

Abstract
In the rod cell of the retina, arrestin is responsible for blocking signaling of the G-protein-coupled receptor rhodopsin. The general visual signal transduction model implies that arrestin must be able to interact with a single light-activated, phosphorylated rhodopsin molecule (Rho*P), as would be generated at physiologically relevant low light levels. However, the elongated bi-lobed structure of arrestin suggests that it might be able to accommodate two rhodopsin molecules. In this study, we directly addressed the question of binding stoichiometry by quantifying arrestin binding to Rho*P in isolated rod outer segment membranes. We manipulated the “photoactivation density,” i.e. the percentage of active receptors in the membrane, with the use of a light flash or by partially regenerating membranes containing phosphorylated opsin with 11-cis-retinal. Curiously, we found that the apparent arrestin-Rho*P binding stoichiometry was linearly dependent on the photoactivation density, with one-to-one binding at low photoactivation density and one-to-two binding at high photoactivation density. We also observed that, irrespective of the photoactivation density, a single arrestin molecule was able to stabilize the active metarhodopsin II conformation of only a single Rho*P. We hypothesize that, although arrestin requires at least a single Rho*P to bind the membrane, a single arrestin can actually interact with a pair of receptors. The ability of arrestin to interact with heterogeneous receptor pairs composed of two different photo-intermediate states would be well suited to the rod cell, which functions at low light intensity but is routinely exposed to several orders of magnitude more light.
Keywords: Fluorescence, G-protein-coupled Receptors (GPCR), Protein-Protein Interactions, Signal Transduction, Vision, Arrestin, Rhodopsin, Stoichiometry
Introduction
The hundreds of different types of G-protein-coupled receptors (GPCRs)2 and their binding partners represent versatile protein families, whose individual members have been modified by nature to accomplish many different functions while preserving a basic structure and activation mechanism (1, 2). GPCRs share a common seven-transmembrane helical structure, which, when activated by ligand or stimuli, binds and activates G-proteins to initiate cell signaling (3). Most GPCRs also share a common mechanism of signal termination, which involves receptor phosphorylation and binding of the protein arrestin. However, the fates of arrestin-bound receptors vary widely depending on the type of receptor and its bound ligand. Arrestin-bound receptors may be internalized and degraded, internalized and recycled, or even initiate G-protein independent signaling (4).
Although detailed structural information on different GPCRs (5–9) and arrestins (10–13) has been available for some time, basic questions regarding their interaction remain unanswered. In particular, the binding stoichiometry (how many active receptors does a single arrestin bind?) is a matter of debate. Historically, one-to-one binding has always been assumed, because many GPCRs are present at low concentrations on the cell surface (1), or in the case of the photoreceptor rhodopsin, active receptors are separated by large distances or dispersed on different disc membranes in conditions of low light flux (14, 15). However, it has also been proposed that a single arrestin molecule could accommodate two receptors, because arrestin is composed of two near-symmetric lobes (see Fig. 1), which together span about 75 Å, although the cytoplasmic face of the receptor is only about 35–40 Å across (16, 17). This speculation has further implications for the ongoing debate regarding whether class A GPCRs, like rhodopsin, form functional dimers (1, 16, 18–20). On the other hand, the fact that arrestin has such a large binding surface area compared with rhodopsin (21) has contributed to the theory that arrestin must undergo a dramatic change in its conformation to bind (22).
FIGURE 1.

Locations of fluorophore attachment. Model of arrestin derived from the crystal structure (12). The N-domain is colored blue; the C-domain is colored green, and the C-terminal tail is colored orange. The residues, which were individually mutated to cysteine and then reacted with the IANBD fluorophore, are indicated. Note that Ala-366 is located in a long flexible loop (dashed) not visible in the crystal structure. The receptor-binding interface of arrestin is oriented toward the top in this figure.
Over the past 20 years, only a handful of studies have directly addressed the arrestin-receptor stoichiometry. Early experiments suggested a one-to-one binding stoichiometry when a small fraction of photoreceptors was activated, by indirectly measuring binding through the arrestin-dependent stabilization of the active metarhodopsin II (Meta II) over the inactive Meta I precursor (22, 23). More recently, transgenic mice were employed to determine how much arrestin translocated to the outer segment in vivo during an hour-long continuous exposure to bright light (24). The authors (24) concluded that every rhodopsin was bound by its own arrestin, although the ratio of translocated arrestin to receptor was only 0.65 in wild-type mice. Similarly, arrestin translocation in Drosophila rhabdomeres was found to be directly proportional to the amount of active photoreceptors, which could be consistent with a one-to-one binding stoichiometry (25). Finally, it has been recently shown by two separate groups that monomeric Rho*P in nanodiscs is sufficient to bind arrestin (26, 27).
Despite the seemingly overwhelming evidence supporting a one-to-one binding stoichiometry, we were surprised to discover that arrestin binding to Rho*P in isolated rod outer segment membranes plateaued at a ratio of one arrestin to two Rho*P when all receptors were photoactivated. After this initial discovery, and in contrast to the studies cited above, we sought to carefully quantify how the binding stoichiometry varied with the photoactivation density or the percentage of activated receptors on the membrane surface. We directly measured arrestin binding using multiple quantitative methods, many of which were time-resolved. These included kinetic light scattering, fluorescence spectroscopy, UV/Vis absorbance spectroscopy, and centrifugal pulldown analysis. We significantly improved the quantitative ability of this last assay by using fluorescently labeled single cysteine arrestin mutants.
As we describe below, we found that the arrestin-to-Rho*P binding ratio varied with the photoactivation density, which was one-to-one at low photoactivation density and one-to-two3 at high photoactivation density. Based on the observations we present here, we believe that arrestin can interact with two receptor molecules, and the interaction is asymmetric with respect to the activation states of the receptors. Given that all experimental evidence published to date shows that a single activated receptor is sufficient to bind arrestin, our study is the first experimental evidence suggesting that other binding modes are possible.
EXPERIMENTAL PROCEDURES
Materials
Bovine retina were obtained from W. L. Lawson in Lincoln, NE. Fatty acid-free bovine serum albumin (BSA) for membrane washes was purchased from Serva. Commercially available all-trans-retinal was used to create the 11-cis-retinal isomer, which was then purified in-house using HPLC (28). Chromatography supplies for arrestin purification were purchased from GE Healthcare, and the cysteine-reactive fluorophore N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine (IANBD) was obtained from Molecular Probes (Invitrogen). The high affinity analog peptide derived from the α-subunit of transducin (Gtα-HAA), VLEDLKSCGLF, was synthesized by Dr. Petra Henklein (Institut für Biochemie, Charité). All other reagents were from Sigma.
Preparation of Phosphorylated Rhodopsin
Rod outer segments (ROS) were isolated from bovine retina as described previously (29). Immediately following isolation, the rhodopsin in these membranes was phosphorylated using the endogenous rhodopsin kinase (30). ROS were diluted to ∼10 μm in ROS-buffer (70 mm potassium phosphate, pH 6.8, 1 mm magnesium acetate, 5 mm 2-mercaptoethanol, 0.5 mm PMSF), to which 3 mm ATP and 30 μm GTP were added, and the membranes were illuminated at room temperature using a standard desk lamp placed about 30 cm above the samples. After 2 h, 20 mm hydroxylamine was added to the quench phosphorylation and convert all rhodopsin photoproducts to phosphorylated opsin (opsP) and retinal oxime, and membranes were pelleted by centrifugation (60,000 × g, 30 min). The membranes were washed three times with ROS-buffer to remove the hydroxylamine and then resuspended in ROS-buffer to a final opsP concentration of ∼50 μm. A 3-fold molar excess of 11-cis-retinal was added, and the membrane suspension was incubated overnight at 4 °C in the dark on a rocking platform. After this incubation, 100 mm hydroxylamine was added, and the regenerated membranes were extensively washed, by resuspension and homogenization, with ROS-buffer containing 2% BSA. Typically, 12 wash steps with BSA, followed by three washes without BSA, were required to remove all contaminating retinal oxime. Following the washing steps, membranes were resuspended in 50 mm HEPES, pH 7, at an approximate concentration of 100 μm. Exact rhodopsin concentration was determined by the loss of 500 nm absorbance (ϵ = 40,800 m−1 cm−1) following illumination of membranes diluted in 100 mm hydroxylamine. To prepare partially regenerated ROS membranes, the fully regenerated membranes described above were diluted to ∼10 μm in ROS-buffer, pH 6, containing 10 mm hydroxylamine in a glass beaker with a stir bar. The sample was exposed to a top-mounted dim light source (>495 nm) for 10–60 s, while being stirred and held at 35 °C. In this way, the ROS membranes were evenly bleached in conditions favoring the formation of opsin. We did not partially regenerate ROS membranes by adding limited amounts of 11-cis-retinal due to the likelihood that membranes would be unevenly regenerated. Contaminating retinal oxime was then removed with BSA washes as described above. The percent of regenerated rhodopsin in the membrane was determined by the absorption spectrum of a detergent-solubilized aliquot (1:10 dilution in 1% dodecyl maltoside, followed by high speed centrifugation). Fully regenerated ROS membranes had a 280:500 absorbance ratio of 2.5, and partially regenerated membranes had correspondingly higher 280:500 ratios. We were confident that the regenerated rhodopsin molecules in our partially regenerated membranes were well dispersed and not clustered, because kinetic light scattering data using a flash were nearly identical to those employing partially regenerated membranes (see Fig. 4B).
FIGURE 4.

Apparent arrestin-Rho*P binding stoichiometry was different at high and low photoactivation densities. A, light scattering signals (green traces) were measured for an arrestin titration against 4 μm Rho*P. The samples were fully photoactivated at t = 0 s. The binding signal between 0 and 10 s was obscured by the photoactivating light and has been removed for clarity. From the bottom to top, LS signals correspond to 0.2, 0.5, 0.75, 1, 1.5, 2, 4, and 8 μm arrestin A366NBD. B, as in A, except that ROS-P membranes containing 4 μm RhoP were exposed to a flash, which photoactivated 23% of the RhoP (0.92 μm Rho*P, red traces). From the bottom to top, LS signals correspond to 0.2, 0.5, 0.75, 1, 2, and 6 μm arrestin A366NBD. Similar binding kinetics were observed for arrestin (0.5 μm) binding to 25% regenerated ROS-P membranes after full photoactivation (1 μm Rho*P, blue trace). Note that the y scales are different for A and B. C, maximum LS amplitudes were plotted as a function of arrestin concentration for 100% photoactivated membranes (green symbols), 23% photoactivated membranes (red symbols), and 25% regenerated, fully photoactivated membranes (blue symbols). Independent experiments are represented as differently shaped symbols, and the color scheme of the titration plots matches the raw data shown in A and B. The fitted curves report that the arrestin-Rho*P binding ratio was one-to-two at high photoactivation density and closer to one-to-one at ∼20% photoactivation density. hν, light; A366NBD, IANBD-labeled arrestin mutant A366C.
Preparation of Fluorescently Labeled Arrestin Mutants
Recombinant bovine mutant arrestin, from which the native cysteine and tryptophan residues had been removed (C63A, C128S, C143A, and W194F), was expressed and purified from Escherichia coli exactly as described previously (31). “Cys-less” arrestin has been previously shown to retain wild-type function (32, 33), and we verified that our mutant construct was functionally identical to native arrestin by centrifugal pulldown, light scattering, and extra Meta II analyses (data not shown). Single cysteine substitutions I72C, S344C, and A366C (see Fig. 1) were created using PCR and verified by DNA sequencing. Labeling of these introduced cysteine residues with IANBD was performed as described previously for bimane (31). The concentration and labeling efficiency were determined using molar extinction coefficients of 0.025 μm−1 cm−1 at 500 nm for IANBD (as reported by the manufacturer) and 0.02076 μm−1 cm−1 at 278 nm for arrestin (33). When bound to the protein, IANBD was found to contribute no absorbance at 278 nm, and all mutants were labeled at near 100% efficiency. Note on terminology: the labeled arrestin mutant I72C is referred to as I72NBD.
Illumination Protocol
For full photoactivation of ROS membranes, samples were illuminated for 10 s with a 150-fiber optic light source filtered through a heat filter (Schott KG2) and a 495 nm long pass filter. We verified by absorbance that this protocol photoactivated >90% of RhoP. For partial photoactivation of ROS membranes, a commercial photo-flash fitted with a green bandpass filter (500 ± 20 nm) was employed. The intensity of the flash was modulated with neutral density filters. The total flash duration was <2 ms, to avoid formation of photochemical side products. For the consecutive flashes shown in Fig. 6B (extra Meta II assay), a single sample containing ROS-P and arrestin was split into 3 aliquots. Each aliquot was exposed to a differently modulated flash intensity (5, 19, or 32% photoactivating). In this way, a full range of activation densities could be obtained while limiting the number of consecutive flashes to four. Flash intensity was quantified by absorbance spectroscopy using a sample of ROS-P diluted in 50 mm hydroxylamine.
FIGURE 6.

Each arrestin molecule stabilized one receptor molecule as Meta II. A, unlabeled arrestin was titrated against three different concentrations of RhoP (5, 10, or 20 μm), and the amount of stabilized Meta II following an activating flash (19%) was measured as shown in the inset. The concentrations of Rho*P in each titration are indicated. The fitted curves reported that 0.7 arrestins bound for every light-activated rhodopsin (Rho*P). Likewise, ∼70% of Rho*P was stabilized as Meta II. Inset, binding to and stabilization of Meta II over its precursor Meta I resulted in an increase in absorbance (380–417 nm). Meta II stabilization by arrestin was quantified by comparing absorption signals to those obtained with the peptide Gtα-HAA, which stabilized all Rho*P as Meta II. Samples contained 10 μm RhoP plus 300 μm Gtα-HAA or 10 μm arrestin and were activated by a flash at t = 0 s. B, fully or partially regenerated ROS membranes containing 10 μm RhoP were sequentially photoactivated. In the presence of Gtα-HAA (300 μm), every Rho*P was stabilized as Meta II (closed diamonds, 100% regenerated ROS-P; gray diamonds, 52% regenerated ROS-P; open diamonds, 23% regenerated ROS-P). In the presence of arrestin (20 μm), different proportions of Rho*P were stabilized as Meta II, depending on the photoactivation density (circles, 100% regenerated ROS-P; triangles, 52% regenerated ROS-P; squares, 23% regenerated ROS-P). An extrapolation of the data points to full photoactivation indicated a linear relationship between the amount of stabilized Meta II and the photoactivation density (inset).
Centrifugal Pulldown (PD) Binding Assay
Briefly, IANBD-labeled arrestin was mixed with ROS-P at the concentrations specified for the experiment (120-μl volume). Samples were fully photoactivated and then immediately transferred to the rotor (Beckman TLA-100). Centrifugation (100,000 × g, 10 min) began exactly 2 min after the onset of illumination. Following centrifugation, 90 μl of the supernatant was carefully removed from each sample, and the absorbance was measured using a Varian Cary 50 spectrometer (small window cuvette, 1 cm path length). The amount of arrestin “pulled down” with the membranes was simply the difference between the total concentration of arrestin in the sample and the concentration of arrestin remaining in the supernatant, which was determined by the IANBD absorbance at 500 nm. Because scattering is minimal at the red end of the spectrum, measurements accurately reported the amount of IANBD-labeled arrestin in the supernatant. This assay represents a large improvement in ease, speed, and accuracy as compared with earlier approaches, which used radiolabeled arrestin or quantitative Coomassie staining and/or Western blot following gel electrophoresis to quantify arrestin (24, 34, 35).
Kinetic Light Scattering (LS)
The intensity of scattered near-infrared light was measured from samples in a 1-cm path length cuvette on a self-made instrument as described previously (36, 37). In this study, arrestin was titrated against a fixed amount of ROS-P, and all binding signals were corrected by subtracting the reference signal obtained from a sample of ROS-P in the absence of arrestin. For some experiments, ROS-P samples were sonicated briefly in a bath sonicator (5–10 s) before use to improve signal quality (sonication of membranes had no effect on apparent arrestin-Rho*P binding stoichiometry as measured by LS, fluorescence, and extra Meta II assays). Arrestin binding was initiated with the illumination protocols described above. The maximum binding signal (ΔI/I) that was obtained after illumination (usually 60–120 s after the onset) was plotted as a function of arrestin concentration to yield the binding curve. Control experiments indicated that the arrestin mutant A366NBD was identical to native arrestin with regard to binding amplitude and kinetics (data not shown).
Fluorescence Spectroscopy (Fluor)
Fluorescence measurements were performed on a SPEX Fluorolog (1680) instrument. The fluorophore IANBD was excited at 360 nm to minimize receptor activation, and emission was collected between 500 and 660 nm (2-nm step size, 0.5-s integration per point). Excitation slits were minimized at 0.1 nm to avoid excessive irradiation of the sample, although emission slits were opened to 4 nm. Samples were measured in a quartz cuvette (80-μl sample volume, 0.3-mm path length) with temperature regulation. Samples were fully photoactivated as described above, and spectra were recorded about 2 min after light activation (to match PD results). Fluorescence intensity was determined by integrating the area below the background-subtracted fluorescence spectrum using the program Sigma Plot 10.0 (Systat Software, Inc.). Note that flash photolysis was not used in fluorescence experiments, because we found that the IANBD fluorophore was significantly quenched due to energy transfer to dark state rhodopsin when arrestin was bound to a partially photoactivated ROS-P membrane.
Extra Meta II Assay (UV/Vis)
The arrestin- or Gtα-HAA peptide-dependent stabilization of Meta II was measured using a two-wavelength spectrophotometer (Shimadzu UV3000) as described previously (22, 23). The absorbance at 380 nm (Meta II) was corrected for light-scattering artifacts inherent to membranous samples by subtracting the absorbance at 417 nm (the isosbestic point between Meta II and its precursor, Meta I). Samples were measured under conditions favoring Meta I (pH 8, 2 °C) in a quartz cuvette (2-mm path length). Absorbance was measured over time, and samples were photoactivated with a flash as described above. In the case of Gtα-HAA peptide, maximal amplitude was reached 60 s after flash, although arrestin-samples required longer times (5 min), especially at low arrestin concentrations. Absorbance values (380–417 nm) were converted to the actual concentration of Meta II with use of the Gtα-HAA peptide. This peptide stabilizes all photoactivated rhodopsin as Meta II, so it can be used to standardize absorbance changes in RhoP samples of known concentration.
Experimental Conditions
PD, LS, and Fluor experiments were carried out in 50 mm HEPES, pH 7, 130 mm NaCl at 20 °C. For the arrestin mutant I72NBD, NaCl concentration was 10 mm. UV/Vis experiments were carried out in 100 mm HEPES, pH 8, at 2 °C.
Mathematical Analysis of Titration Curves
Titration data were fit to an expression derived from the definition of the dissociation constant, KD. Briefly, KD = [A][R]/[AR], where [A] is the concentration of free arrestin, [R] is the concentration of free receptor, and [AR] is the concentration of the arrestin-receptor complex. The total arrestin and receptor concentrations are, respectively, [Atot] = [A] + [AR] and [Rtot] = [R] + [AR]. By solving for [A] and [R] in these equations and substituting into the KD expression, the concentration of [AR] at any given [Atot], [Rtot], and KD is given by the solution to the resulting quadratic Equation 1,
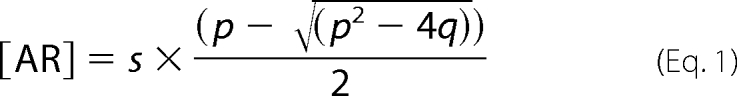 |
where p = KD + [Atot] + [Rtot] and q = [Atot] × [Rtot]. The factor “s” scales raw experimental data to concentration units. For the arrestin titrations presented in this study, [Atot] was the independent variable, and KD and [Rtot] were treated as dependent variables during the fitting procedure. The value determined for [Rtot] was indicative of how much arrestin bound and hence the stoichiometry. For the fitting of PD, Fluor, and UV/Vis experimental data, nonlinear regression algorithms were written within the graphing program Sigma Plot 10.0 (Systat Software, Inc.).
In the case of UV/Vis data, the fitting procedure yielded an apparent KD, which was influenced by the Meta I/Meta II equilibrium. The true KD value of arrestin binding to Meta II was derived from the apparent KD value as described previously (22), using a Meta II/Meta I ratio of 0.09 (38).
For LS, the data for full photoactivation and partial photoactivation (by flash and partial regeneration) were subjected to a global fit, where a single scalar factor s was determined for all three fits, using the program MicroMath Scientist (St. Louis, MO).
RESULTS
One Arrestin Bound for Every Two Rho*P at High Photoactivation Density
We first quantified arrestin binding to Rho*P using the centrifugal PD assay. The ROS-P membranes used were fully regenerated with 11-cis-retinal and then completely photoactivated. Hence, these experiments represent the upper limit of active receptor density in the native membrane.
Arrestin binding to 4 μm Rho*P was measured for a range of arrestin concentrations (Fig. 2A). The titration data saturated at a value of about 2 μm arrestin, i.e. a maximum of 2 μm arrestin could bind to membranes containing 4 μm Rho*P. Superimposed on the data are binding curves representing different binding stoichiometries. Clearly, the experimental data are most consistent with a binding ratio of one arrestin per two Rho*P. We also fit numerous experimental data sets to Equation 1 and obtained an apparent stoichiometry of one arrestin per ∼1.9 Rho*P (Table 1). We next performed the reverse titration; RhoP of increasing concentrations was mixed with 2 μm arrestin, and arrestin binding was measured in the dark and after full photoactivation (Fig. 2B). As for the arrestin titration, the experimental data are most consistent with a binding stoichiometry of one arrestin per two Rho*P. In addition, we could infer from the data that our arrestin preparations were fully functional, because all arrestin could be pulled down with excess rhodopsin.
FIGURE 2.

Apparent arrestin-Rho*P binding stoichiometry was one-to-two at high photoactivation density. A, arrestin was titrated against 4 μm Rho*P. B, Rho*P was titrated against 2 μm arrestin. For each titration, binding of arrestin was measured by the centrifugal pulldown assay in the dark (open symbols) and after full photoactivation (closed symbols) using fluorescently labeled arrestin mutants (A366NBD, red; S344NBD, blue; I72NBD, green). Data points from independent experiments are indicated by differently shaped symbols. Note that the high amount of dark binding for arrestin I72NBD was due to the special experimental conditions used for this mutant (see text for details). The dashed traces represent binding curves corresponding to stoichiometries of one arrestin to one Rho*P (one-to-one) or one arrestin to two Rho*P (one-to-two). For the arrestin titration in A, we have included binding curves corresponding to the published KD of 20 nm (23) and our experimentally determined value of 365 nm (Table 1). For the Rho*P titration in B, we have included curves calculated using a KD of 365 nm. Both the arrestin and Rho*P titrations are consistent with a one-to-two stoichiometry.
TABLE 1.
Summary of binding parameters derived from arrestin titrations
| Method | Photoactivation density |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Full (100% activation)a |
Flash (∼20% activation)a |
Partial regeneration (∼20% activation)a |
|||||||
| KD | Apparent stoichiometry (arrestin/Rho*P) | nb | KD | Apparent stoichiometry (arrestin/Rho*P) | n | KD | Apparent stoichiometry (arrestin/Rho*P) | n | |
| nm | nm | nm | |||||||
| PDc | 365 ± 55 | 0.53 ± 0.02 | 10 | NDd | ND | ND | ND | ||
| LSe | 83 ± 28 | 0.46 ± 0.02 | 4 | 71 ± 23 | 0.85 ± 0.04 | 4 | 92 ± 32 | 0.68 ± 0.04 | 2 |
| Fluorf | 310 ± 37 | 0.51 ± 0.02 | 8 | ND | ND | 181 ± 24 | 0.73 ± 0.02 | 5 | |
| UV/Visg | ND | ND | 10.9 ± 1.8 | 0.73 ± 0.01 | 3 | ND | ND | ||
a Binding was measured at different photoactivation densities, as described in Fig. 3.
b Number of independent experiments is given. For PD, Fluor, and UV/Vis data, data from independent experiments were fit individually to Equation 1, and KD and stoichiometry values are reported as the mean ± S.E. In the case of LS data, data points from independent experiments were combined for a global fit, in which the same scalar factor s was used for each curve, and the reported error is the standard deviation of the fit.
c Centrifugal pulldown analysis. Experiments employed three different labeled arrestin mutants as follows: A366NBD (n = 5), S344NBD (n = 3), and I72NBD (n = 2). Experiments were performed as described in Fig. 2A and under “Experimental Procedures.”
d ND means not determined.
e Kinetic light scattering is shown. Experiments employed arrestin mutant A366NBD, as described in Fig. 4 and under “Experimental Procedures.”
f Fluorescence spectroscopy is shown. Experiments employed arrestin mutants I72NBD and S344NBD and were carried out as described in Fig. 5 and under “Experimental Procedures.”
g Extra Meta II assay is shown. Experiments employed expressed unlabeled arrestin lacking native cysteine and tryptophan residues (see “Experimental Procedures” for details) and three different concentrations of ROS-P, as described in Fig. 6A.
Note that similar titration results with Rho*P were obtained for all IANBD-labeled arrestin mutants used in this study (see Fig. 1). With regard to binding kinetics and ability to stabilize Meta II, the “backside”-labeled arrestin mutant A366NBD functioned essentially the same as expressed wild-type or native arrestin (data not shown). For arrestin mutants A366NBD and S344NBD, a low amount of binding to dark ROS-P in isotonic buffer occurred at higher arrestin concentrations (Fig. 2A), as has been reported previously (39). Because the binding of arrestin mutant I72NBD to Rho*P was found to be inhibited by salt, we limited the concentration of NaCl to 10 mm in experiments involving arrestin I72NBD. Consequently, a significant amount of arrestin I72NBD binding to dark ROS-P was observed (Fig. 2, A and B).
Nearly One Arrestin Bound for Every Rho*P at Low Photoactivation Density
To understand how arrestin binds Rho*P under conditions of low photoactivation density, we manipulated the percent of active receptor in our ROS-P samples using two different approaches (Fig. 3). In the first approach, ROS-P membranes that had been fully regenerated with 11-cis-retinal were exposed to a light flash of known and reproducible intensity, which activated only a certain percentage of the receptors. In the second approach, ROS-P membranes were only partially regenerated with 11-cis-retinal to a known level. The RhoP in these membranes was then fully photoactivated. Both approaches produced the same photoactivation density. The first approach produced a mixture of dark RhoP and Rho*P, and the second approach produced a mixture of opsP and Rho*P.
FIGURE 3.

Schematic showing how different photoactivation densities were achieved using either partial regeneration or light flash. Starting from left- hand side, isolated and washed ROS membranes containing phosphorylated opsin (white circles) can either be fully (top) or partially (bottom) regenerated with 11-cis-retinal to reform dark state RhoP (black circles). 1) Full photoactivation of fully regenerated ROS membranes leads to high active receptor density. Activated receptors (Rho*P) are represented by an inscribed star. 2) Exposing full-regenerated membranes to a calibrated light flash leads to low active receptor density. 3) Same photoactivation density as described in 2 can be achieved by full photoactivation of partially regenerated membranes.
Arrestin binding was measured by kinetic light scattering, which measures the change in scattered light that occurs when a soluble protein like arrestin binds the ROS-P membrane (37). LS has the advantage of being time-resolved, so that the maximum amount of arrestin bound after photoactivation could be accurately determined (Fig. 4, A and B). Arrestin A366NBD was titrated against ROS-P membranes containing different densities of active receptor (Fig. 4C), and the fitting parameters, including KD and the apparent stoichiometry, are reported in Table 1. For membranes containing the highest density of photoactive receptor, the fitted curve indicated a stoichiometry of 1 arrestin per ∼2 Rho*P. This finding agrees perfectly with the stoichiometry determined by PD (Fig. 2A). For fully regenerated membranes where only ∼23% of RhoP was activated by a flash, the fitted curve indicated a stoichiometry of 1 arrestin per ∼1.2 Rho*P. Interestingly, similar results (1 arrestin per ∼1.4 Rho*P) were obtained when the same photoactivation density was created by partial regeneration. Note that, because of the significant concentration of opsP in partially regenerated membranes, some arrestin was bound to the membrane before photoactivation (i.e. dark binding). Because LS only measures the change in membrane-bound mass, the stoichiometry may have been underestimated as compared with flash data.
Overall, the LS results suggest that at lower densities of photoactive receptor, the apparent arrestin-Rho*P stoichiometry approached one-to-one. Moreover, neither the stoichiometry nor the binding kinetics (Fig. 4B) appeared to be dependent on whether dark RhoP or opsP were interspersed between the photoactivated receptors.
Apparent Stoichiometry Was Linearly Dependent on the Photoactivation Density
We further explored the effect of varying the photoactivation density using fluorescently labeled arrestin mutants (Fig. 5). The solvent-sensitive IANBD fluorophore, when attached to a cysteine residue at site 72 or 344, reports arrestin binding to Rho*P as a large increase in fluorescence (Fig. 5A, inset). This effect is similar to that previously reported for bimane-labeled arrestin (40). Arrestin-Rho*P binding stoichiometry was determined by titrating IANBD-labeled arrestin against a fixed amount of receptor and measuring the fluorescence intensity before and after photoactivation (Fig. 5A). In these experiments, the total amount of Rho*P present in each titration was 4 μm. However, the photoactivation density was varied by using ROS-P membranes, which had been regenerated to different levels (100, 52, and 23%). The fitted curves indicated that binding saturated at a ratio of one arrestin to two Rho*P at high photoactivation density, although one arrestin bound per 1.4 Rho*P at the lowest photoactivation density (Table 1). Strikingly, there was an apparent linear relationship between the binding stoichiometry, as determined from the fitted curves, and the photoactivation density (Fig. 5B). Likewise, there was a linear relationship between the final amplitudes of the curves, which were directly proportional to the total amount of arrestin binding, and the photoactivation density (Fig. 5C). This linear relationship was seen using both mutants I72NBD and S344NBD (Fig. 5, B and C) and also native arrestin as measured by LS using consecutive flashes to generate a range of arrestin/Rho*P ratios (data not shown).
FIGURE 5.

Apparent arrestin-Rho*P binding stoichiometry was linearly related to the photoactivation density. A, arrestin I72NBD was titrated against 4 μm Rho*P at different photoactivation densities, which were achieved by using ROS-P membranes that were regenerated to different levels (100%, circles; 52%, triangles; and 23%, squares). Note that for each binding curve, the amount of activated receptor was the same (4 μm), although the amount of total opsP varied depending on the regeneration level. Inset, fluorescence was measured in the dark (solid trace) and after full photoactivation (dashed trace). The Δ Fluorescence represents the difference between the buffer-subtracted integrated fluorescence intensities of the dark and +light spectra. Fluorescence data were fit to binding curves (see Table 1). B, relationship between the photoactivation density and the stoichiometry, which was determined from the fitted binding curves. Data points represent the average of two independent experiments. Note that both fluorescently labeled arrestin mutants I72NBD (closed symbols) and S344NBD (open symbols) report nearly the same stoichiometry at each photoactivation density and the same linear dependence of stoichiometry on photoactivation density. C, relationship between the photoactivation density and the maximum fluorescence amplitude, which is directly related to the total amount of arrestin binding. Both I72NBD and S344NBD report the same linear dependence.
Interestingly, an extrapolation of the line relating stoichiometry to photoactivation density intercepts the y axis at a ratio of arrestin to Rho*P of 0.8 (or one arrestin per 1.25 Rho*P) (Fig. 5B). In other words, even at the lowest theoretical photoactivation density, only one arrestin would bind per every 1.25 Rho*P.
Each Arrestin Molecule Stabilized One Receptor as Meta II
Following light activation, Rho*P exists in a pH- and temperature-dependent equilibrium between the photoproducts Meta I and Meta II, which differ in the protonation state of the retinal Schiff-base and protein conformation (3, 38). In the presence of a binding partner that binds Meta II, like transducin or arrestin, the Meta I/Meta II equilibrium is shifted in the direction of Meta II at the expense of Meta I (22, 41). We investigated how the amount of this stabilized Meta II, referred to as extra Meta II, depended on the arrestin concentration and on the photoactivation density (Fig. 6).
First, we performed arrestin titrations using different concentrations of RhoP (5, 10, or 20 μm), where only 19% of receptors were activated by flash (Fig. 6A). The absorbance change after flash was measured at each arrestin concentration, and the Gtα-HAA peptide was used to convert absorbance units to the concentration of Meta II (see “Experimental Procedures”). The data were fit to Equation 1 to yield KD values and apparent binding stoichiometry (Table 1). Intriguingly, every curve reported a binding stoichiometry of one arrestin per ∼1.4 Rho*P, i.e. for every molecule of Rho*P created by the flash, 0.7 arrestins bound and stabilized 0.7 activated receptors as Meta II.
Second, we quantified how much extra Meta II was stabilized under conditions of excess arrestin and varying photoactivation density (Fig. 6B). Fully and partially regenerated ROS-P membranes, which contained a total of 10 μm regenerated receptor (RhoP), were flashed multiple times. In the presence of the peptide Gtα-HAA, all photoactivated Rho*P was stabilized as Meta II. When the same protocol was carried out with an excess of arrestin, a very different effect was observed. For each fully or partially regenerated ROS-P sample, arrestin stabilized 70–80% of the photoactivated Rho*P as Meta II when a low percentage (<20%) of receptors was activated by flash. As more and more of the receptors were activated, the amount of stabilized Meta II depended on the fraction of regenerated RhoP in the membranes. We estimated the amount of stabilized Meta II at full photoactivation by extrapolating the data points. For fully regenerated ROS-P membranes, half (49%) of the light-activated Rho*P would be stabilized as Meta II. For ROS-P membranes where only half of the receptors were regenerated, 65% of light-activated Rho*P would be stabilized as Meta II. For ROS-P membranes where only a quarter of receptors were regenerated, 70% of light-activated Rho*P would be stabilized as Meta II. Strikingly, these extrapolated values, which represent the percentage of Rho*P stabilized as Meta II by an excess of arrestin if all receptors in the membrane were photoactivated, were linearly related to the photoactivation density (Fig. 6B, inset). The line relating extra Meta II and the photoactivation density intercepts the y axis at 8 μm, suggesting that 80% of light-activated Rho*P would be stabilized as Meta II at the lowest theoretical photoactivation density. This result exactly mirrors that seen when arrestin binding was measured using the fluorescence changes of probes located on arrestin mutants I72NBD and S344NBD (Fig. 5B).
Different Methods Reported Different Binding Affinities
Our PD and Fluor methods reported KD values 10–20 times higher than the 20–50 nm that has been reported for arrestin binding to Rho*P in native ROS membranes (22, 23). The cause of this discrepancy is likely temperature. Whereas the studies cited above employed low temperature (2–10 °C), our experiments were performed at 20 °C. Meta II decay at 20 °C is fast enough such that a significant portion of Rho*P decays before arrestin binding is complete, especially when the ratio of arrestin to Rho*P is stoichiometric.4 Furthermore, the data points near the stoichiometric ratio most heavily influence the overall shape and hence the KD of the fitted curve. This effect can be clearly seen in how the arrestin titration data points deviate from the 20 nm binding curve in Fig. 2A. These effects were most pronounced in our PD and Fluor experiments, because binding was measured 2–3 min after photoactivation. LS yielded lower KD values than PD and Fluor, because the time resolution of LS afforded accurate quantification of arrestin binding. The influence of Meta II decay on arrestin binding at different concentrations is apparent in the LS signals presented in Fig. 4, A and B.
Incidentally, the reason the extra Meta II experiments reported here by us and by others (23) yielded KD values in the range of 10–20 nm is related to the effect described above. At 2 °C, the rate of Meta II decay (hours) (42) is vastly slower than the rate of arrestin binding at micromolar concentrations (minutes) (Fig. 6A, inset). Hence, Meta II decay does not influence the quantification of arrestin binding, and consequently, binding curves with low KD values are obtained (Fig. 6A).
Binding Stoichiometry Was Underestimated
It is likely our experimental procedures led to an underestimation of arrestin binding when partially regenerated membranes were used (Figs. 4C, 5, and 6B). The 10-s illumination generated ∼10% isorhodopsin (data not shown), which results when Meta I absorbs activating light (43). In addition, some portion of our ROS-P preparations may have been non- or under-phosphorylated.5 Our phosphorylation procedure, based on one previously established, yielded an average of ∼6 phosphates per rhodopsin (40). Although we did not directly analyze the different phosphorylated species present in our ROS-P samples, a previous report (44) suggests that up to 10% of the rhodopsin in our samples may have had less than the required two phosphates necessary for arrestin activation and binding (45). The formation of isorhodopsin and/or under-phosphorylation of ROS-P samples might explain why a portion of the Rho-P in our samples appeared unable to bind arrestin at low photoactivation density. Two independent methods (Fluor and UV/Vis) reported that at the lower limit of the photoactivation density, only 0.8 arrestins would bind for every active receptor (Figs. 5B and 6B, inset). Interestingly, LS experiments employing a photoactivating flash consistently reported a stoichiometry closer to one-to-one (Table 1), probably because the flash generated minimal isorhodopsin.
DISCUSSION
As far as we are aware, our study represents the first attempt to quantify how the arrestin-Rho*P binding stoichiometry depends on the photoactivation density or the percentage of activated receptors. Our in vitro approach using isolated native membranes was well suited to such an investigation, because the naturally high receptor density of the rod outer segment disc membrane was preserved. We were able to accurately manipulate the photoactivation density with the use of light flashes or partial regeneration. In a living system, the exact percentage of photoactivated receptors varies over time and cannot be determined with the necessary accuracy (14, 24, 46).
We quantified arrestin binding to Rho*P using a variety of biochemical and biophysical methods. In summary, all methods reported the following. 1) When a low percentage of receptors was activated, about one arrestin bound for every active receptor. 2) As more and more receptors were activated, the binding ratio decreased until, at full photoactivation density, one arrestin bound for every two active receptors. 3) The amount of arrestin that could bind was linearly related to the photoactivation density. 4) At every photoactivation density, the concentration of stabilized Meta II was equal to the concentration of bound arrestin.
The key finding of this study is that the apparent binding stoichiometry shifts with the relative photoactivation density in the native rod disc membrane. The one-to-one binding seen at low photoactivation density agrees with the historically accepted paradigm, in which one arrestin is required to “shut off” one activated receptor (1, 22). Indeed, it has recently been shown that a single receptor in a nanodisc is sufficient to bind arrestin (26, 27).
In contrast, the one-to-two binding seen when the entire rod disc membrane was photoactivated is harder to reconcile with the accepted paradigm. First of all, we believe it is unlikely that the one-to-two binding stoichiometry was due to only half the rhodopsin molecules being capable of binding arrestin. Rho*P binding was not obscured by inside-out vesicle arrangement or membrane aggregation, because sonicated ROS-P samples yielded identical results as unsonicated samples (see “Experimental Procedures”). Moreover, if half the rhodopsin in our samples was nonfunctional, then we would have observed a one-to-two apparent stoichiometry at low photoactivation density as well. Because the stoichiometry shifted with respect to how many receptors were activated, there must be another underlying cause for the one-to-two binding seen when the entire membrane was photoactivated.
Two possibilities arise as follows. The high density of rhodopsin molecules in the rod disc precludes one-to-one binding because of molecular crowding. Alternatively, it is possible that arrestin interacts with two receptors. These two possibilities are explored below.
Can Arrestin Interact with Two Receptors?
Two models of arrestin binding have been proposed in recent years. In the first, a single arrestin binds a single activated receptor, and interaction with the receptor induces the two lobes of arrestin to close like a clam shell to bring distant receptor-binding elements together (1, 21). In the second model, a single arrestin molecule remains more or less in the elongated form seen in the crystal structure (12), and the two lobes of arrestin each engage one receptor (16, 17, 47).
If the first model is correct, then the one-to-two binding stoichiometry we observe at high photoactivation density would result from crowding. The binding of an arrestin molecule to a single Rho*P would be envisioned to block or shield neighboring receptors from arrestin binding. Rhodopsin molecules are tightly packed on the membrane surface at about 25,000 molecules per μm2, resulting in a membrane that is 50% protein (14, 15). The cytoplasmic surface of the activated receptor has an area of ∼10 nm2 (flat projection) (6), although a cross-section of the receptor-binding surface in the crystallized conformation of arrestin is about 25 nm2 (12). Assuming that half of the membrane surface is receptor and half is lipid, there would be just about enough space to accommodate one arrestin for every receptor, even more so if arrestin folds like a clam shell when binding the activated receptor (21). Still, reducing the rhodopsin content in mouse rod outer segments (by knocking out one rhodopsin allele) has been observed to increase the ratio of translocated arrestin to rhodopsin to 0.83, compared with 0.65 in wild-type mice (24), which might reflect the influence of crowding on arrestin binding in the native ROS membrane. Our results are superficially similar, in that we observed that arrestin binding increased with the space between the activated receptors.
However, our quantitative data indicated a linear correlation between apparent stoichiometry and photoactivation density, as opposed to a sharp transition between one-to-one binding and one-to-two binding at the point where space becomes limiting on the membrane. For example, if arrestin binding at high photoactivation density was simply limited by crowding, then a 2-fold increase in the space between activated receptors should have allowed one-to-one binding. However, we observed an intermediate binding stoichiometry (1 arrestin to 1.5 Rho*P) with our 50% regenerated membranes (Fig. 5B). The crowding hypothesis cannot explain this result.
Thus our results support the second binding model in which the bi-lobed arrestin molecule can simultaneously interact with two receptor molecules. If every membrane-bound arrestin molecule can stabilize only one Rho*P as Meta II, how can we then infer that arrestin actually engages two receptors? We believe the answer lies in the way the apparent binding stoichiometry gradually approached one-to-two as the photoactivation density increased. This trend implies that the apparent stoichiometry at intermediate photoactivation densities was a composite of one-to-one and one-to-two binding ratios. For the subpopulation of arrestin molecules involved in one-to-two binding, a relatively long lived interaction with both activated receptors can be inferred, because the “second” receptor molecule was not free to diffuse away and bind another arrestin. Otherwise, a one-to-one stoichiometry would have been observed with excess arrestin at intermediate photoactivation densities.
Assuming arrestin functionally engages two Rho*P molecules, arrestin appears asymmetric in its ability to stabilize Meta II. We hypothesize that this asymmetry applies not only to the photoactivated receptor states (Rho*P) but also to the inactive states (RhoP and opsP), i.e. at low photoactivation density, arrestin generally interacts with mixed pairs of receptors, Rho*P paired with dark RhoP or opsP. We propose a binding model, in which one part of arrestin interacts specifically with the active Meta II state of the receptor, and the other part of arrestin is less stringent and serves to trap a variety of active or inactive states. Moreover, because arrestin is fully capable of binding monomeric Rho*P in nanodiscs (26, 27), arrestin would not even require a second receptor to bind a Rho*P.
The interaction of arrestin with two receptors is probably asymmetric with regard to binding affinity as well. The interaction that stabilizes Meta II has sufficient energy to shift the Meta I/Meta II equilibrium and hence represents the high affinity interaction. The less specific interaction with the other receptor is of lower affinity yet does not translate to a higher off-rate, because the two halves of the arrestin molecule are bound together. In this way, the Meta II-stabilizing interaction provides the binding energy for the less specific interaction that traps the neighboring receptor.
Interestingly, arrestin has been computationally docked onto a mixed receptor pair composed of one active and one inactive rhodopsin (17). When a single arrestin was docked onto a rhodopsin dimer, the authors noted that the two lobes of arrestin were unequal in their interaction with regard to binding energy and requirement for an activated receptor.
The ability of arrestin to engage mixed receptor pairs probably gives rise to the linear relationship between apparent stoichiometry and photoactivation density. At low photoactivation density, arrestin interacts primarily with mixed pairs of Rho*P-RhoP or Rho*P-opsP, leading to an apparent binding stoichiometry of one-to-one. As more and more receptors are activated, the probability of photoactivating adjacent RhoP molecules increases; arrestin binds more Rho*P-Rho*P pairs, and the apparent stoichiometry consequently approaches one-to-two.
Finally, our results may help reconcile the two different theories regarding class A GPCR dimerization. According to our interpretation, receptors could well exist as freely diffusing monomers before light activation (48–50), and arrestin binding could stabilize a Rho*P dimer or dimer containing one active and one inactive receptor. Arrestin binding would then provide the energy required for receptor dimerization, as it has been argued that the interaction of two rhodopsin molecules cannot provide enough binding energy to make a dimer that is long lived enough to be considered functional (51). Notably, we observed no difference in binding kinetics or stoichiometry at low photoactivation density when opsP was interspersed between active receptors instead of dark RhoP (Fig. 4B). This result shows that regenerated rhodopsin molecules were not clustered within the larger pool of opsP, and large preformed arrays of opsP dimers (16) probably did not exist.
Physiological Implications
An arrestin-Rho*P binding stoichiometry that shifts with the photoactivation density might be highly useful for the rod cell, which is routinely exposed to a wide range of light intensities (14). Regardless of whether arrestin can functionally bind only one or two receptors, the fact remains that arrestin binding saturates at an overall binding stoichiometry of one arrestin to two receptors. Thus, a single arrestin is expected to functionally block two receptors from interacting with G-protein, even if the two receptors exist at different photoactivation or decay states. Furthermore, because the amount of rhodopsin in the rod cell exceeds the amount of available arrestin (24), this binding paradigm makes sense biologically.
Although the rod cell is capable of sensing a single photon (52), rod signaling after dark adaptation saturates at light levels corresponding to ∼0.003% photoactivation (53). It is generally assumed that at higher levels of photoactivation, rods are turned off as sensors and exist essentially in survival mode. However, evidence has been presented that rods may affect color perception and hence can function as sensors in bright light (54). In any case, the photoactivation conditions used in our study are physiologically relevant to our daily lives in sunshine or bright artificial lighting. Under these conditions, simultaneous light absorption and rhodopsin regeneration lead to a situation where rhodopsin, light-activated metarhodopsin, and its decay products are in steady state (14, 55). Thus, it is likely that the actual arrestin-receptor stoichiometry usually lies between the one-to-one and one-to-two values we observe here. Indeed, such stoichiometry values were observed in living mice exposed to bright continuous light (24). Under these conditions, the bleached pigment consists of various metarhodopsin species as well as the apoprotein opsin. If arrestin is able to simultaneously engage two different functional forms of the receptor, as we suggest above, such an attribute would be advantageous in a rod disc membrane, because it would increase the effective blocking power of arrestin. Arrestin binding would not only block a single activated receptor but also dark rhodopsin, opsin, and the various metarhodopsin species in-between.
Supplementary Material
Acknowledgments
We thank Helena Seibel, Anja Koch, and Brian Bauer for assistance in creating and expressing arrestin mutants.
This work was supported by International Research Fellowship 0700410 from the National Science Foundation and Grants SFB 740, HE 2704/1-1, and SO 1037/1-1 from the Deutsche Forschungsgemeinschaft.
This article was selected as a Paper of the Week.
Our descriptions of stoichiometry follow the template arrestin-to-Rho*P, i.e. “one-to-two” binding refers to a binding ratio of one arrestin to two Rho*P.
Meta II decay is not so influential at the extremes of the titration curve. When Rho*P is in excess, any arrestin released from decayed Rho*P can quickly bind another Rho*P, and when arrestin is in excess, binding is fast enough to limit the influence of Meta II decay.
V. Gurevich, personal communication.
- GPCR
- G-protein-coupled receptor
- Fluor
- fluorescence spectroscopy
- Gtα-HAA
- high affinity peptide analog derived from the C terminus of the α-subunit of transducin
- I72NBD
- IANBD-labeled arrestin mutant I72C
- LS
- kinetic light scattering
- Meta II
- metarhodopsin II
- opsP
- phosphorylated opsin
- PD
- centrifugal pull down
- RhoP
- phosphorylated rhodopsin
- Rho*P
- light-activated phosphorylated rhodopsin
- ROS
- rod outer segment membranes
- ROS-P
- ROS containing phosphorylated rhodopsin
- UV/Vis
- absorbance spectroscopy or the extra Meta II assay
- IANBD
- N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine.
REFERENCES
- 1. Gurevich V. V., Gurevich E. V. (2008) Trends Neurosci. 31, 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kobilka B. K. (2007) Biochim. Biophys. Acta 1768, 794–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hofmann K. P., Scheerer P., Hildebrand P. W., Choe H. W., Park J. H., Heck M., Ernst O. P. (2009) Trends Biochem. Sci. 34, 540–552 [DOI] [PubMed] [Google Scholar]
- 4. Gurevich E. V., Gurevich V. V. (2006) Genome Biol. 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 6. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., Ernst O. P. (2008) Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 7. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K. (2007) Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 8. Schertler G. F. (2005) Curr. Opin Struct. Biol. 15, 408–415 [DOI] [PubMed] [Google Scholar]
- 9. Jaakola V. P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., Stevens R. C. (2008) Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granzin J., Wilden U., Choe H. W., Labahn J., Krafft B., Büldt G. (1998) Nature 391, 918–921 [DOI] [PubMed] [Google Scholar]
- 11. Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., Schubert C. (2001) Structure 9, 869–880 [DOI] [PubMed] [Google Scholar]
- 12. Hirsch J. A., Schubert C., Gurevich V. V., Sigler P. B. (1999) Cell 97, 257–269 [DOI] [PubMed] [Google Scholar]
- 13. Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., Gurevich V. V. (2005) J. Mol. Biol. 354, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 14. Lamb T. D., Pugh E. N., Jr. (2004) Prog. Retin. Eye Res. 23, 307–380 [DOI] [PubMed] [Google Scholar]
- 15. Molday R. S. (1998) Invest. Ophthalmol. Vis. Sci. 39, 2491–2513 [PubMed] [Google Scholar]
- 16. Liang Y., Fotiadis D., Filipek S., Saperstein D. A., Palczewski K., Engel A. (2003) J. Biol. Chem. 278, 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Modzelewska A., Filipek S., Palczewski K., Park P. S. (2006) Cell Biochem. Biophys. 46, 1–15 [DOI] [PubMed] [Google Scholar]
- 18. Chabre M., le Maire M. (2005) Biochemistry 44, 9395–9403 [DOI] [PubMed] [Google Scholar]
- 19. Park P. S., Filipek S., Wells J. W., Palczewski K. (2004) Biochemistry 43, 15643–15656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Milligan G. (2007) Biochim. Biophys. Acta 1768, 825–835 [DOI] [PubMed] [Google Scholar]
- 21. Gurevich V. V., Gurevich E. V. (2004) Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 22. Schleicher A., Kühn H., Hofmann K. P. (1989) Biochemistry 28, 1770–1775 [DOI] [PubMed] [Google Scholar]
- 23. Pulvermüller A., Maretzki D., Rudnicka-Nawrot M., Smith W. C., Palczewski K., Hofmann K. P. (1997) Biochemistry 36, 9253–9260 [DOI] [PubMed] [Google Scholar]
- 24. Hanson S. M., Gurevich E. V., Vishnivetskiy S. A., Ahmed M. R., Song X., Gurevich V. V. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3125–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Satoh A. K., Xia H., Yan L., Liu C. H., Hardie R. C., Ready D. F. (2010) Neuron 67, 997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsukamoto H., Sinha A., DeWitt M., Farrens D. L. (2010) J. Mol. Biol. 399, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bayburt T. H., Vishnivetskiy S. A., McLean M. A., Morizumi T., Huang C. C., Tesmer J. J., Ernst O. P., Sligar S. G., Gurevich V. V. (2011) J. Biol. Chem. 286, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garwin G. G., Saari J. C. (2000) Methods Enzymol. 316, 313–324 [DOI] [PubMed] [Google Scholar]
- 29. Papermaster D. S. (1982) Methods Enzymol. 81, 48–52 [DOI] [PubMed] [Google Scholar]
- 30. Kühn H., Wilden U. (1982) Methods Enzymol. 81, 489–496 [DOI] [PubMed] [Google Scholar]
- 31. Sommer M. E., Smith W. C., Farrens D. L. (2006) J. Biol. Chem. 281, 9407–9417 [DOI] [PubMed] [Google Scholar]
- 32. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., Gurevich V. V. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sommer M. E., Farrens D. L., McDowell J. H., Weber L. A., Smith W. C. (2007) J. Biol. Chem. 282, 25560–25568 [DOI] [PubMed] [Google Scholar]
- 34. Gurevich V. V., Benovic J. L. (2000) Methods Enzymol. 315, 422–437 [DOI] [PubMed] [Google Scholar]
- 35. Hofmann K. P., Pulvermüller A., Buczyłko J., Van Hooser P., Palczewski K. (1992) J. Biol. Chem. 267, 15701–15706 [PubMed] [Google Scholar]
- 36. Heck M., Hofmann K. P. (2001) J. Biol. Chem. 276, 10000–10009 [DOI] [PubMed] [Google Scholar]
- 37. Heck M., Pulvermüller A., Hofmann K. P. (2000) Methods Enzymol. 315, 329–347 [DOI] [PubMed] [Google Scholar]
- 38. Parkes J. H., Liebman P. A. (1984) Biochemistry 23, 5054–5061 [DOI] [PubMed] [Google Scholar]
- 39. Gurevich V. V., Benovic J. L. (1993) J. Biol. Chem. 268, 11628–11638 [PubMed] [Google Scholar]
- 40. Sommer M. E., Smith W. C., Farrens D. L. (2005) J. Biol. Chem. 280, 6861–6871 [DOI] [PubMed] [Google Scholar]
- 41. Emeis D., Kühn H., Reichert J., Hofmann K. P. (1982) FEBS Lett. 143, 29–34 [DOI] [PubMed] [Google Scholar]
- 42. Janz J. M., Fay J. F., Farrens D. L. (2003) J. Biol. Chem. 278, 16982–16991 [DOI] [PubMed] [Google Scholar]
- 43. Lewis J. W., van Kuijk F. J., Carruthers J. A., Kliger D. S. (1997) Vision Res. 37, 1–8 [DOI] [PubMed] [Google Scholar]
- 44. Wilden U., Kühn H. (1982) Biochemistry 21, 3014–3022 [DOI] [PubMed] [Google Scholar]
- 45. Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., Gurevich V. V. (2007) J. Biol. Chem. 282, 32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nair K. S., Hanson S. M., Mendez A., Gurevich E. V., Kennedy M. J., Shestopalov V. I., Vishnivetskiy S. A., Chen J., Hurley J. B., Gurevich V. V., Slepak V. Z. (2005) Neuron 46, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skegro D., Pulvermüller A., Krafft B., Granzin J., Hofmann K. P., Büldt G., Schlesinger R. (2007) Photochem. Photobiol. 83, 385–392 [DOI] [PubMed] [Google Scholar]
- 48. Edrington T. C., 5th, Bennett M., Albert A. D. (2008) Biophys. J. 95, 2859–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cone R. A. (1972) Nat. New Biol. 236, 39–43 [DOI] [PubMed] [Google Scholar]
- 50. Liebman P. A., Entine G. (1974) Science 185, 457–459 [DOI] [PubMed] [Google Scholar]
- 51. Gurevich V. V., Gurevich E. V. (2008) Trends Pharmacol. Sci. 29, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burns M. E., Arshavsky V. Y. (2005) Neuron 48, 387–401 [DOI] [PubMed] [Google Scholar]
- 53. Naarendorp F., Esdaille T. M., Banden S. M., Andrews-Labenski J., Gross O. P., Pugh E. N., Jr. (2010) J. Neurosci. 30, 12495–12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wachtler T., Dohrmann U., Hertel R. (2004) Vision Res. 44, 2843–2855 [DOI] [PubMed] [Google Scholar]
- 55. McBee J. K., Palczewski K., Baehr W., Pepperberg D. R. (2001) Prog. Retin. Eye Res. 20, 469–529 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.