

Abstract
The vitamin K oxidoreductase (VKOR) reduces vitamin K to support the carboxylation and consequent activation of vitamin K-dependent proteins, but the mechanism of reduction is poorly understood. VKOR is an integral membrane protein that reduces vitamin K using membrane-embedded thiols (Cys-132 and Cys-135), which become oxidized with concomitant VKOR inactivation. VKOR is subsequently reactivated by an unknown redox protein that is currently thought to act directly on the Cys132–Cys135 residues. However, VKOR contains evolutionarily conserved Cys residues (Cys-43 and Cys-51) that reside in a loop outside of the membrane, raising the question of whether they mediate electron transfer from a redox protein to Cys-132/Cys-135. To assess a possible role, the activities of mutants with Ala substituted for Cys (C43A and C51A) were analyzed in intact membranes using reductants that were either membrane-permeable or -impermeable. Both reductants resulted in wild type VKOR reduction of vitamin K epoxide; however, the C43A and C51A mutants only showed activity with the membrane-permeant reductant. We obtained similar results when testing the ability of wild type and mutant VKORs to support carboxylation, using intact membranes from cells coexpressing VKOR and carboxylase. These results indicate a role for Cys-43 and Cys-51 in catalysis, suggesting a relay mechanism in which a redox protein transfers electrons to these loop residues, which in turn reduce the membrane-embedded Cys132–Cys135 disulfide bond to activate VKOR. The results have implications for the mechanism of warfarin resistance, the topology of VKOR in the membrane, and the interaction of VKOR with the carboxylase.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Membrane Enzymes, Oxidation-Reduction, Vitamin K, Gamma-Glutamyl Carboxylase, VKOR
Introduction
The vitamin K oxidoreductase (VKOR)3 supports carboxylation and consequent activation of vitamin K-dependent (VKD) proteins. Carboxylation occurs in the endoplasmic reticulum, where the γ-glutamyl carboxylase converts clusters of Glu to carboxylated Glu (Gla), transforming the Gla domain of VKD proteins into a calcium-binding module that functions either at cell surfaces or in the extracellular matrix (1, 2). The carboxylase drives Glu modification by using the energy of oxygenation of reduced vitamin K, generating a vitamin K epoxide product. VKOR then recycles the vitamin K epoxide back to the reduced form to support continuous carboxylation (Fig. 1a). The combined actions of VKOR and the carboxylase are important to hemostasis, as naturally occurring mutations cause combined VKD clotting factor deficiency (3–7), and mice lacking the genes for each enzyme show a severe bleeding phenotype (8, 9). VKOR is the target of oral anticoagulants like warfarin, which dampens coagulation by limiting the supply of reduced vitamin K. VKOR and the carboxylase have biological impact that extends beyond hemostasis, as VKD proteins are also known to function in calcium homeostasis, apoptosis, signal transduction, and growth control (1, 10). The broad physiological impact of these enzymes makes it critical to understand their mechanism of action.
FIGURE 1.

VKOR and the VKD carboxylase mediate the vitamin K cycle. a, simplified schematic of the reactions carried out by VKOR and the VKD carboxylase (reviewed in Ref. 1). Brackets indicate the portion of the cycle carried out by each enzyme. The carboxylase uses oxygenation of vitamin K hydroquinone (KH2) to vitamin K epoxide (KO) to drive the incorporation of CO2 into glutamyl (Glu) residues to produce carboxylated Glu (Gla) in VKD proteins. VKOR subsequently reduces vitamin K epoxide (KO) to vitamin K quinone (K) and then to vitamin K hydroquinone to support carboxylase activity. b, vitamin K epoxide is reduced by VKOR residues Cys-132 and Cys-135, which become oxidized, inactivating VKOR. Whether electron flow through VKOR to regenerate active enzyme involves the evolutionarily conserved residues Cys-43 and Cys-51 is the focus of this study.
How VKOR reduces vitamin K epoxide is not currently well defined. The mechanism is known to involve active site thiols that become oxidized to a disulfide bond during the reaction, inactivating VKOR (11–13). These cystines are then ultimately reduced by a redox protein, which reactivates VKOR. Potentially relevant Cys residues were indicated by the identification of the gene for VKOR (14, 15), which also revealed that VKOR has a much broader evolutionary distribution than that of the carboxylase, including the presence of VKOR in plants and bacteria. Bioinformatic analysis showed that four Cys residues are evolutionarily conserved in all known VKOR homologs (16), and two of these residues (Cys-132 and Cys-135) are in a CXXC motif that has been implicated in redox reactions in other proteins (17). VKOR is an integral membrane protein, and these two Cys residues are membrane-embedded (18, 19). The remaining two Cys residues (Cys-43 and Cys-51) are present in a loop that is thought to be located in the cytoplasm based on topology studies (18). Mutagenesis studies support the role of Cys-132 and Cys-135 in reducing vitamin K epoxide, as substitutions with either Ser or Ala completely abolish VKOR activity (20, 21). In contrast, cognate substitutions in Cys-43 or Cys-51 result in VKOR mutants that retain significant levels of activity. These data led to the conclusion that this region of VKOR is dispensable for function, with a redox protein acting directly on the Cys132–Cys135 disulfide bond to regenerate VKOR activity (20, 21).
The caveat with the conclusion that Cys-43 and Cys-51 are not required for function is that the experimental design may not have assessed their normal role. Thus, the activity of the VKOR mutants was measured with DTT, a reductant that is commonly used for in vitro VKOR assays to substitute for physiologically relevant redox proteins. DTT is membrane-permeable and can therefore directly access Cys-132 and Cys-135, and so its use could bypass a normal requirement of Cys-43 and Cys-51 in the reaction. To assess the function of these two residues in a more unambiguous manner, we developed a different approach that used a membrane-impermeant reductant in combination with intact membranes that contain individual VKOR variants. As presented below, our results indicate that both Cys-43 and Cys-51 are required for function and suggest that they participate in an electron transfer mechanism in which a redox protein first acts on these two residues, which in turn reduce the Cys132–Cys135 disulfide bond to activate VKOR for vitamin K epoxide reduction (Fig. 1b).
EXPERIMENTAL PROCEDURES
Generation and Expression of VKOR Variants
Wild type human VKOR was generated by PCR using r-VKORC1/ZEM229 as the template and the primers VKRC1S and VKRC1AS1, all of which have previously been described (22). The PCR product was digested with BglII and then gel isolated and subcloned into the BamHI site of BacPak8 to generate pBacPak8-VKOR. A cDNA encoding VKOR with a Cys to Ala substitution at residue 43 (C43A) was generated using overlap PCR; two sets of primers, VKRC1S plus C43A1AS (GGTGCCGACGTCGGCGAGCGCGCGGTAATCCC) and C43A1S (CTCGCCGACGTCGGCACCGCCATCAGCTGTTC) plus VKRC1AS1, were used in the first round of PCR, and primers VKRC1S and VKRC1AS1 were used in the second round. A cDNA mutated in Cys-51 (C51A) was generated by the same approach, using different primers in the first round of PCR, i.e. C51A1AS (CGCGCGAAGCGCTGATGGCGGTGCCCAC) and C51A1S (CCATCAGCGCTTCGCGCGTCTTCTCCTCC). The primers introduced AatII or AfeI restriction endonuclease sites into the C43A or C51A cDNAs, respectively, which were used to screen for the mutants. The cDNAs in positively identified clones were subsequently sequenced on both strands to confirm that the only alteration introduced was that of the mutation. These plasmids (pBacPak8-VKOR, pBacPak8-VKORC43A, and pBacPak8-VKORC51A) were then cotransfected into SF21 cells with BakPak6 (Clontech), and large scale virus preparations were generated following plaque isolation, as described previously (23).
Preparation of Microsomes
SF21 cells (6 × 108) were infected with baculovirus containing wild type or mutant VKORs or with a baculovirus containing the carboxylase, using a multiplicity of infection of 5. Coinfection with baculoviruses containing the carboxylase and wild type or mutant VKORs was also performed, using a multiplicity of infection of 5 for each virus. Cells were harvested 42 h post-infection and washed twice with phosphate-buffered saline and then lysed in 250 mm sucrose, 25 mm Tris, pH 7.4, and 2 mm PMSF (buffer A) by sonication and subsequent Dounce homogenization. All procedures were performed at 4 °C. Following centrifugation at 1100 × g for 15 min, the post-mitochondrial supernatant was centrifuged at 105 × g for 1 h; the supernatant was discarded, and the microsomal pellets were either quick-frozen in liquid nitrogen or resuspended in 9 ml of buffer A and then stored at −80 °C.
In most experiments, microsomes were first subjected to a washing step before VKOR activity was measured to remove peripheral proteins. Microsomal pellets were thawed and resuspended in a volume (∼10 ml) of 100 mm sodium carbonate, pH 10.5, 1 m KCl, 0.025% sodium deoxycholate sufficient to yield a protein concentration of 6–7 mg/ml, as determined by a BCA assay (Pierce). The suspension was Dounce-homogenized, centrifuged at 105 × g for 1 h, and the supernatant discarded. The pellets were surface washed twice with 500 μl of buffer A and then resuspended in 4 ml of the same buffer. All steps were at 4 °C. The protein concentration of the washed microsomes was then determined, and the microsomes were adjusted with buffer A to give a final concentration of 4 mg/ml protein. Equivalent amounts of protein from washed and unwashed microsomes were tested for vitamin K reduction and VKOR-supported peptide carboxylation (described below) to determine whether the washing had any effect on VKOR activity.
Determination of Thioredoxin (Trx) and Thioredoxin Reductase (TrxR) Activity
To assess the activities of Trx and TrxR preparations for their ability to donate electrons in vitro, Trx plus TrxR activity was measured using a spectrophotometric assay that monitors insulin reduction and consequent turbidity. A 100-μl mixture of 100 mm sodium phosphate, pH 7.5, 0.1 mm NADPH (Sigma), 100 μm insulin (Sigma), and 5 μm Escherichia coli Trx (American Diagnostica Inc.) was placed in a cuvette. An aliquot (4 μl) of a 1.8 mg/ml stock solution of E. coli TrxR (American Diagnostica Inc.) was diluted in 196 μl of 50 mm Tris, pH 7.4, 0.1 mg/ml BSA (Pierce), and 1 mm EDTA, and 10 μl of the diluted TrxR was then added to the cuvette to initiate the reaction. The decrease in absorbance at 340 nm was then immediately monitored to determine the activity. Mammalian Trx plus TrxR was assayed using a similar method in a mixture containing 100 mm sodium phosphate, pH 7.0, 0.2 mm NADPH, 160 μm insulin, 2 mm EDTA, 5 μm human Trx (American Diagnostica Inc.), and 50 μg/ml rat TrxR (American Diagnostica Inc.). The mammalian Trx/TrxR showed a lag in activity, consistent with previous studies on Trx/TrxR (24), and so the change in absorbance at 340 nm was monitored after 20 min. Both the bacterial and mammalian Trx/TrxR activities were determined immediately before performing the VKOR and carboxylase assays (described below).
VKOR Activity Assays
The ability of VKOR to support carboxylase activity was measured in microsomes prepared from insect cells infected with baculovirus containing the carboxylase or coinfected with baculoviruses containing the carboxylase and either wild type or mutant VKORs. In some experiments, both washed and unwashed microsomes were analyzed. In other experiments, the microsomes were first solubilized by adjusting the protein concentration to 4 mg/ml, followed by the addition of CHAPS to a final concentration of 0.5% and nutation for 30 min at 4 °C. The specific microsomal preparations used in individual experiments are indicated in the appropriate figure legends. In all experiments that used washed microsomes, the microsomes were treated immediately before the enzymatic analyses.
To assay carboxylation, microsomes (1 mg/ml final concentration) were incubated in 200 μl of 0.5 m ammonium sulfate, 50 mm Tris, pH 7.4, 2.5 mm Phe-Leu-Glu-Glu-Leu, 10 μm factor X propeptide, and 1.8 mm [14C]sodium bicarbonate. Four different reaction mixtures were analyzed, which contained the following: (a) no reductant; (b) DTT at a 5 mm final concentration; (c) E. coli Trx, E. coli TrxR, and NADPH at final concentrations of 25 μm, 90 μg/ml, and 1 mm, respectively; and (d) human Trx, rat TrxR, and NADPH at final concentrations of 10 μm, 140 μg/ml, and 1 mm, respectively. All samples were incubated at 21 °C for 20 min prior to the addition of vitamin K, to eliminate potential complication due to activity lags associated with the Trx/TrxR system (24). The reactions were then initiated by the addition of either vitamin K epoxide (11 μm) or vitamin K quinone (11 μm), or in some experiments, vitamin K hydroquinone (80 μm). The samples were incubated for 90 min, over which time activity was found to be linear (data not shown), and the reactions were then quenched by the addition of 1 ml of 10% trichloroacetic acid. The incorporation of [14C]CO2 into Phe-Leu-Glu-Glu-Leu was then quantitated by scintillation counting, and the observed counts/min were converted into picomoles based on a specific activity of 90 cpm/pmol for the [14C]CO2, as determined previously (25). All experiments were performed in duplicate.
VKOR was also assayed for the reduction of vitamin K epoxide, using microsomes from cells that were either mock-infected or infected with baculovirus containing wild type or mutant VKORs. The reaction conditions were similar to those described above, i.e. a 90-min reaction of 1 mg/ml microsomes in the carboxylation mixture with no reductant, DTT, or E. coli or mammalian Trx/TrxR. The only differences were that unlabeled rather than radioactive sodium bicarbonate was used, and the only form of vitamin K analyzed was vitamin K epoxide. Using these conditions to study vitamin K epoxide reduction allowed a direct comparison with the results of the carboxylation assay. All of the vitamin K epoxide reduction assays were performed in duplicate. Vitamin K epoxide reduction by wild type and mutant VKORs was also analyzed using a reaction mixture that contained buffer A, 11 μm vitamin K epoxide, and the various reductants indicated above, and the results from these experiments were essentially the same as those obtained using the carboxylation mixture (data not shown). In both cases, the reaction was quenched by adding 500 μl of a 1:1 mixture of ethanol and hexane, followed by vortexing for several seconds. A synthetic vitamin K standard (K25, a phylloquinone analog with five isoprenyl groups, GL Synthesis, 2 nmol) was then added to correct for sample recovery during the remaining procedure. The samples were vortexed for 1 min; the organic phase was removed and dried under nitrogen, and the residual vitamin K was resuspended in 60 μl of ethanol, followed by HPLC using a C18 column and detection and quantitation by absorbance. Vitamin K hydroquinone is quantitatively oxidized to the quinone form during the course of preparing the samples for HPLC, and so the assay measures the sum of vitamin K hydroquinone and quinone.
Western Analysis
Washed microsomes were subjected to SDS-PAGE using the NuPAGE system (Invitrogen) and manufacturer's instructions to prevent thiol oxidation of proteins. Western blots were analyzed using an anti-VKOR antibody (0.4 μg/ml) and a goat polyclonal antibody conjugated to IR dye 800CW (0.2 μg/ml, LiCor Biosciences). The amount of VKOR was quantitated using the Odyssey LiCor and a FLAG-tagged VKOR whose concentration was predetermined by reference to known amounts of FLAG-tagged bacterial alkaline phosphatase (BAP-FLAG Sigma).
RESULTS
Membrane-impermeant Reductant Activates VKOR in Intact Membranes
To assess whether Cys-43 and Cys-51 have a role in vitamin K epoxide reduction, the activity of VKOR was measured using intact membranes and a reducing system that cannot directly interact with the membrane-embedded Cys-132 and Cys-135 residues that react with vitamin K epoxide. The use of intact membranes was advantageous in maintaining VKOR in the most native state possible and also proved to be valuable because it could be extended to studying VKOR-supported carboxylation (as described below). To study VKOR activation, i.e. the reduction of cystines in VKOR that leads to activity, recombinant wild type VKOR was expressed in SF21 cells, which do not contain endogenous carboxylase or VKD proteins (26), and then tested for activity using Trx, TrxR, and NADPH as the membrane-impermeant source of reducing power. This reducing system was chosen based on previous studies with liver microsomes showing Trx/TrxR-mediated VKOR activation (27, 28). It is important to note that Trx/TrxR was tested because it is clearly membrane-impermeant, and its use is not meant to imply that it is the physiologically relevant redox protein for VKOR in vivo.
The previous in vitro Trx/TrxR studies used solubilized microsomes. When we tested the ability of Trx/TrxR to activate VKOR in solubilized microsomes from insect cells, substantial VKOR activity was observed (Fig. 2a). However, Trx/TrxR did not activate VKOR in unsolubilized membranes, despite significant levels of VKOR activation by the membrane-permeant reductant DTT (Fig. 2b).
FIGURE 2.
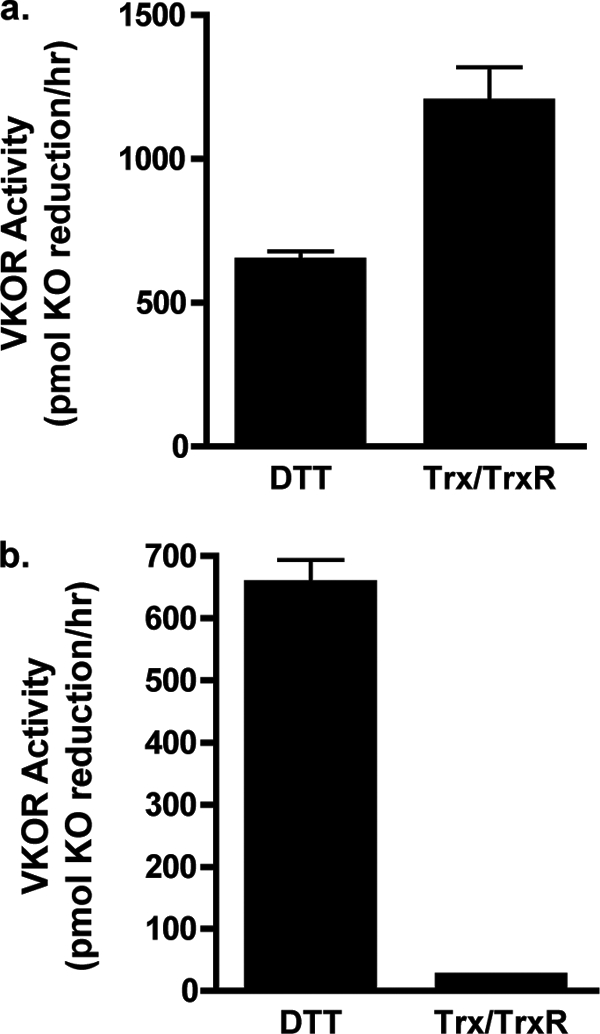
VKOR activation by Trx and TrxR. Solubilized (a) or unsolubilized (b) microsomes prepared from insect cells infected with baculovirus containing wild type VKOR were incubated in a vitamin K epoxide reduction reaction containing buffer A, vitamin K epoxide, and either DTT or E. coli Trx, TrxR, and NADPH. After 30 min of incubation, the vitamin K forms were isolated by organic extraction and quantitated by absorbance using HPLC. The range in values that were obtained is indicated by the error bars, which in the case of the Trx/TrxR assay in b is too small to be observable.
One possible explanation for the difference in activation in unsolubilized versus solubilized microsomes is interference by peripheral proteins that are released when the microsomes are solubilized. Alternatively, the reason why activity was observed only after solubilization may be due to loss of polarity of the membrane; during microsomal isolation, polarity is retained with the cytoplasmic face exposed on the exterior of the membrane vesicle and the luminal contents buried within the interior (29), and this orientation may have blocked accessibility of VKOR-reactive Cys residues to Trx/TrxR. We therefore repeated the test of Trx/TrxR activation of VKOR using a well established washing method (30, 31) that removes peripheral proteins and converts the vesicle to a laminar membrane that is intact but has lost polarity. As described in detail under “Experimental Procedures,” a high pH sodium carbonate wash was performed on the microsomes, which removed ∼60% of the protein while still retaining VKOR activity. When washed, unsolubilized microsomes were incubated with Trx and TrxR, VKOR activation was observed (Fig. 3a). This activity was VKOR-dependent, as only trace amounts of activity were observed when microsomes from mock-infected insect cells were assayed (Fig. 3a). Activation of VKOR was obtained with both mammalian and E. coli Trx/TrxR, consistent with results previously obtained with solubilized rat liver microsomes (28). The signal observed with Trx/TrxR was less than that obtained with DTT (Fig. 3a); however, it should be noted that the concentrations of mammalian and E. coli Trx were in the micromolar range versus millimolar amounts of DTT, indicating that the Trx/TrxR system was quite potent in activating VKOR.
FIGURE 3.

Trx/TrxR activates VKOR in intact membranes. a, microsomes prepared from insect cells infected with baculovirus containing wild type VKOR or from uninfected cells (mock) were washed with sodium carbonate as described under “Experimental Procedures” and then assayed for vitamin K epoxide (KO) reduction, as in the legend to Fig. 2. b, washed intact microsomes from wild type VKOR-expressing cells were assayed for 30 min in 200 μl of buffer A containing vitamin K epoxide (11 μm), human Trx (10 μm) and/or rat TrxR (140 μg/ml) and 1 mm NADPH. The results in a and b were obtained using different microsomal preparations, and the reason why the VKOR activity in the Trx plus TrxR reaction differs is due to the variation in the levels of VKOR expression that occur during individual viral infections.
The simultaneous requirement of both Trx and TrxR for VKOR activation was also tested because the mammalian TrxR is known to have broad substrate specificity and was previously reported to directly reduce menadione (32). This vitamin K form has two significant differences from the physiologically relevant vitamin K family members, i.e. the phylloquinone and menaquinone epoxides that are normally reduced by VKOR in vivo. First, menadione is a quinone and not an epoxide. Second, it lacks the phytyl group that allows the insertion of phylloquinone and menaquinones into the membrane where VKOR and the carboxylase act. These differences raised the question of whether TrxR could directly activate the phylloquinone epoxide used in the in vitro assays. When intact washed microsomes from insect cells infected with baculovirus containing VKOR were incubated with mammalian TrxR and/or Trx, VKOR activation only occurred if both TrxR and Trx were present (Fig. 3b). Thus, TrxR does not directly reduce vitamin K epoxide, and its ability to activate VKOR is indirect through Trx.
A second reducing system, i.e. protein disulfide isomerase in combination with reduced RNase, has also been shown to activate VKOR in liver microsomes (33). We therefore performed studies similar to those described above to test the suitability of this reductant for in vitro analysis. Significantly lower VKOR activity was observed, and so all of the subsequent studies were performed using the Trx/TrxR redox system.
Mutations in VKOR Residues Cys43 and Cys51 Disrupt Trx/TrxR-mediated VKOR Activation
The ability of Trx/TrxR to activate VKOR in intact membranes would be unlikely to occur by direct reduction of the membrane-embedded Cys-132 and Cys-135 residues, raising the question of whether activation is indirect through the evolutionarily conserved Cys-43 and Cys-51 residues in VKOR that are present in a loop outside of the membrane. Mutants with individual Cys substituted by Ala were therefore generated and expressed in insect cells (Fig. 4). When intact washed microsomes from insect cells infected with baculoviruses, containing either VKOR C43A or C51A, were assayed using DTT, both preparations were active in reducing vitamin K epoxide (Fig. 5), as expected based on previous studies (20, 21). The data shown in Fig. 5 represent the actual values obtained, to illustrate the absolute level of signal. When normalized to the amount of VKOR protein expressed, the specific activity of DTT-supported activity for both mutants was 27% that of wild type enzyme. This value for C43A is similar to that previously reported for Ser- or Ala-substituted Cys-43 (20, 21), while the value for C51A lies between the values obtained for C51S (0% (21)) and C51A (100% (20)). One possible contributing factor to this variance is the use of different preparations of VKOR, i.e. intact membranes in this study versus solubilized microsomes or purified protein in the previous studies.
FIGURE 4.

VKOR expression in insect cells. Insect cells infected with baculoviruses containing either wild type or mutant VKORs were analyzed in a Western blot using antibody against the C terminus of VKOR, as described under “Experimental Procedures.”
FIGURE 5.

C43A and C51A VKOR variants are not activated by Trx/TrxR. Intact microsomes from insect cells infected with baculovirus containing either wild type or mutant VKORs were washed with sodium carbonate, and vitamin K epoxide reduction was then measured using the different reductants that are indicated. The details of this assay are described under “Experimental Procedures.” Wild type and mutant VKOR preparations were washed and analyzed on the same day, and the entire experiment was performed two times, giving similar results.
The results obtained using Trx/TrxR as the reductant instead of DTT were strikingly different. Wild type VKOR was clearly activated by both E. coli and mammalian Trx/TrxR (Fig. 5), as before (Fig. 3). In contrast, activities of the VKOR variants C43A and C51A with this reductant were undetectable. These results thus indicate that Cys-43 and Cys-51 are required to transfer reducing potential from Trx/TrxR to VKOR for activation.
Trx and TrxR Support VKOR-mediated Carboxylation in Intact Membranes
The consequence of VKOR activation on downstream carboxylation was also assessed using the Trx/TrxR reducing system. These analyses monitored the complete reduction of vitamin K epoxide, i.e. to the vitamin K hydroquinone cofactor form that is required by the carboxylase. VKOR and the carboxylase were coexpressed using baculoviruses containing the wild type form of each enzyme. Coexpression of these carboxylation components has not been previously described, and so we first determined whether the presence of the carboxylase affected VKOR expression and activity. This test showed that coexpression of the carboxylase did not alter either the specific activity of VKOR (Fig. 6a) or its level of expression (data not shown). VKOR-supported carboxylation was observed (Fig. 6b), using DTT as the reductant and a reaction time point when the activities of both VKOR and the carboxylase were in the linear range. These results were obtained using intact microsomes, which was the only way the analysis could be performed because we found that solubilization disrupted VKOR-supported carboxylase activity (data not shown).
FIGURE 6.

VKOR-supported carboxylation in insect cell membranes. a, solubilized microsomes from insect cells infected with baculovirus containing VKOR or coinfected with baculoviruses containing VKOR and the carboxylase (Carb) were prepared and quantitated for VKOR by a Western blot and LiCor analysis. Equivalent amounts of VKOR protein were then assayed for vitamin K epoxide (KO) reduction using DTT as the reductant. To assay vitamin K epoxide reduction in the absence of carboxylase activity that converts the vitamin K hydroquinone back to vitamin K epoxide, the sample containing both carboxylase and VKOR was placed under a stream of dry nitrogen for 5 min prior to the addition of vitamin K epoxide, and the entire reaction was then carried out in a glove bag filled with pure nitrogen. Parallel treatment of the sample containing only VKOR showed that oxygen depletion had no effect on the ability of VKOR to reduce vitamin K epoxide (data not shown). b and c, microsomes were prepared from insect cells coinfected with baculoviruses containing VKOR and the carboxylase. Both unwashed and sodium carbonate-washed preparations were then assayed for carboxylation of a Glu-containing peptide. The reaction with vitamin K epoxide in b used the reductant DTT. d, solubilized microsomes from insect cells infected with baculovirus-containing carboxylase were assayed in a carboxylation reaction that contained 80 μm vitamin K hydroquinone (KH2) and the indicated range of DTT concentrations.
The effect of membrane washing on carboxylase activity was assessed to determine carboxylase stability and retention of VKOR-supported carboxylation. When microsomes containing VKOR and the carboxylase were incubated in a reaction with vitamin K hydroquinone, which is used directly by the carboxylase, a small decrease in activity was observed after sodium carbonate treatment (Fig. 6c). However, when the same microsomal preparation was assayed using vitamin K epoxide to measure VKOR-supported carboxylation, activity was slightly improved by membrane washing (Fig. 6b), indicating the suitability of the washed microsomes for subsequent studies with Trx/TrxR. The amount of vitamin K epoxide-supported carboxylation was less than that observed with vitamin K hydroquinone (Fig. 6, b versus c) but appreciable, considering that saturating amounts of vitamin K hydroquinone (80 μm) were used in one assay, while in the other the vitamin K hydroquinone was generated by VKOR.
The ability of Trx/TrxR to support VKOR and consequent carboxylase activity was then monitored. We first tested whether DTT had any effect upon carboxylase activity, because previous studies with liver microsomes indicated large differences in activity that might complicate interpretation in comparing DTT- versus Trx/TrxR-supported carboxylation. Carboxylase activity in insect cells did not show a significant response to DTT; when microsomes containing carboxylase (but not VKOR) were assayed using vitamin K hydroquinone and a range of DTT concentrations, carboxylase activity was essentially the same in the presence or absence of DTT (Fig. 6d). Washed microsomes containing both wild type carboxylase and VKOR were then tested using Trx/TrxR, which showed that this reductant resulted in carboxylase activity (Fig. 7). The effect of the DTT and Trx/TrxR reductants on carboxylation was due to VKOR reduction of vitamin K epoxide, as carboxylation was barely detectable with washed microsomes containing only the carboxylase (Fig. 7a). The amount of VKOR-supported carboxylation was greater with the E. coli Trx/TrxR reductant than with mammalian Trx and TrxR. This difference was due at least in part to the use of higher concentrations of E. coli Trx than mammalian Trx (25 and 10 μm, respectively), which in the case of the E. coli Trx was close to saturating concentrations (Fig. 8). Interestingly, the amount of Trx/TrxR-supported VKOR activity relative to that of DTT was higher in these studies with carboxylase than in those measuring vitamin K epoxide reduction by VKOR alone; the amount of E. coli and mammalian Trx/TrxR-driven carboxylation was 90 and 50% that of DTT, respectively (Fig. 7a), versus 27% (E. coli) and 18% (mammalian) for VKOR reduction of vitamin K epoxide (Fig. 3).
FIGURE 7.

Trx/TrxR drives VKOR-supported carboxylation. Intact washed microsomes were prepared from insect cells infected with baculovirus containing carboxylase (Carb) or coinfected with baculoviruses containing carboxylase and VKOR (VKOR·Carb). The microsomes were then assayed in a carboxylation reaction using the indicated reductants and either vitamin K epoxide (KO) (a) or vitamin K quinone (K) (b). The details of the assay are described under “Experimental Procedures.” The lack of activity in the carboxylase samples was not due to low carboxylase levels, as Western analysis and detection by LiCor showed that carboxylase expression was actually 2-fold higher in the “Carb” microsomes than in the “VKOR·Carb” microsomes (data not shown).
FIGURE 8.

Response of VKOR-supported carboxylation to levels of Trx. Intact washed microsomes from insect cells coinfected with baculoviruses containing VKOR and the carboxylase were assayed in a carboxylation reaction that contained vitamin K epoxide (KO) and either mammalian TrxR (140 μg/ml) and varying concentrations of mammalian Trx (a) or E. coli TrxR (90 μg/ml) and varying concentrations of E. coli Trx (b). Saturating VKOR activity with the human Trx was not obtainable because it required prohibitively large amounts of Trx.
Trx/TrxR supported carboxylation through VKOR reduction of either vitamin K quinone (Fig. 7b) or vitamin K epoxide (Fig. 7a), showing that VKOR performs quinone reduction as well as epoxide reduction. The results were similar with one significant difference. In the case of vitamin K quinone-supported carboxylation, activity was observed with DTT in the microsomes containing only the carboxylase. This result was not surprising because vitamin K quinone, but not vitamin K epoxide, is known to be reduced by DTT. Such reduction also likely accounts for the ∼20% higher level of DTT-supported carboxylation observed with vitamin K quinone versus vitamin K epoxide in the microsomes containing VKOR and the carboxylase (Fig. 7, a versus b). In contrast to DTT, mammalian TrxR did not drive either vitamin K epoxide or vitamin K quinone-supported carboxylation in the absence of VKOR. This result is consistent with that obtained by measuring vitamin K epoxide reduction (Fig. 3b) and indicates that the broad substrate specificity of TrxR does not include reducing appreciable amounts of phylloquinone to drive carboxylation.
Mutations in VKOR Residues Cys43 and Cys51 Abolish VKOR-supported Carboxylation
The effect of substitutions in VKOR residues Cys-43 or Cys-51 on Trx/TrxR-supported carboxylation was measured in microsomes from insect cells coexpressing carboxylase and either wild type or C43A or C51A mutants. Western analysis indicated that the levels of both VKOR and the carboxylase were similar in cells expressing each of the VKOR variants (Fig. 9). Vitamin K hydroquinone-supported carboxylase activity was also measured, indicating similar levels in microsomes from C43A, C51A, or wild type VKOR-expressing cells (data not shown). When these microsomes were incubated with vitamin K epoxide and DTT, which can activate VKOR by directly reducing the Cys132–Cys135 disulfide bond, significant levels of carboxylation were observed for the C43A and C51A mutants (Fig. 10a). The specific activities were 28% (C43A) and 47% (C51A) that of wild type enzyme, and these values are similar to those obtained when the mutants were assayed for vitamin K epoxide reduction (Fig. 5). Similar results were obtained when vitamin K quinone-supported carboxylation was monitored (Fig. 10b), except that slightly higher levels of activity were observed with DTT as the reductant, due presumably to some reduction of the vitamin K quinone by DTT as described above.
FIGURE 9.
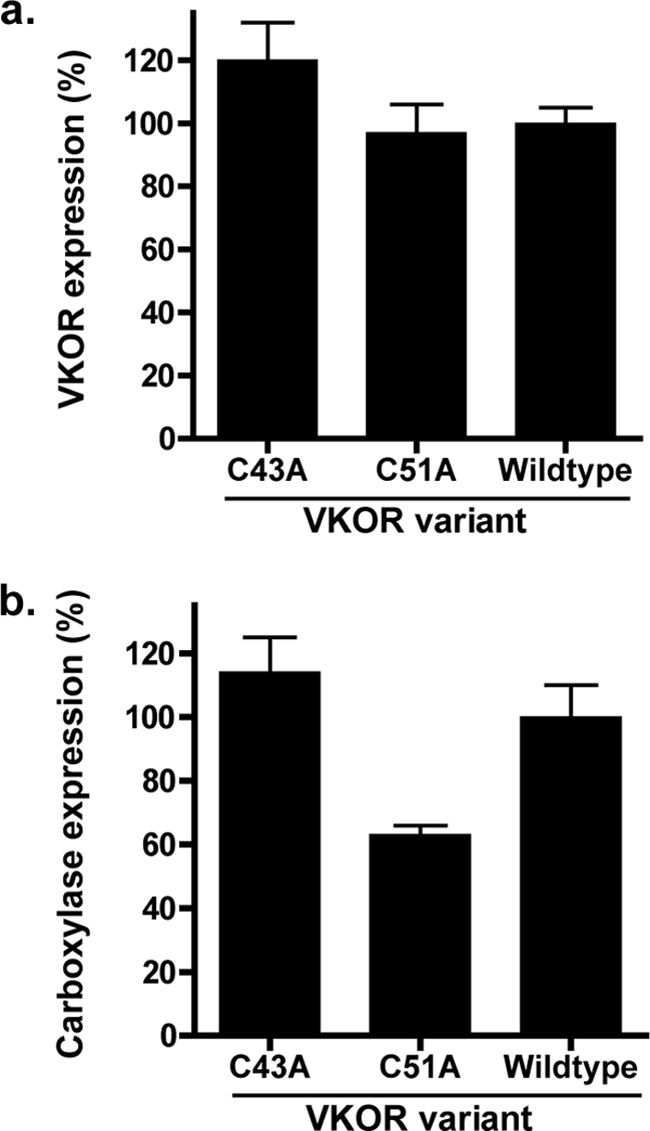
Expression of VKOR and the carboxylase in coinfected cells. Duplicate samples of intact washed microsomes from insect cells coinfected with baculoviruses containing the carboxylase and either wild type or mutant VKORs were quantitated by LiCor analysis on Western blots that detected VKOR using an antibody against the C terminus of VKOR (a) and carboxylase using an antibody against the C-terminal FLAG epitope (b).
FIGURE 10.

Mutations in VKOR residues Cys43 and Cys51 disrupt Trx/TrxR-mediated, VKOR-supported carboxylation. Microsomes from insect cells coinfected with baculoviruses containing the carboxylase and wild type or mutant VKORs were washed with sodium carbonate and then assayed in a carboxylation reaction in the presence of vitamin K epoxide (KO) (a) or vitamin K quinone (K) (b) and the indicated reductants as described under “Experimental Procedures.” The activities observed are presented to indicate the absolute level of signal. The values observed for DTT-supported C43A and C51A activities corresponded to 8000 or 12,000 cpm in the assay, respectively, and these values are 20–30-fold over background. The entire experiment was performed twice, giving similar results.
E. coli Trx/TrxR was an effective reductant for wild type VKOR (Fig. 10), as before (Fig. 7), resulting in higher levels of carboxylase activity than that obtained with DTT. In contrast, E. coli Trx/TrxR incubation with either C43A or C51A resulted in only trace amounts of vitamin K epoxide- or vitamin K quinone-supported carboxylation (Fig. 10). Similarly, the level of mammalian Trx/TrxR-supported carboxylation was substantial with wild type VKOR but essentially undetectable with the two VKOR mutants. These data strongly indicate that residues Cys-43 and Cys-51 are not dispensable for function but rather facilitate activation of VKOR and subsequent carboxylation.
DISCUSSION
VKOR becomes activated when thiols are generated by reduction and previous studies on four evolutionarily conserved Cys residues led to the conclusion that two of the thiols (Cys-132 and Cys-135) are responsible for reducing vitamin K epoxide, while the remaining two (Cys-43 and Cys-51) are not required for function (20, 21). In this model of VKOR activation, a redox protein would donate electrons directly to the Cys132–Cys135 disulfide bond to generate thiols, with access to these membrane-embedded residues occurring either because the redox protein is an integral membrane protein or has a hydrophobic domain that penetrates into the phospholipid bilayer. The studies that led to this model used the membrane-permeant reductant DTT to substitute for physiologically relevant redox proteins. Our studies using a membrane-impermeant redox system (Trx/TrxR) show a different mechanism for VKOR activation. We found that Trx/TrxR activates VKOR in intact membranes (Figs. 3, 5, and 10), which would be unlikely to occur through direct interaction with the membrane-embedded Cys-132 and Cys-135 residues. Mutants with substitutions in Cys-43 or Cys-51 could not be activated by Trx/TrxR even though they retained the ability to be activated by DTT (Figs. 5 and 10). These data suggest that Cys-43 and Cys-51, which are located in a loop outside of the membrane, participate in an electron relay mechanism in which a redox protein first reduces the Cys43–Cys51 disulfide bond to generate free thiols that then reduce the Cys132–Cys135 disulfide bond, activating VKOR. This mechanism affords efficient delivery of electrons into the membrane, because the loop facilitates VKOR interaction with a redox protein outside of the membrane, and the location of Cys pairs on the same molecule results in high local concentrations and consequent efficient electron transfer. The mechanism would allow two alternative pathways for VKOR activation in reactions supported by DTT; the Cys132–Cys135 bond can be directly reduced by DTT that penetrates into the membrane or can be indirectly reduced by DTT acting on the Cys43–Cys51 disulfide bond. These individual pathways may not be equivalent in contributing to the total amount of DTT-supported activity. Only one pathway is available in the case of the C43A and C51A variants, i.e. that involving direct DTT reduction of Cys132–Cys135, and this difference from wild type VKOR may explain why these mutants had activities that were ∼30% that of wild type enzyme (Fig. 5).
Implications for Warfarin Resistance
A role for Cys-43 and Cys-51 in the reaction mechanism is of interest with regard to warfarin resistance. Patients undergoing warfarin therapy exhibit considerable inter-individual variability in the amount of warfarin required to control hemostasis, which has been attributed to both environmental and genetic factors that include VKOR mutations. The mutations necessitate higher doses of warfarin, but how these mutations cause warfarin resistance is unknown, due in part to the fact that the mechanism by which warfarin inhibits VKOR is poorly understood. Warfarin inhibition is noncompetitive and irreversible (34), indicating nonidentical binding sites for warfarin and vitamin K, and the binding site for warfarin has not been identified. Warfarin binds to the oxidized but not the reduced form of VKOR (11), which means that there is only a certain window of time during the multiple steps of the reaction when warfarin can bind and block VKOR activity. Previously, it was generally assumed that oxidation of Cys-132 and Cys-135 leads to warfarin binding; however, our results raise the possibility that such binding may instead occur as a consequence of oxidation of Cys-43 and Cys-51. This possibility may be relevant to the observation that most of the known warfarin-resistant mutations map in the loop that contains Cys-43 and Cys-51 and alter amino acids that are evolutionarily conserved in all organisms that perform carboxylation (Fig. 11) (15, 35–41). Our results implicating Cys-43 and Cys-51 within this region as important to VKOR catalysis raise the intriguing possibility that residues in the loop facilitate electron transfer between Cys-43/Cys-51 and Cys-132/Cys-135 and that warfarin resistance arises from disruption in the normal flow of electrons. Mutations in the loop then could lead to warfarin resistance from multiple mechanisms. For example, redox protein binding to VKOR may block accessibility to warfarin, and in the case of the mutations slower redox protein catalysis or release from VKOR would limit the amount of free VKOR that warfarin can bind and inhibit. The interference with normal electron transfer would predict impaired VKOR activity in the warfarin-resistant mutants, which in fact is what has been observed for those loop mutants that have been analyzed (15). A puzzling aspect of these analyses is that mutations that cause warfarin resistance in vivo actually show warfarin sensitivity when assayed in vitro. Importantly, those assays were performed using DTT as the reductant, and so an impact of a redox protein on the reaction would be missed. Reassessing the activity of these mutants with a natural redox protein will likely provide valuable information regarding the mechanism of warfarin resistance.
FIGURE 11.

Cys43 and Cys51 reside within a VKOR region that is highly conserved in organisms that carboxylate VKD proteins. The protein sequences for VKOR from different vertebrates were aligned using the ClustalW function of the MacVector program (version 7.2.2). Amino acid residues that are identical in five or more species are shown as white letters in a black box, and those that are chemically similar are shown as boldface letters in a gray box. The species and the sequence accession numbers used to produce the alignment are human (NP_076869), cow (NP_001003903), rat (NP_976080), mouse (NP_848715), chicken (NP_996530), the toad Xenopus (AAH43742), the fish Tetraodon (CAG07588), and the pufferfish Takifugu (AAR82912). The mutations below the alignment are associated with warfarin resistance.
Implications for Topology
Electron relay between Cys-43/Cys-51 and Cys-132/Cys-135 is also of interest with regard to the topology of VKOR in the membrane. Human VKOR is currently thought to have three transmembrane sequences, based on membrane insertion studies that examined full-length or variant VKORs deleted in predicted transmembrane sequences (18). Those studies also indicated that the VKOR N terminus is located in the lumen of the endoplasmic reticulum, which would place Cys-43 and Cys-51 in the cytoplasm where reducing power could be harvested by a redox protein that activates VKOR. We only observed Trx/TrxR-supported VKOR activity in intact membranes that had been washed with sodium carbonate (Fig. 2 versus 3), which destroys the polarity of the membrane, and so our studies do not provide insight into the location of redox proteins in the lumen or cytoplasm. They do, however, indicate interaction between Cys-43/Cys-51 and Cys-132/Cys-135, and so it is notable that in the current model of VKOR topology these residues are far apart, being positioned on opposite sides of the membrane (Fig. 12a).
FIGURE 12.

Alternative three- versus four-transmembrane topologies of human VKOR. Shown are the three-transmembrane topology of human VKOR based on biochemical studies (18) (a) and the four-transmembrane topology suggested by the crystal structure of a bacterial VKOR homolog (42) (b). The N-terminal lumenal location for human VKOR determined by Tie et al. (18) places Cys-43/Cys-51 (−SH) in the cytoplasm, although cognate Cys residues in the bacterial homolog are in the periplasm, where the oxidative environment is analogous to the lumen of the endoplasmic reticulum. Cys-132/Cys-135 (−SH in the membrane), which react with vitamin K, are also shown. Note that in a and b, these Cys residues are on the opposite or same side of the membrane as Cys-43 and Cys-51, respectively. The red dots represent warfarin-resistant mutants, whose identity and position are detailed in Fig. 11.
This model of VKOR topology was recently challenged. As presented in the Introduction, the identification of the gene for VKOR indicated bacterial VKOR homologs, and a crystal structure was determined for a homolog from Synechococcus, which revealed a four-transmembrane helical bundle that wraps around the quinone (42). Hydropathy algorithms suggest a fourth predicted transmembrane sequence in mammalian VKORs (16), and it was suggested that the mammalian homologs are the same as the bacterial VKORs in having four rather than three transmembrane sequences (42). It is important to note that there are significant differences between the mammalian and bacterial homologs. For example, the bacterial VKOR homolog was isolated bound to ubiquinone rather than vitamin K, is a quinone reductase rather than an epoxide reductase, and has a different cellular environment, i.e. bacterial periplasm versus the endoplasmic reticulum. Nonetheless, the suggestion of a four-transmembrane topology for human VKOR is intriguing because it would position the Cys-43/Cys-51 residues closer to Cys-132 and Cys-135. The Cys-132/Cys-135 and Cys-43/Cys-51 pairs are still predicted to be membrane-embedded or present in an extramembrane loop, respectively (Fig. 12b), but the two pairs of cysteines would now be localized to the same side of the membrane, facilitating their interaction. A four-transmembrane structure with the lumenal N terminus indicated by the previous topology studies (18) has the opposite orientation from that of the bacterial homolog with N and C termini in the cytoplasm (42). The orientations have important implications both for a redox protein that activates VKOR and for whether mammalian VKOR plays a role in protein oxidation like the bacterial homolog (discussed below). These considerations and the apparent differences between the mammalian and bacterial homologs highlight the need for additional studies that compellingly define the topology of mammalian VKOR.
The Synechococcus VKOR structure supports our proposal that Cys-43 and Cys-51 in human VKOR relay electrons between a redox protein and Cys-132/Cys-135. The bacterial VKOR homologs are thought to have a role in disulfide bond formation in the periplasm as they are structurally similar to DsbB, an integral membrane protein that shuttles electrons generated by disulfide bond formation to quinones in the membrane, and a VKOR homolog (from Mycobacterium tuberculosis) complements DsbB function in vivo (42, 43). Both DsbB and the VKOR homologs contain two membrane-embedded Cys residues and two residues in a loop located in the periplasm. The Synechococcus VKOR homolog additionally contains a C-terminal Trx-like domain that functions analogously to a redox protein for mammalian VKOR (16). The crystal structure was determined with a mutant, C56S, where the Cys-56 residue corresponds to the human VKOR Cys-51 residue analyzed in this study. This structure revealed a disulfide bond between the remaining loop Cys (Cys-50 in the bacterial homolog, corresponding to human VKOR residue Cys-43) and a Cys in the Trx-like domain, and this intramolecular disulfide bond was suggested to be a trapped intermediate of the redox reaction (42). This interpretation is consistent with our studies indicating a functional role for Cys-43 and Cys-51 in the human VKOR reaction.
Implications for VKOR-Carboxylase Interaction
A comparison of Trx/TrxR-supported VKOR activity in the presence versus absence of carboxylase is relevant with regard to how vitamin K is shuttled between VKOR and the carboxylase. At present, it is not known whether these two integral membrane proteins exist in a complex or as separate components, with vitamin K shuttling between them by diffusion through the phospholipid bilayer. Carboxylase solubilized from membrane remains bound to VKD proteins (1), and this tight association ensures processive carboxylation of Glu residues (44, 45). In the case of the carboxylase and VKOR, it is difficult to assess whether a complex exists. Neither VKOR-supported carboxylation nor coimmunoprecipitation of the two proteins is detected in solubilized membranes (data not shown); however, solubilization could be disrupting normal association. The dissection of VKOR activity in the presence or absence of carboxylase in intact membranes therefore provides particularly useful information. When the level of Trx/TrxR-supported VKOR reduction of vitamin K is compared with that of DTT-supported reduction, VKOR activity in the presence of carboxylase (Fig. 10) is 3–4-fold higher than VKOR activity in the absence of carboxylase (Figs. 3 and 5). The ability of the carboxylase to modulate the reaction of Trx/TrxR with VKOR would not be expected unless there is physical interaction between VKOR and the carboxylase, and the results therefore suggest that a complex exists. Such association has the potential to increase the efficiency of carboxylation, consistent with the observation that the level of carboxylation driven by VKOR and vitamin K epoxide was not that much less than that obtained using saturating concentrations of vitamin K hydroquinone (Fig. 6, b and c).
In conclusion, these studies indicating that VKOR is activated by electron relay between loop and membrane Cys pairs provide novel insight into both the mechanism of VKOR and its interaction with the carboxylase. The expression of VKOR and the carboxylase in virtually every tissue in humans and the diverse functions of the VKD proteins highlight the importance of understanding the mechanism of VKOR activation and the significance of this study.
Acknowledgment
We thank Kurt Runge for helpful discussions during the course of this work.
This work was supported, in whole or in part, by National Institutes of Health Grant 5RO1HL081093 (to K. L. B.).
- VKOR
- vitamin K oxidoreductase
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- KO
- vitamin K epoxide
- K
- vitamin K quinone
- VKD
- vitamin K-dependent.
REFERENCES
- 1. Berkner K. L. (2008) Vitam. Horm. 78, 131–156 [DOI] [PubMed] [Google Scholar]
- 2. Tie J. K., Stafford D. W. (2008) Vitam. Horm. 78, 103–130 [DOI] [PubMed] [Google Scholar]
- 3. Darghouth D., Hallgren K. W., Shtofman R. L., Mrad A., Gharbi Y., Maherzi A., Kastally R., LeRicousse S., Berkner K. L., Rosa J. P. (2006) Blood 108, 1925–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rost S., Fregin A., Koch D., Compes M., Müller C. R., Oldenburg J. (2004) Br. J. Haematol. 126, 546–549 [DOI] [PubMed] [Google Scholar]
- 5. Brenner B., Sánchez-Vega B., Wu S. M., Lanir N., Stafford D. W., Solera J. (1998) Blood 92, 4554–4559 [PubMed] [Google Scholar]
- 6. Spronk H. M., Farah R. A., Buchanan G. R., Vermeer C., Soute B. A. (2000) Blood 96, 3650–3652 [PubMed] [Google Scholar]
- 7. Oldenburg J., von Brederlow B., Fregin A., Rost S., Wolz W., Eberl W., Eber S., Lenz E., Schwaab R., Brackmann H. H., Effenberger W., Harbrecht U., Schurgers L. J., Vermeer C., Müller C. R. (2000) Thromb. Haemost. 84, 937–941 [PubMed] [Google Scholar]
- 8. Spohn G., Kleinridders A., Wunderlich F. T., Watzka M., Zaucke F., Blumbach K., Geisen C., Seifried E., Müller C., Paulsson M., Brüning J. C., Oldenburg J. (2009) Thromb. Haemost. 101, 1044–1050 [PubMed] [Google Scholar]
- 9. Zhu A., Sun H., Raymond R. M., Jr., Furie B. C., Furie B., Bronstein M., Kaufman R. J., Westrick R., Ginsburg D. (2007) Blood 109, 5270–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berkner K. L. (2005) Annu. Rev. Nutr. 25, 127–149 [DOI] [PubMed] [Google Scholar]
- 11. Fasco M. J., Principe L. M., Walsh W. A., Friedman P. A. (1983) Biochemistry 22, 5655–5660 [DOI] [PubMed] [Google Scholar]
- 12. Lee J. J., Fasco M. J. (1984) Biochemistry 23, 2246–2252 [DOI] [PubMed] [Google Scholar]
- 13. Whitlon D. S., Sadowski J. A., Suttie J. W. (1978) Biochemistry 17, 1371–1377 [DOI] [PubMed] [Google Scholar]
- 14. Li T., Chang C. Y., Jin D. Y., Lin P. J., Khvorova A., Stafford D. W. (2004) Nature 427, 541–544 [DOI] [PubMed] [Google Scholar]
- 15. Rost S., Fregin A., Ivaskevicius V., Conzelmann E., Hörtnagel K., Pelz H. J., Lappegard K., Seifried E., Scharrer I., Tuddenham E. G., Müller C. R., Strom T. M., Oldenburg J. (2004) Nature 427, 537–541 [DOI] [PubMed] [Google Scholar]
- 16. Goodstadt L., Ponting C. P. (2004) Trends Biochem. Sci. 29, 289–292 [DOI] [PubMed] [Google Scholar]
- 17. Holmgren A. (1985) Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 18. Tie J. K., Nicchitta C., von Heijne G., Stafford D. W. (2005) J. Biol. Chem. 280, 16410–16416 [DOI] [PubMed] [Google Scholar]
- 19. Wallin R., Sane D. C., Hutson S. M. (2002) Thromb. Res. 108, 221–226 [DOI] [PubMed] [Google Scholar]
- 20. Jin D. Y., Tie J. K., Stafford D. W. (2007) Biochemistry 46, 7279–7283 [DOI] [PubMed] [Google Scholar]
- 21. Rost S., Fregin A., Hünerberg M., Bevans C. G., Müller C. R., Oldenburg J. (2005) Thromb. Haemost. 94, 780–786 [DOI] [PubMed] [Google Scholar]
- 22. Hallgren K. W., Qian W., Yakubenko A. V., Runge K. W., Berkner K. L. (2006) Biochemistry 45, 5587–5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berkner K. L., McNally B. A. (1997) Methods Enzymol. 282, 313–333 [DOI] [PubMed] [Google Scholar]
- 24. Holmgren A. (1979) J. Biol. Chem. 254, 9627–9632 [PubMed] [Google Scholar]
- 25. Rishavy M. A., Berkner K. L. (2008) Biochemistry 47, 9836–9846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roth D. A., Rehemtulla A., Kaufman R. J., Walsh C. T., Furie B., Furie B. C. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8372–8376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Haarlem J. L. M., Soute B. A., Vermeer C. (1987) FEBS Lett. 222, 353–357 [DOI] [PubMed] [Google Scholar]
- 28. Soute B. A., Groenen J. L., van Dooren M. M., Holmgren A., Lundström J., Vermeer C. (1992) Biochem. J. 281, 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katz F. N., Rothman J. E., Lingappa V. R., Blobel G., Lodish H. F. (1977) Proc. Natl. Acad. Sci. U.S.A. 74, 3278–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kreibich G., Debey P., Sabatini D. D. (1973) J. Cell Biol. 58, 436–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luthman M., Holmgren A. (1982) Biochemistry 21, 6628–6633 [DOI] [PubMed] [Google Scholar]
- 33. Wajih N., Hutson S. M., Wallin R. (2007) J. Biol. Chem. 282, 2626–2635 [DOI] [PubMed] [Google Scholar]
- 34. Fasco M. J., Principe L. M. (1982) J. Biol. Chem. 257, 4894–4901 [PubMed] [Google Scholar]
- 35. Harrington D. J., Gorska R., Wheeler R., Davidson S., Murden S., Morse C., Shearer M. J., Mumford A. D. (2008) J. Thromb. Haemost. 6, 1663–1670 [DOI] [PubMed] [Google Scholar]
- 36. Harrington D. J., Underwood S., Morse C., Shearer M. J., Tuddenham E. G., Mumford A. D. (2005) Thromb. Haemost. 93, 23–26 [DOI] [PubMed] [Google Scholar]
- 37. Loebstein R., Dvoskin I., Halkin H., Vecsler M., Lubetsky A., Rechavi G., Amariglio N., Cohen Y., Ken-Dror G., Almog S., Gak E. (2007) Blood 109, 2477–2480 [DOI] [PubMed] [Google Scholar]
- 38. Bodin L., Perdu J., Diry M., Horellou M. H., Loriot M. A. (2008) J. Thromb. Haemost. 6, 1436–1439 [DOI] [PubMed] [Google Scholar]
- 39. Wilms E. B., Touw D. J., Conemans J. M., Veldkamp R., Hermans M. (2008) J. Thromb. Haemost. 6, 1224–1226 [DOI] [PubMed] [Google Scholar]
- 40. Schmeits P. C., Hermans M. H., van Geest-Daalderop J. H., Poodt J., de Sauvage Nolting P. R., Conemans J. M. (2010) Br. J. Haematol. 148, 955–957 [DOI] [PubMed] [Google Scholar]
- 41. D'Ambrosio R. L., D'Andrea G., Cafolla A., Faillace F., Margaglione M. (2007) J. Thromb. Haemost. 5, 191–193 [DOI] [PubMed] [Google Scholar]
- 42. Li W., Schulman S., Dutton R. J., Boyd D., Beckwith J., Rapoport T. A. (2010) Nature 463, 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dutton R. J., Boyd D., Berkmen M., Beckwith J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stenina O., Pudota B. N., McNally B. A., Hommema E. L., Berkner K. L. (2001) Biochemistry 40, 10301–10309 [DOI] [PubMed] [Google Scholar]
- 45. Morris D. P., Stevens R. D., Wright D. J., Stafford D. W. (1995) J. Biol. Chem. 270, 30491–30498 [DOI] [PubMed] [Google Scholar]