagenesis primers: TTC0976-E523Q (5′-ATCCTGGACCAGGCCACGG-3′), TTC0977-D500N (5′-CTCATCCTGGACAACGCCTTAAGC-3′), and TTC0977-D500E (5′-TCATCCTGGACGAAG CCTTAAGC-3′). All of the constructs were confirmed by DNA sequencing. The expression vectors were transformed into E. coli strain BL21 (DE3) (Novagen). The cells were grown in LB medium at 37 °C and 180 rpm to the mid-logarithmic phase. Expression was induced at an A600 of 0.8 by addition of isopropyl β-d-thiogalactopyranoside (0.5 mm). After induction, the cells were incubated for 3 h at 37 °C before harvesting. To analyze the expression level, E. coli cells were supplemented with HP-protease inhibitor mix (Serva) and broken by 10 cycles of freeze and thaw. After benzonase treatment (20 units/ml; Merck), the cell extract was analyzed by SDS-PAGE (10%, Coomassie staining) and quantitative immunoblotting using an anti-His antibody (Novagen).
Purification of TmrAB
E. coli cell pellets were resuspended in lysis buffer (20 mm HEPES, pH 7.5, 150 mm NaCl) containing HP protease inhibitor mix (Serva) and benzonase (3 units/ml; Merck). Subsequently, the cells were broken by a cell disrupter (1.7 kbar; Basic Z, Constant Systems). Cell lysates were supplemented with 10 mm EDTA, pH 8.0, followed by removal of cell debris via a low speed centrifugation step (6,800 × g; 10 min). The supernatant was centrifuged at 115,000 × g for 45 min to obtain E. coli membranes. For solubilization, the membranes were resuspended in 1% DDM (w/v), 20 mm HEPES, pH 7.5, 150 mm NaCl, and 2 mm imidazole, pH 7.5, to a final concentration of 5 mg/ml total protein and incubated for 1 h at 4 °C under gentle shaking. Solubilized proteins were heated up to 70 °C for 20 min. Precipitated E. coli proteins were removed by high speed centrifugation at 115,000 × g for 30 min, and the supernatant was applied on a nickel-loaded HiTrapChelating HP-column (1 ml; GE Healthcare) pre-equilibrated with purification buffer (0.02% DDM, 20 mm HEPES, 150 mm NaCl, and 2 mm imidazole, pH 7.5). The column was washed with 10 mm imidazole (20 column volumes) in purification buffer. Specifically bound proteins were eluted with 250 mm imidazole in purification buffer (5 column volumes). Fractions containing TmrAB were pooled and stored at 4 °C. The proteins (wild type and mutants) were purified to greater than 98% homogeneity as demonstrated by SDS-PAGE (10%, Coomassie staining) and identified by MALDI-MS.
For MALDI-MS, 2 μm TmrAB (10 mm HEPES, pH 7.5, 0.02% DDM) were diluted 1:10 in solvent A (H2O/acetonitrile (70/30) supplemented with 0.1% TFA). The matrix used was a solution of 20 mg/ml sDHB (mixture of 2,5-dihydroxybenzoic acid and 5-methoxysalicylic acid; Bruker Daltonics) in solvent A. 1 μl of protein solution and 1 μl of matrix solution were mixed directly on the sample target and dried in a stream of cold air. MALDI-MS was carried out in the linear, positive ion mode on a Voyager DE-Pro (Applied Biosystems).
Complex Formation of TmrAB
Oligomerization of TmrAB was analyzed by size exclusion chromatography (TSK gel G3000SW; Tossoh Bioscience LLC). 50 μl of TmrAB (1 μm) were separated in 20 mm HEPES, 150 ml NaCl, pH 7.0, and 0.02% DDM at 4 °C with a flow rate of 0.5 ml/min. For calibration, thyroglobulin (669 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), and carbonic anhydrase (29 kDa) were used.
LILBID-MS
The quaternary structure of TmrAB was investigated by ultra soft LILBID-MS (25–27). Briefly, tiny droplets of native solution (65 pl) were injected into high vacuum where they were irradiated by mid-IR laser pulses. During the subsequent explosion, the analyte molecules were set free and analyzed by a home-built TOF mass spectrometer. For the LILBID-MS analysis, an aliquot of the stock solution (75 μm) was diluted to 10% using a 10 mm HEPES buffer, pH 7.5, containing 0.02% DDM. The spectra were obtained by averaging over 300 droplets after loading a total volume of ∼5 μl.
Drug Translocation in Inside-out Vesicles
E. coli cells expressing TmrAB (T7 Express; New England BioLabs) were harvested by centrifugation at 6,000 × g for 15 min at 4 °C and resuspended in lysis buffer (250 mm sucrose, 150 mm NaCl, 2.5 mm MgSO4, 20 mm HEPES, pH 7.5) containing HP protease inhibitor mix (Serva) and benzonase (4 units/ml). The cells were lysed by three passages trough a cell disrupter (1.7 kbar; Basic Z; Constant Systems). Subsequently, 15 mm EDTA (pH 8.0) was added, and cell debris was removed by centrifugation at 8,000 × g for 10 min at 4 °C. IOVs were harvested at 100,000 × g for 1 h at 4 °C and resuspended in 50 mm potassium phosphate buffer, pH 7.0, supplemented with 10% glycerol to a protein concentration of ∼10 mg/ml. The membrane vesicles were frozen in liquid nitrogen and stored at −80 °C.
To analyze the orientation of membrane insertion of TmrAB, the accessibility of the His10 tag in IOVs was determined before and after TEV cleavage. IOVs (10 μg of total protein) were incubated overnight at 30 °C in the presence of TEV protease (2 units; MobiTec). As positive control, the protein was treated with TEV protease and 2% Triton X-100 to guarantee accessibility of the vesicle lumen. Proteins were separated via SDS-PAGE (10%), and the orientation of TmrAB was analyzed by subsequent immunoblotting using an anti-His antibody (Novagen). Quantification of luminescence signal was carried out with a Lumi-Imager F1 (Roche Applied Science) and the LumiAnalyst software (version 3.1).
For Hoechst 33342 uptake, IOVs were diluted to a concentration of 0.6 mg/ml in potassium phosphate buffer (pH 7.0) containing 2 mm MgSO4. After incubation for 80 s at 37 °C and 50 °C, respectively, Hoechst 33342 (2-(4-ethoxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5-bi-1H-benzimidazole; Molecular Probes) was added to a final concentration of 0.75 μm. After equilibration, IOVs were energized with 1.5 mm ATP, and changes of Hoechst 33342 fluorescence were monitored over time at excitation and emission wavelengths of 355 and 457 nm, respectively, using the FP-6500 spectrofluorometer (Jasco). Proton motive force was dissipated by the addition of the ionophore nigericin (1 μm) and the F-type ATPase inhibitor NaN3 (10 mm). Verapamil known as a competitor for Hoechst 33342 was used in a concentration range from 0 to 1 mm. IC50 value was determined using the following equation (whereas F represents the fluorescence intensities).
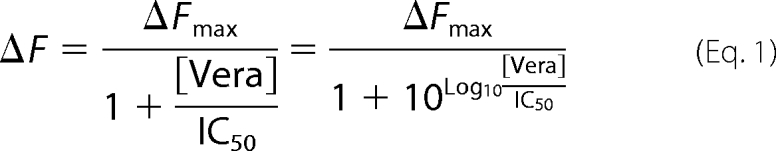 |
ATPase Assay
ATPase activity was determined by the release of inorganic phosphate as described previously (28, 29). 25 μl of purified TmrAB (0.12 μm) were mixed with 25 μl of ATP buffer (20 mm HEPES, 150 mm NaCl, 0.02% DDM, 20 mm MgCl2 pH 7.5) containing 10 mm of ATP, supplemented with [γ-32P]ATP (5 μCi). The reactions were performed for 4 min at 68 °C. The addition of the ABC ATPase inhibitors BeFx (5 mm) or orthovanadate (5 mm, pH 8.0) and the F-type ATPase inhibitor NaN3 (10 mm) verified the specificity of the ATPase activity. ATP hydrolysis was quenched by addition of 1 ml of reagent A (10 mm ammonium molybdate in 1 n HCl). Subsequently, 15 μl of reagent B (20 mm H2PO4) and 2 ml of reagent C (isobutanol, cyclohexane, acetone, and reagent A in a ratio of 5:5:1:0.1) were added. The samples were mixed by vigorous vortexing for 30 s. After phase separation, 1 ml of organic phase was added to 2 ml of scintillation fluid (Rotiszint eco plus; Carl Roth), and the amount of released inorganic phosphate was quantified by β-counting (Beckman LS 6500 liquid scintillation counter). The IC50 value for inhibition of ATPase by Hoechst 33342 was determined by fitting data to the dose-response curve (Equation 1).
To determine the vmax and Km values, the data were fitted to the Michaelis-Menten equation,
 |
where v represents the rate of ATP hydrolysis, and [S] represents the concentration of ATP.
8-Azido-ATP Photo-labeling
Nucleotide binding and trapping was visualized by 8-azido-ATP photo-cross-linking experiments. Purified wild type TmrAB (0.5 μm) was incubated with 4 μm of either 8-N3-[α-32P]ATP (9.8 Ci/mmol) or 8-N3-[γ-32P]ATP (11.7 Ci/mmol) in the presence of 1 mm MgCl2 and 0.4 mm orthovanadate. Experiments with the ATPase-deficient E523Q/WT mutant were performed in the absence of orthovanadate. To analyze ATP binding, the samples were incubated for 4 min at room temperature followed by 4 min of UV irradiation (366 nm; Atlas Fluotest® forte). For vanadate trapping experiments probing the posthydrolysis state, protein samples were heated up to 68 °C for 45 s in the absence or presence of 5 mm ATP, to show the specificity of cross-linking reaction. Subsequently, 4 μm of radiolabeled azido-ATP was added, and the samples were incubated for 2 min on ice. Prior to cross-linking, the samples were supplemented with an excess of cold ATP (2 mm), to demonstrate tight occlusion of nucleotides in the NBDs. UV cross-linking was carried out for 4 min. The cross-linked samples were analyzed by SDS-PAGE (10%) and autoradiography.
Stoichiometry of Nucleotide Binding
Bound nucleotides were quantified by trapping experiments. To this end, the TmrAB complex (2 μm) was incubated for 3.5 min at 4 or 68 °C in the presence of 5 mm MgCl2 and 700 μm ATP, traced with 70 nm of either [α-32P]ATP or [γ-32P]ATP (3 Ci/μmol). 5 mm orthovanadate, pH 8.0, was added to wild type TmrAB and ATPase-active mutants to trap the nucleotides in the posthydrolysis state. To demonstrate specificity of nucleotide trapping, the samples were incubated in the absence or presence of 10 mm ATP and AMP, respectively. After incubation, the samples were cooled down to RT for 2 min and supplemented with an excess of ATP (2 mm) to probe for tight trapping of the nucleotides in the NBDs. To separate bound from free nucleotides, 50 μl of reaction mixture was loaded onto a Micro Bio-Spin® 30 gel filtration column, pre-equilibrated in purification buffer containing 2 mm MgCl2, and centrifuged for 4 min at room temperature. To quantify trapped nucleotides, the eluate was mixed with 2 ml of scintillation fluid (Rotiszint®eco plus; Carl Roth). The bound nucleotides were quantified by β-counting (Beckman LS 6500 liquid scintillation counter).
The identity of trapped nucleotides was determined by thin layer chromatography. After spin-down, ATP-trapped protein samples were immediately incubated with 10 mm EDTA and 1% SDS for 5 min. Afterward 1 μl of the sample was applied onto a polyethyleneimine cellulose plate (Merck). The nucleotides were separated in 0.75 m KH2PO4, pH 3.4, and analyzed by phosphorus imaging (Phosphor-Imager 445Si; Molecular Dynamics).
RESULTS
Identification of ABC Half-transporters TmrA and TmrB
Analysis of the genome of the thermophilic Gram-negative bacterium. T. thermophilus HB27 (30) revealed the presence of ∼46 different ABC systems, predicted by homology to be involved in various cellular processes, including nutrient uptake or extrusion of cytotoxic compounds. The genes TTC0976 (tmrA) and TTC0977 (tmrB) code for two half-site ABC transporters, which are clustered in the same operon and share overlapping open reading frames. Thus, expression of both genes should be tightly regulated (31). The two gene products feature the typical architecture of ABC half-transporters, with a TMD followed by a NBD (Fig. 1A). The analysis of the membrane topology using HMMTOP 2.0 (32) revealed that the TMDs of TmrA and TmrB are composed of six transmembrane segments. All of the characteristic ABC motifs, Walker A/B, and C- and H-loops, which are involved in ATP binding and hydrolysis, are conserved in TmrA and TmrB (Table 1). However, in TmrB the putative catalytic glutamate next to the Walker B motif is substituted by aspartate (Asp-500), leading to a noncanonic ATP-binding site (Fig. 1A). In bacteria and eukaryotes, several ABC half-transporters with asymmetric NBDs have been identified, such as LmrCD from L. lactis (24), human TAP1/2 (17), and ABCG5/G8 (33). In contrast to TmrAB, these heterodimeric ABC complexes exhibit additional variations in conserved ABC sequence motifs, such as the C- and H-loop. However, the way in which a canonic and noncanonic NBD allosterically coordinate ATP binding and hydrolysis is still unresolved. Because the asymmetry in TmrAB appears to be focused on a single residue (Asp-500 in TmrB), the ABC complex represents an interesting model system. Thus, we mutated the putative catalytic base in both TmrAB subunits to the respective amides, resulting in E523Q/WT and WT/D500N, respectively. Furthermore, we introduced a glutamate residue in the noncanonic TmrB site, resulting in WT/D500E. Additionally, double mutants E523Q/D500N and E523Q/D500E were generated.
FIGURE 1.

Expression and purification of the TmrAB complex. A, topology model of the ABC transporters TmrA and TmrB. Each polypeptide is composed of a TMD and a NBD. Both TMDs consist of six transmembrane helices as predicted by HMMTOP 2.0 (32). Conserved ABC motifs (Walker A/B and C-loop) are found in the NBDs of both subunits. The position of the putative catalytic residue next to the Walker B motif is indicated. In TmrB, the catalytic glutamate is substituted by an aspartate. At the C terminus of TmrA, a TEV cleavage site followed by a His10 tag was introduced for purification of the ABC transport complex. B, expression of TmrA and TmrB. After isopropyl β-d-thiogalactopyranoside induction, the expression of TmrAB in E. coli BL21 (DE3) was analyzed by SDS-PAGE (10%, Coomassie staining, upper panel) and immunoblotting using the anti-His antibody (lower panel). 5 μg of total protein were applied per lane. C, purification of the TmrAB complex. Wild type and various mutants (E523Q/WT and WT/D500N) were solubilized in 1% DDM and purified via metal affinity chromatography. The heterodimeric ABC transport complex was analyzed by SDS-PAGE (10%, Coomassie staining).
TABLE 1.
Conserved ABC motifs of TmrAB
X and Φ indicate random amino acid and hydrophobic residues, respectively. Letters in bold type illustrate key residues within each motif involved in ATP binding/hydrolysis.
| Walker A | Walker B | C-loop | D-loop | H-switch | Q-loop | |
|---|---|---|---|---|---|---|
| Consensus | (G/A)XXXXGK(T/S) | ΦΦΦΦDE | LSGGQ | SALD | H | Q |
| TmrA | GATGAGKT | LLILDE | LSTGE | ASVD | AHRL | VLQE |
| TmrB | GRTGSGKS | ILILDD | LSGGQ | SAVD | SHRT | APQE |
Expression and Purification of TmrAB
For functional studies, we expressed TmrAB in E. coli BL21 (DE3). The expression levels were analyzed by SDS-PAGE (10%, Coomassie staining) and immunoblotting using an anti-His antibody directed against the C-terminal His10 tag of TmrA (Fig. 1B). Expression of TmrA was time-dependent and reached its maximum 3 h after induction by isopropyl β-d-thiogalactopyranoside (0.5 mm) at 37 °C. The membranes were solubilized with DDM and His-tagged proteins were purified by metal-chelate affinity chromatography. The eluate was analyzed by SDS-PAGE (10%, Coomassie staining), revealing that both subunits were co-purified in stoichiometric amounts. TmrA and TmrB migrate at a lower molecular mass as predicted for very hydrophobic membrane proteins. TmrA-His10 (Mtheor = 70.5 kDa) and TmrB (Mtheor = 64.4 kDa) were detected at 56 and 52 kDa, respectively (Fig. 1C). All single and double mutants were expressed and purified in amounts comparable to wild type TmrAB. As an example, purified E523Q/WT and WT/D500N mutants are depicted in Fig. 1C.
Quaternary Structure of the TmrAB Complex
The integrity of TmrA and TmrB was first investigated by MALDI-MS in positive ion mode (Fig. 2A). The single peaks of 69.1 and 64.4 kDa correspond to TmrA after TEV cleavage of the His10 tag and TmrB, respectively. Notably, in the range of 20–75 kDa, no additional masses were detected, confirming the identity and composition of the purified TmrAB complex. We next analyzed the oligomeric state of the ABC complex by analytical gel filtration (Fig. 2B). TmrA and TmrB co-elute as a monodisperse peak with an apparent molecular mass of approximately 250 kDa including the extra mass of the DDM micelle.
FIGURE 2.

TmrAB forms a stable heterodimeric complex. A, MALDI-TOF-MS of purified TmrAB recorded in the linear positive ion mode. Peak A and B have masses of 69.1 and 64.4 kDa, corresponding to TmrA after TEV treatment and TmrB, respectively. Single and multiple charged states are indicated (sum of 100 laser shots). B, gel filtration of purified TmrAB. Purified TmrAB was separated on a TSK gel 3000SW column in the presence of 0.02% DDM (w/v). TmrAB elutes at an apparent mass of approximately 250 kDa. The void volume of the column is indicated with V0, and the retention volume is indicated with Vt. C, LILBID-MS anion spectra of TmrAB after TEV cleavage recorded at low laser (upper panel) or high laser intensity (lower panel). At low laser intensity the charge distribution corresponds to a mass of 148 kDa indicated by the dotted lines. The respective charge states are labeled. At high laser intensity, an additional charge distribution, corresponding to a mass of 133 kDa, is visible. The charge distribution and the corresponding charge states are labeled.
To analyze the quaternary structure of the TmrAB complex in its native state, we applied the newly developed ultra soft LILBID-MS (26). Droplets of TmrAB solution were injected into high vacuum and irradiated by mid-IR laser pulses. Subsequently, the ionized TmrAB complex was set free and analyzed by TOF mass spectrometry. LILBID measurements were performed in two modes. In general, low laser intensity (ultra soft mode) is used to make whole protein complexes “fly.” In the dissociation mode using elevated laser power, macromolecular complexes are typically thermolyzed into their subunits. In the ultra soft negative ion mode, TmrA and TmrB fly as a negatively charged complex with a molecular mass of 148 kDa (Fig. 2C, upper panel). Of note, the onset of the mass peak corresponds to the exact mass of the complex. The width of the peak does not correspond to the mass resolution R (m/Δm) = 400, but it is caused by solvation and adduct formation with buffer as can be seen under more intense laser conditions in the lower panel of Fig. 2C. The native TmrAB complex (after TEV cleavage) has a molecular mass of 133.5 kDa, leading to the suggestion that additional molecules are associated with the purified protein complex. By gradual increase of the infrared laser power, associated molecules are released and a second mass peak at 133 kDa becomes apparent, corresponding to the exact molecular mass of the TmrAB complex (Fig. 2C, lower panel). Importantly, even at high laser intensities the protein complex stays intact, demonstrating a tight noncovalent interaction of both half-site ABC transporters. In summary, these results demonstrate that TmrAB forms a very stable heterodimeric ABC complex.
TmrAB Functions as Multidrug Transporter
Hoechst 33342 is a well characterized substrate for multidrug resistance proteins and its transmembrane movement can be monitored by fluorescence spectroscopy. We therefore examined its membrane translocation in IOVs containing either wild type or ATPase-deficient E523Q/D500N complex (Table 2). The correct membrane orientation of the ABC complex in IOVs was analyzed by TEV protease accessibility based on a site-specific TEV-His10 tag at the C terminus of TmrA (Fig. 3A). Immunodetection using an anti-His antibody showed that wild type and E523Q/D500N are equally expressed and that >95% of TmrAB complexes are oriented with their NBDs to the outside, optimal for uptake assays.
TABLE 2.
ATP hydrolysis of WT and mutant TmrAB
Km,ATP and kcat values of wild type and TmrAB mutants (60 nm) were determined at 68 °C. ND, nondetectable.
| TmrA/TmrB | kcat | Km,ATP |
|---|---|---|
| 1/s | mm | |
| WT/WT | 8.83 ± 0.87 | 0.92 ± 0.24 |
| E523Q/WT | 0.16 ± 0.01 | 1.66 ± 0.20 |
| WT/D500N | 10.63 ± 1.58 | 1.51 ± 0.51 |
| WT/D500E | 10.31 ± 1.81 | 1.81 ± 0.37 |
| E523Q/D500N | 0.0 | ND |
| E523Q/D500E | 0.0 | ND |
FIGURE 3.

Hoechst 33342 uptake by TmrAB in inside-out vesicles. A, orientation of wild type and mutant TmrAB in IOVs. IOVs were prepared from wild type and E523Q/D500N TmrAB expressing E. coli cells and incubated overnight at 30 °C in the absence or presence of TEV protease. As a positive control, IOVs were solubilized with 2% Triton X-100 (TX-100) before TEV cleavage. 20 μg of total protein were separated by SDS-PAGE and orientation of TmrAB was determined via immunoblotting using an anti-His antibody. B, wild type TmrAB mediates Hoechst 33342 uptake in IOVs. IOVs derived from E. coli cells expressing wild type TmrAB (black line), E523Q/D500N TmrAB (green line), and the empty pET22b+ vector (cyan line), respectively, were incubated with 0.75 μm of Hoechst 33342. Membrane translocation was initiated after 2 min by the addition of MgATP (1.5 mm). The fluorescence was monitored over time at 50 °C with excitation and emission wavelengths of 355 and 457 nm, respectively. As control, Hoechst 33342 uptake was measured at 37 °C using IOVs containing wild type TmrAB (gray line). C, inhibition of Hoechst 33342 uptake by NaN3 and nigericin. The experiments were performed as described in C using IOVs containing wild type TmrAB (black line). The reaction was carried out in the presence of 10 mm NaN3 (gray line) to inhibit the F1F0-ATPase. To dissipate proton motive force, 1 μm of nigericin was added after 5 min (blue line). D, inhibition of Hoechst 33342 uptake by verapamil. Hoechst 33342 uptake was carried out in wild type IOVs as described above. The reaction was inhibited by titration of verapamil, resulting in an IC50 value of 83 μm.
To monitor the translocation activity, IOVs containing wild type TmrAB were incubated with Hoechst 33342 at 37 and 50 °C (Fig. 3B). The addition of ATP leads to a movement of the fluorophore into the aqueous medium at 50 °C as reflected by rapid fluorescence quenching. However, only minor changes in the fluorescence signal were observed at 37 °C, where TmrAB is ATPase inactive (Fig. 4A), suggesting that membrane movement of the fluorophore is tightly coupled to the ATPase activity of TmrAB. Transmembrane movement of Hoechst 33342 was impaired in IOVs lacking TmrAB (ΔtmrAB). In addition, IOVs containing the ATPase inactive E523Q/D500N complex do not translocate Hoechst 33342, indicating that ATP hydrolysis is necessary to mediate movement of the fluorescent dye.
FIGURE 4.

ATP hydrolysis of purified TmrAB. A, temperature dependence of the ATPase of TmrAB. The basal ATPase activity of DDM-solubilized wild type TmrAB (60 nm) was analyzed by monitoring the release of inorganic phosphate at different temperatures. The reaction was carried out for 5 min in the presence of 5 mm ATP. B, inhibition of ATPase activity. The ATPase of wild type TmrAB (60 nm) was analyzed for 4 min at 68 °C in the presence of BeFx (5 mm), vanadate (5 mm), and NaN3 (10 mm). C, Hoechst 33342 inhibits ATP hydrolysis by TmrAB. ATP hydrolysis of purified TmrAB (WT) was carried out as described in B in the presence of increasing concentrations of Hoechst 33342. ATPase activity was inhibited in a dose-dependent manner resulting in an IC50 value of 95 μm. D, determination of Km,ATP and kcat values. The ATPase activity of TmrAB (60 nm) was measured as a function of ATP concentration for 4 min at 68 °C. Wild type TmrAB (filled circles) was ATPase active, resulting in a kcat of 8.8 s−1 and a Km,ATP of 0.92 mm. The E523Q/D500N mutant (filled triangles) shows no detectable ATPase activity above background.
Recent studies on the ABC transporter LmrA from L. lactis showed that ATP hydrolysis is not the direct driving force for Hoechst 33342 translocation (34, 35). The authors concluded that LmrA-specific Hoechst 33342 extrusion rather depends on changes in the proton-motive force that is coupled to the activity of F1F0-ATPase. To examine the effect of the proton-motive force on the translocation activity of TmrAB, the ionophore nigericin was added after reaching maximum fluorescence change (Fig. 3C). Breakdown of the membrane potential completely reverses the Hoechst 33342 translocation activity. Additionally, Hoechst 33342 extrusion was prevented by the addition of the F1F0-ATPase inhibitor NaN3, indicating that the readout may also depend on the proton gradient generated by the F1F0-ATPase. Importantly, increasing concentrations of verapamil inhibit TmrAB-mediated Hoechst 33342 translocation, revealing an IC50 value of 83 μm (Fig. 3D). This result is in good agreement with observations that verapamil inhibits P-gp-, LmrA-, and Sav1866-mediated multidrug resistance (11, 36, 37) as well as LmrA- and Sav1866-specific Hoechst 33342 uptake (11, 13). These data suggest an interaction of TmrAB with Hoechst 33342 and verapamil. The TmrAB complex mediates a temperature-dependent movement of the fluorophore, leading to the conclusion that conformational changes and ATP hydrolysis are required for transport function of TmrAB.
ATP Hydrolysis of TmrAB
We next analyzed the ATPase activity of the purified TmrAB complex at different temperatures (Fig. 4A). No ATP hydrolysis was detectable at 37 °C, the physiological growth temperature of E. coli. ATP turnover of TmrAB was observed at temperatures from 50 to 95 °C, reaching a maximum at 75 °C with a turnover rate of 7.5 ATP/s. The temperature optimum correlates to the physiological growth temperature of T. thermophilus (49–72 °C). In conclusion, the function of the TmrAB complex is maintained after heterologous expression and purification. We further examined the influence of various ATPase inhibitors (Fig. 4B). The ATPase activity was 10- and 100-fold decreased by orthovanadate and BeFx, respectively. No inhibition was observed with the F1F0-ATPase inhibitor NaN3. To provide direct evidence that the purified TmrAB complex interacts with Hoechst 33342, we analyzed the ATPase activity at 68 °C in the presence of increasing drug concentrations (Fig. 4C). Interestingly, ATP hydrolysis of TmrAB was inhibited in a dose-dependent manner (IC50 of 95 μm).
We next analyzed the ATPase activity of the TmrAB complex as a function of ATP concentration, yielding a Km value of 0.9 ± 0.2 mm and a turnover number kcat of 8.8 ± 0.9 s−1 for the wild type complex (Fig. 4D). Mutation of the putative catalytic base in TmrA (E523Q) resulted in a 70-fold reduction of the ATPase activity, whereas the corresponding D500N mutation in TmrB did not significantly alter the Km and kcat values compared with the wild type complex (Table 2). Notably, introduction of a catalytic glutamate in TmrB (D500E) does not alter the enzymatic fingerprint of the ABC complex, suggesting that functional asymmetry in the NBDs cannot be outweighed by generating an additional second catalytically active site. As expected, the E523Q/D500N complex lacks a detectable ATPase activity, indicating that glutamate 523 in TmrA is essential for ATP hydrolysis in the asymmetric ABC complex.
Nucleotide Trapping by the TmrAB Complex
To gain mechanistic insight in the ATP hydrolysis cycle of the TmrAB complex, we examined the binding and stable trapping of nucleotides in both ATP-binding sites. Purified TmrAB complexes were photo-cross-linked by azido-[α-32P]ATP or azido-[γ-32P]ATP. At nonpermissive temperatures (ATPase inactive), photo-cross-linking of azido-[α-32P]ATP or [γ-32P]ATP of TmrA in the wild type and the E523Q/WT complex is observed. Thus, mutation of the catalytic residue does not alter the ATP binding properties of the complex (Fig. 5A). An excess of cold ATP abolished the photo-cross-linking, demonstrating that the reaction is ATP-specific. Surprisingly, no cross-linking of TmrB was observed.
FIGURE 5.

Nucleotide trapping by wild type and E523Q/WT TmrAB complex. A, binding of 8-azido-[α-32P]ATP and 8-azido-[γ-32P]ATP by wild type and E523Q/WT TmrAB. Purified wild type and E523Q/WT complexes (0.5 μm of each) were incubated with radiolabeled 8-azido-ATP (4 μm) for 4 min at room temperature in the presence or absence of an excess of cold ATP (5 mm). Additionally, 0.4 mm orthovanadate (Vi) was added to the wild type complex. Photo-cross-linking was performed for 4 min on ice. The proteins (0.6 μg/lane) were separated by SDS-PAGE (10%), and the photo-cross-linked proteins were visualized by autoradiography. B, 8-azido-ATP photo-cross-linking under hydrolysis conditions. Purified wild type and E523Q/WT mutant TmrAB (0.5 μm) were incubated at 68 °C, followed by the addition of either 8-N3-[α-32P]ATP or 8-N3-[γ-32P]ATP. The wild type complex was supplemented with 0.4 mm vanadate (Vi) to trap nucleotides in the posthydrolysis state. Cold ATP (5 mm) was used to probe for specific cross-linking. After cooling down, the samples were incubated with an excess of cold ATP to demonstrate tight occlusion of nucleotides. Subsequently, the cross-link reaction was performed for 4 min on ice. As a control, binding of labeled azido-ATP at room temperature (RT) is shown for the wild type complex. Nucleotide binding was analyzed as described above.
At permissive temperatures (68 °C), orthovanadate causes a posthydrolysis trapping of 8-azido-ADP in the TmrA half-site (Fig. 5B). Similar results were obtained with BeFx (data not shown). Notably, the E523Q/WT complex traps ATP in the prehydrolysis state in a vanadate-independent manner, verifying that Glu-523 in TmrA is essential for ATP hydrolysis. Therefore, a stable occlusion of ATP was observed in the ATPase inactive E523Q/WT complex. Again, no ATP cross-linking of TmrB was detected, indicating that site II either does not bind ATP or that the cross-linking (insertion of the reactive nitrene) is inefficient at this site.
Stoichiometry of Bound Nucleotides in a Pre- and Posthydrolysis State
Because no azido-ATP photo-cross-linking of TmrB was observed, we examined the stoichiometry of nucleotide binding before and after ATP hydrolysis by rapid (spin-down) gel filtration. At ATPase-permissive temperatures (68 °C), we show that in the presence of orthovanadate, two α-32P-labeled nucleotides are trapped in the wild type complex (Fig. 6A). Prior to separation of free and bound nucleotides, the samples were supplemented with an excess of cold ATP to demonstrate stable occlusion of nucleotides in the TmrAB complex. Strikingly, only substoichiometric amounts of nucleotides (0.1 mol/complex) were occluded at 4 °C (Fig. 6A). These findings show that permissive temperatures (>50 °C) are required to drive the TmrAB complex into an occluded, vanadate-trapped posthydrolysis state by conformational changes as derived from the outward and inward facing conformations of ABC exporters (38, 39). Thus, all further experiments were performed at ATPase-permissive temperatures. Under these conditions, stoichiometric trapping of [α-32P]ATP and [γ-32P]ATP in the wild type complex is strictly vanadate-dependent (Fig. 6B). In the vanadate-trapped state, two α-32P-labeled nucleotides were bound per TmrAB, whereas only one [γ-32P]-ATP was trapped in the ABC complex, reflecting the occlusion of one posthydrolysis ADP-Vi and one ATP. The composition of the trapped nucleotides was verified by TLC analysis revealing an equimolar ratio of ATP and ADP (Fig. 6D). Nucleotide trapping by TmrAB can be competed with an excess of ATP but not with AMP (Fig. 6B), showing the specificity of the vanadate trapping experiments. The results give evidence that both ATP-binding sites of TmrAB bind ATP, whereas only one site is active in ATP hydrolysis. Because the E523Q mutation in TmrA leads to hydrolysis-deficient TmrAB complex, which is trapped in a prehydrolysis state, we propose that the canonic site I forms the stable ADP-Vi posthydrolysis state (Fig. 6E). To exclude that slow hydrolysis occurs in the noncanonic site II, trapping experiments were performed for 3 min at ATPase-permissive temperatures. Under these conditions no significant hydrolysis was observed in the noncanonic site, verifying that site II is indeed ATP hydrolysis-deficient. Because nucleotide occlusion was observed in both sites, we conclude that ATP is not permanently bound at site II.
FIGURE 6.

Stoichiometry of bound nucleotides in the pre- and posthydrolysis state. A, nucleotide trapping in TmrAB is temperature-dependent. Wild type TmrAB was incubated in the presence of ATP (0.7 mm) and 5 mm orthovanadate (Vi) for 3.5 min at 4 and 68 °C, respectively. Tracer amounts of [α-32P]ATP were added during reaction. Prior to spin-down, the samples were supplemented with an excess of cold ATP to verify stable trapping of nucleotides. After separation by rapid gel filtration, the amount of bound nucleotides was determined by β-counting. B, vanadate-dependent nucleotide trapping under hydrolysis conditions. Trapping experiments with wild type TmrAB were performed for 3.5 min at 68 °C with ATP (0.7 mm) using tracer amounts of [α-32P]ATP (black bars) and [γ-32P]ATP (white bars), respectively. Protein samples were incubated in the absence or presence of 5 mm orthovanadate (Vi). As competitor, either ATP or AMP (10 mm of each) was added. After cooling down to room temperature, the samples were supplemented with 2 mm ATP to demonstrate stable occlusion of nucleotides. Free ATP was removed via rapid gel filtration (spin-down). The amount of trapped nucleotides was determined as described above. C, effects of mutations on nucleotide binding and hydrolysis. Trapping of [α-32P]ATP (black bars) and [γ-32P]ATP (white bars) by wild type and mutant TmrAB was analyzed as described in B. Nucleotide trapping of the ATPase-deficient E523Q/WT, E523Q/D500N, and E523Q/D500E mutants was performed in the absence of vanadate. The calculated stoichiometries result from at least three independent experiments. D, determination of nucleotide composition by TLC. Trapped nucleotides were analyzed by thin layer chromatography. The amount of either [α-32P]ATP or [α-32P]ADP was quantified via phosphorus imaging, and the amounts of ATP (black bars) and ADP (white bars) were calculated for wild type and mutant TmrAB. The data are represented as the means ± S.E. of at least three independent experiments. E, model of ATP hydrolysis in the TmrAB complex. Vanadate traps ATP in WT TmrAB, forming a posthydrolysis state, with one ADP-Vi bound in site I and one ATP bound in site II. Mutation of the catalytic base residue Glu-523 mediates the vanadate-independent occlusion of two ATP in the prehydrolysis state. Introduction of a new catalytic base residue (Asp-500) in TmrB leads to an ATPase active site II (slow hydrolysis), which depends on formation of the posthydrolysis state in site I. The exact data are shown in C and D. The vanadate-trapped posthydrolysis state is indicated in red. Mutations of the putative catalytic base residues are labeled in orange.
Allosteric Communication between the Canonic and Noncanonic ATP-binding Site
To get further insight into the communication between the two asymmetric sites, we characterized nucleotide trapping of various TmrAB mutants (Fig. 6C). The E532Q mutation of the catalytic base in TmrA results in an ATPase inactive complex that traps two ATP molecules (either [α-32P]ATP or [γ-32P]ATP) in the prehydrolysis state. Similar results were obtained for the double mutants E523Q/D500N and E523Q/D500E. Although samples were immediately treated with EDTA and SDS after rapid spin-down, the TLC analysis of the bound nucleotides in the E523Q/WT complex revealed that 25% of the bound [α-32P]ATP are hydrolyzed to [α-32P]ADP. Interestingly, 2 mol of the γ-32P label are still detectable after separation of the TmrAB complex, implying that the occluded ATP is slowly hydrolyzed in the E523Q site at ATPase-permissive temperatures followed by retarded phosphate release.
The D500N substitution in the noncanonic TmrB site does not change the Km value of ATP hydrolysis. Similar to wild type, the WT/D500N complex traps two [α-32P]ATP molecules in a vanadate-dependent manner, whereas only half that amount of [γ-32P]ATP label is bound. Thus, exactly 50% of bound nucleotides are hydrolyzed, indicating that removal of the negative charge at the noncanonic site II does not affect ATP binding of the transport complex and ATP hydrolysis in the canonic site I.
Introduction of a canonic catalytic residue (D500E) in TmrB leads to a decreased occlusion of [γ-32P]ATP compared with the wild type complex, whereas still two α-32P-labeled nucleotides are trapped by vanadate. These findings show that a second hydrolytically active site can be introduced in the TmrAB complex, leading to more than 80% hydrolysis of the bound ATP in the WT/D500E complex. Notably, the double mutant E523Q/D500E occludes two ATP molecules in its prehydrolysis state. This striking difference (WT/D500E versus E523Q/D500E) points to an allosteric communication between the two active sites. Thus, ATP hydrolysis in the D500E site depends on ATP hydrolysis in the consensus site I (vanadate trapping of the posthydrolysis state). In contrast, no ATP hydrolysis is observed if the multidrug ABC complex is trapped in its prehydrolysis state by the E523Q mutation. However, the overall hydrolysis rate of the WT/D500E complex is not significantly altered as compared with the wild type TmrAB complex (Table 2), implying that the ATP hydrolysis in the newly generated D500E site is not rate-limiting for the ATP hydrolysis cycle. Furthermore, the asymmetry in the NBDs of the ABC complex cannot be overcome by introducing a second catalytic glutamate in the noncanonic site, revealing that asymmetry is a built-in feature of the TmrB NBD or the entire complex and not based on a single noncanonic residue.
DISCUSSION
Bacteria evolved a wide range of strategies to resist to the effects of toxic compounds. One mechanism to prevent accumulation of noxious agents is the active extrusion by different integral membrane proteins, including members of the ABC superfamily. These multidrug transporters catalyze the export of various chemically unrelated compounds and thereby mediate the cellular resistance to a broad range of antibiotics and other toxic drugs (4). In the last years, several prokaryotic ABC exporters, such as LmrA, LmrCD, and Sav1866 (11, 13, 14), have been linked to multidrug resistance. Multidrug resistance proteins can cause severe problems in the treatment of infectious diseases (40). In this study, we identified and characterized the first and only ABC export system present in the thermophilic Gram-negative eubacterium T. thermophilus, which we named TmrAB.
To verify TmrAB as a member of the multidrug protein family, the ABC transporter was expressed in E. coli. Hoechst 33342 uptake studies in TmrAB containing inside-out vesicles revealed a Hoechst 33342 transmembrane movement that is temperature-dependent and TmrAB-specific. We further demonstrate that Hoechst 33342 extrusion by TmrAB is inhibited by verapamil, a well known substrate of the multidrug resistance proteins, such as LmrA, Sav1866, and P-gp (11, 36, 37). Verapamil has been shown to bind to transmembrane segment six of P-gp and LmrA and thereby inhibit Hoechst 33342 transport (13, 36, 41–43). Because TmrAB was inhibited by verapamil, the mechanism of drug binding in these multidrug resistance ABC proteins might be conserved.
To derive mechanistic insight in the ATP hydrolysis cycle, the heterodimeric ABC transport complex from the thermophilic Gram-negative bacterium TmrAB was expressed in E. coli and purified via metal affinity chromatography. Both subunits were isolated in stoichiometric amounts via the C-terminal His10 tag of TmrA, indicating a tight association of TmrA and TmrB that is maintained throughout the purification procedure. This behavior is similar to the heterodimeric ABC transporters LmrCD, YheI/H, ABCG5/8, or TAP1/2 (15, 44–46). Formation of a heterodimeric complex was verified by size exclusion chromatography and the newly established in its ultra-soft mode LILBID-MS.
The ABC export system shows a temperature-dependent ATPase activity reaching a maximum turnover rate at 75 °C, correlating to the growth temperature of T. thermophilus. ATP hydrolysis is inhibited by BeFx and vanadate, which traps ADP in the active site (aligned by Walker A/B of TmrA). Determination of the Michaelis-Menten kinetics revealed a Km,MgATP value of 0.9 mm, which is in good agreement to other ABC proteins (e.g. the homodimeric mitochondrial ABC transport complex MDL1 from Saccharomyces cerevisiae (29)). Comparable with other multidrug resistance proteins like LmrCD or BmrA (45, 47), TmrAB exhibits a high basal ATPase activity of 9 s−1. In conclusion, TmrAB is purified as functionally active heterodimeric transport complex and can be used as a tool for further mechanistic studies.
Remarkably, Hoechst 33342 inhibits ATP hydrolysis by TmrAB in a dose-dependent manner. A similar substrate inhibition has been observed for PDR5, a member of the pleiotropic drug resistance network in S. cerevisiae (48). It is proposed that multidrug ABC transporters are uncoupled and therefore exhibit a high basal ATP turnover. Assuming that the solute release is the rate-limiting step in the catalytic cycle, high solute concentrations might lock the transport system in the outward facing conformation and thereby inhibit ATPase activity (48). Although the exact mechanism of drug inhibition remains to be captured, our data indicate that the solute binding activity is maintained after solubilization and purification of the TmrAB complex. The data further demonstrate that Hoechst 33342 interacts directly with the Thermus ABC complex, confirming that drug transport observed in IOVs is indeed mediated by TmrAB.
Previously, different models of the catalytic cycle have been proposed, regarding the timing and allosteric coupling of ATP binding and hydrolysis by both NBDs to conformational changes of the TMDs. Structural analysis of the homodimeric ABC transporters Sav1866 and MsbA always revealed two bound nucleotides (ATP or AMPPNP) at the interface of both NBDs closed in the outward-facing conformation (49, 50). Notably, ABC proteins have not been crystallized in an asymmetric ATP/ADP-bound state. Furthermore, characterization of ATP hydrolysis in symmetric ABC proteins, e.g. MDL1 or P-gp, showed that both NBDs are able to hydrolyze ATP either in a sequential (processive clamp) or an alternating mode (51–55). In case of prokaryotic heterodimeric multidrug transporters, like LmrCD or YheI/H, the cycle of nucleotide binding and hydrolysis is poorly understood. The NBDs of these ABC proteins are characterized by several nonconsensus motifs in one NBD and are therefore proposed to bind and hydrolyze nucleotides in an asymmetric mode. In contrast, the asymmetry in the TmrAB complex is traced to a single residue. In the TmrB, the catalytic glutamate next to the Walker B motif is replaced by aspartate, leading to a noncanonic site II. To address the question of how a substitution of the glutamate residue influences the catalytic cycle, we characterized the nucleotide binding and hydrolysis properties of various TmrAB mutants. Removal of the catalytic base (Glu-523) of TmrA did not change the nucleotide binding properties of the protein complex. We demonstrate that two ATP molecules are bound per complex, suggesting that both NBDs are functional regarding nucleotide binding. However, in the E523Q/WT complex, ATP hydrolysis is drastically impaired. Comparable results were obtained for the heterodimeric ABC transporter LmrCD, suggesting that hydrolysis in the canonic ATP-binding site is essential for ATPase activity in heterodimeric transport complexes (24). For LmrCD, it was shown that mutation of the aspartate (D495N) in the noncanonic site (LmrC) leads to instability of the protein complex, impeding further characterization of the protein activity. Interestingly, the D500N mutation in TmrB does not alter the expression and complex assembly. We show for the first time that removal of the negative charge (Asp-500) does not change the binding and hydrolysis properties of the TmrAB complex. Nucleotide trapping experiments revealed exactly two nucleotides in the NBDs in the case of the wild type and the WT/D500N complex. One site traps ADP by vanadate (site I), whereas ATP is occluded in the site II. Because Glu-523 in TmrA is essential for ATP hydrolysis, we conclude that site I is responsible for ATPase function. Our data provide strong and direct evidence that the subunits of heterodimeric TmrAB are functionally nonequivalent.
To prove whether symmetry can be rescued, we introduced a second catalytic glutamate (D500E in TmrB) in the noncanonic site II. However, the D500E mutation did not alter the ATP turnover of the TmrAB complex. Additionally, no ATPase activity was observed in the double E523Q/D500E mutant, indicating that the glutamate in the noncanonic site cannot restore the activity of the ABC transporter. Interestingly, in the posthydrolysis state (vanadate-trapped) in the WT/D500E complex, the introduced glutamate mediates slow hydrolysis in site II. In contrast, two ATP molecules are occluded in the prehydrolysis state in the E523Q/D500E mutant. We therefore conclude that hydrolysis in the newly generated canonic site depends on formation of the posthydrolysis state in site I, supporting a sequential hydrolysis model. However, the overall ATP hydrolysis rate of the TmrAB complex is not influenced by introduction of a catalytic glutamate in TmrB, demonstrating that asymmetry in the TmrAB NBDs is structurally imprinted in the entire complex and not based on an exchange of a single noncanonic residue.
In this study, we analyzed for the first time the basic function and ATP hydrolysis cycle of a heterodimeric multidrug ABC transporter from a hyperthermophilic Gram-negative bacterium. We further demonstrated that the asymmetry of the NBDs regarding nucleotide binding and hydrolysis is a general feature of heterodimeric ABC proteins, suggesting a common mechanism of the catalytic cycle. In contrast to homodimeric ABC proteins, only one site is essential for ATP hydrolysis, indicating different drug transport mechanisms in symmetric and asymmetric ABC proteins. In conclusion, we introduced TmrAB as a new multidrug ABC transporter that represents a valuable model of asymmetric heterodimeric ABC complexes for the examination of the mechanism of multidrug transport.
Acknowledgments
We thank Christian Schölz and Drs. Rupert Abele and David Parcej for helpful discussions and Dr. Daniela Maneg and Werner Müller for initial approaches in the project.
This work was supported by German Research Foundation Grants (SFB807-Transport and Communication across Biological Membranes, TA157/7-1, and AB149/1-1) and funds from the European Community Seventh Framework Program as part of the European Drug Initiative on Channels and Transporters.
- ABC
- ATP-binding cassette
- DDM
- n-dodecyl-β-d-maltoside
- IOV
- inside-out vesicles
- LILBID-MS
- laser-induced liquid bead ion desorption mass spectrometry
- NBD
- nucleotide-binding domain
- TMD
- transmembrane domain
- TmrAB
- T. thermophilus multidrug resistance protein A and B
- AMPPNP
- adenosine 5′-(β,γ-imino)triphosphate
- TEV
- Tobacco Etch Virus Protease.
REFERENCES
- 1. Higgins C. F. (1992) Annu. Rev. Cell Biol. 8, 67–113 [DOI] [PubMed] [Google Scholar]
- 2. Dean M., Rzhetsky A., Allikmets R. (2001) Genome Res. 11, 1156–1166 [DOI] [PubMed] [Google Scholar]
- 3. Locher K. P. (2009) Philos Trans. R. Soc. Lond B Biol. Sci. 364, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davidson A. L., Dassa E., Orelle C., Chen J. (2008) Microbiol. Mol. Biol. Rev. 72, 317–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lubelski J., Konings W. N., Driessen A. J. (2007) Microbiol. Mol. Biol. Rev. 71, 463–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leonard G. D., Fojo T., Bates S. E. (2003) Oncologist 8, 411–424 [DOI] [PubMed] [Google Scholar]
- 7. Huang Y., Sadée W. (2006) Cancer Lett. 239, 168–182 [DOI] [PubMed] [Google Scholar]
- 8. Leslie E. M., Deeley R. G., Cole S. P. (2001) Toxicology 167, 3–23 [DOI] [PubMed] [Google Scholar]
- 9. Gottesman M. M., Mickisch G. H., Pastan I. (1994) Cancer Treat. Res. 73, 107–128 [DOI] [PubMed] [Google Scholar]
- 10. Robey R. W., Medina-Pérez W. Y., Nishiyama K., Lahusen T., Miyake K., Litman T., Senderowicz A. M., Ross D. D., Bates S. E. (2001) Clin. Cancer Res. 7, 145–152 [PubMed] [Google Scholar]
- 11. Velamakanni S., Yao Y., Gutmann D. A., van Veen H. W. (2008) Biochemistry 47, 9300–9308 [DOI] [PubMed] [Google Scholar]
- 12. Steinfels E., Orelle C., Fantino J. R., Dalmas O., Rigaud J. L., Denizot F., Di Pietro A., Jault J. M. (2004) Biochemistry 43, 7491–7502 [DOI] [PubMed] [Google Scholar]
- 13. Margolles A., Putman M., van Veen H. W., Konings W. N. (1999) Biochemistry 38, 16298–16306 [DOI] [PubMed] [Google Scholar]
- 14. Lubelski J., de Jong A., van Merkerk R., Agustiandari H., Kuipers O. P., Kok J., Driessen A. J. (2006) Mol. Microbiol. 61, 771–781 [DOI] [PubMed] [Google Scholar]
- 15. Torres C., Galián C., Freiberg C., Fantino J. R., Jault J. M. (2009) Biochim. Biophys. Acta 1788, 615–622 [DOI] [PubMed] [Google Scholar]
- 16. Matsuo T., Chen J., Minato Y., Ogawa W., Mizushima T., Kuroda T., Tsuchiya T. (2008) J. Bacteriol. 190, 648–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen M., Abele R., Tampé R. (2004) J. Biol. Chem. 279, 46073–46081 [DOI] [PubMed] [Google Scholar]
- 18. Hou Y. X., Cui L., Riordan J. R., Chang X. B. (2002) J. Biol. Chem. 277, 5110–5119 [DOI] [PubMed] [Google Scholar]
- 19. Sheppard D. N., Welsh M. J. (1999) Physiol. Rev. 79, S23–S45 [DOI] [PubMed] [Google Scholar]
- 20. Beaudet L., Urbatsch I. L., Gros P. (1998) Biochemistry 37, 9073–9082 [DOI] [PubMed] [Google Scholar]
- 21. Qin L., Zheng J., Grant C. E., Jia Z., Cole S. P., Deeley R. G. (2008) Biochemistry 47, 13952–13965 [DOI] [PubMed] [Google Scholar]
- 22. Payen L. F., Gao M., Westlake C. J., Cole S. P., Deeley R. G. (2003) J. Biol. Chem. 278, 38537–38547 [DOI] [PubMed] [Google Scholar]
- 23. Procko E., Ferrin-O'Connell I., Ng S. L., Gaudet R. (2006) Mol. Cell 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 24. Lubelski J., van Merkerk R., Konings W. N., Driessen A. J. (2006) Biochemistry 45, 648–656 [DOI] [PubMed] [Google Scholar]
- 25. Morgner N., Barth H. D., Brutschy B. (2006) Aust. J. Chem. 59, 109–114 [Google Scholar]
- 26. Morgner N., Kleinschroth T., Barth H. D., Ludwig B., Brutschy B. (2007) J. Am. Soc. Mass Spectrom 18, 1429–1438 [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann J., Aslimovska L., Bamann C., Glaubitz C., Bamberg E., Brutschy B. (2010) Phys. Chem. Chem. Phys. 12, 3480–3485 [DOI] [PubMed] [Google Scholar]
- 28. Gorbulev S., Abele R., Tampé R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3732–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hofacker M., Gompf S., Zutz A., Presenti C., Haase W., van der Does C., Model K., Tampé R. (2007) J. Biol. Chem. 282, 3951–3961 [DOI] [PubMed] [Google Scholar]
- 30. Henne A., Brüggemann H., Raasch C., Wiezer A., Hartsch T., Liesegang H., Johann A., Lienard T., Gohl O., Martinez-Arias R., Jacobi C., Starkuviene V., Schlenczeck S., Dencker S., Huber R., Klenk H. P., Kramer W., Merkl R., Gottschalk G., Fritz H. J. (2004) Nat. Biotechnol. 22, 547–553 [DOI] [PubMed] [Google Scholar]
- 31. Johnson Z. I., Chisholm S. W. (2004) Genome Res. 14, 2268–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tusnády G. E., Simon I. (1998) J. Mol. Biol. 283, 489–506 [DOI] [PubMed] [Google Scholar]
- 33. Zhang D. W., Graf G. A., Gerard R. D., Cohen J. C., Hobbs H. H. (2006) J. Biol. Chem. 281, 4507–4516 [DOI] [PubMed] [Google Scholar]
- 34. van den Berg van Saparoea H. B., Lubelski J., van Merkerk R., Mazurkiewicz P. S., Driessen A. J. (2005) Biochemistry 44, 16931–16938 [DOI] [PubMed] [Google Scholar]
- 35. Venter H., Velamakanni S., Balakrishnan L., van Veen H. W. (2008) Biochem. Pharmacol. 75, 866–874 [DOI] [PubMed] [Google Scholar]
- 36. van Veen H. W., Callaghan R., Soceneantu L., Sardini A., Konings W. N., Higgins C. F. (1998) Nature 391, 291–295 [DOI] [PubMed] [Google Scholar]
- 37. Endicott J. A., Ling V. (1989) Annu. Rev. Biochem. 58, 137–171 [DOI] [PubMed] [Google Scholar]
- 38. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dawson R. J., Locher K. P. (2006) Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 40. Jenkinson H. F. (1996) J. Dent. Res. 75, 736–742 [DOI] [PubMed] [Google Scholar]
- 41. Poelarends G. J., Konings W. N. (2002) J. Biol. Chem. 277, 42891–42898 [DOI] [PubMed] [Google Scholar]
- 42. Ecker G. F., Pleban K., Kopp S., Csaszar E., Poelarends G. J., Putman M., Kaiser D., Konings W. N., Chiba P. (2004) Mol. Pharmacol. 66, 1169–1179 [DOI] [PubMed] [Google Scholar]
- 43. Loo T. W., Clarke D. M. (1994) Biochemistry 33, 14049–14057 [DOI] [PubMed] [Google Scholar]
- 44. Herget M., Kreissig N., Kolbe C., Schölz C., Tampé R., Abele R. (2009) J. Biol. Chem. 284, 33740–33749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lubelski J., Mazurkiewicz P., van Merkerk R., Konings W. N., Driessen A. J. (2004) J. Biol. Chem. 279, 34449–34455 [DOI] [PubMed] [Google Scholar]
- 46. Johnson B. J., Lee J. Y., Pickert A., Urbatsch I. L. (2010) Biochemistry 49, 3403–3411 [DOI] [PubMed] [Google Scholar]
- 47. Orelle C., Dalmas O., Gros P., Di Pietro A., Jault J. M. (2003) J. Biol. Chem. 278, 47002–47008 [DOI] [PubMed] [Google Scholar]
- 48. Ernst R., Kueppers P., Klein C. M., Schwarzmueller T., Kuchler K., Schmitt L. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 5069–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dawson R. J., Locher K. P. (2007) FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 50. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janas E., Hofacker M., Chen M., Gompf S., van der Does C., Tampé R. (2003) J. Biol. Chem. 278, 26862–26869 [DOI] [PubMed] [Google Scholar]
- 52. Tombline G., Muharemagić A., White L. B., Senior A. E. (2005) Biochemistry 44, 12879–12886 [DOI] [PubMed] [Google Scholar]
- 53. Sauna Z. E., Nandigama K., Ambudkar S. V. (2006) J. Biol. Chem. 281, 26501–26511 [DOI] [PubMed] [Google Scholar]
- 54. Siarheyeva A., Liu R., Sharom F. J. (2010) J. Biol. Chem. 285, 7575–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Linton K. J., Higgins C. F. (2007) Pflugers Arch. 453, 555–567 [DOI] [PubMed] [Google Scholar]