

Abstract
Because Varp (VPS9-ankyrin-repeat protein)/Ankrd27 specifically binds two small GTPases, Rab32 and Rab38, which redundantly regulate the trafficking of melanogenic enzymes in mammalian epidermal melanocytes, it has recently been implicated in the regulation of trafficking of a melanogenic enzyme tyrosinase-related protein 1 (Tyrp1) to melanosomes. However, the functional interaction between Rab32/38 and Varp and the involvement of the VPS9 domain (i.e. Rab21-GEF domain) in Tyrp1 trafficking have never been elucidated. In this study, we succeeded in identifying critical residues of Rab32/38 and Varp that are critical for the formation of the Rab32/38·Varp complex by performing Ala-based site-directed mutagenesis, and we discovered that a conserved Val residue in the switch II region of Rab32(Val-92) and Rab38(Val-78) is required for Varp binding activity and that its point mutant, Rab38(V78A), does not support Tyrp1 trafficking in Rab32/38-deficient melanocytes. We also identified two critical residues for Rab32/38 binding in the Varp ANKR1 domain and demonstrated that their point mutants, Varp(Q509A) and Varp(Y550A), do not support peripheral melanosomal distribution of Tyrp1 in Varp-deficient cells. Interestingly, the VPS9 domain point mutants, Varp(D310A) and Varp(Y350A), did support Tyrp1 trafficking in Varp-deficient cells, and knockdown of Rab21 had no effect on Tyrp1 distribution. We also found evidence for the functional interaction between a vesicle SNARE VAMP7/TI-VAMP and Varp in Tyrp1 trafficking. These results collectively indicated that both the Rab32/38 binding activity and VAMP7 binding activity of Varp are essential for trafficking of Tyrp1 in melanocytes but that activation of Rab21 by the VPS9 domain is not necessary for Tyrp1 trafficking.
Keywords: G Proteins, Low Molecular Weight G Proteins, Membrane Trafficking, Site-directed Mutagenesis, Subcellular Organelles, Rab, VPS9 Domain, Varp, Ankyrin Repeat Domain, Tyrosinase-related Protein 1
Introduction
Pigmentation of mammalian hair and skin requires proper formation and transport of melanosomes, one of the lysosome-related organelles that specifically synthesize and store melanin pigments, in melanocytes (reviewed in Refs. 1, 2). Defects in the formation and/or transport of melanosomes often cause pigmentary disorders, e.g. albinism and pigmentary dilution, in mammals (reviewed in Ref. 3). The formation and transport of melanosomes involve a variety of intracellular membrane trafficking events, and several distinct Rab-type small GTPases, which are conserved membrane trafficking proteins in all eukaryotic cells (reviewed in Refs. 4–6), have been shown to regulate the maturation and transport of melanosomes in mammalian epidermal melanocytes.
The best characterized Rab isoform that is abundant on mature melanosomes in melanocytes is Rab27A (7–9). Rab27A regulates actin-based melanosome transport through interaction with its specific effector, Slac2-a/melanophilin (10–12), and melanosome anchoring to the plasma membrane through interaction with another effector, Slp2-a (13). As a result, Rab27A deficiency causes human type 2 Griscelli syndrome, which is characterized by silvery hair (i.e. partial albinism) (reviewed in Ref. 14 and references therein). Two other Rab isoforms, Rab32 and Rab38, regulate an early step in melanogenesis, i.e. the transport of melanogenic enzymes to melanosomes (15–17). Actually, dysfunction of Rab38 causes the diluted coat color of chocolate mice, presumably because of impairment of the targeting of tyrosinase-related protein 1 (Tyrp1) to melanosomes (15). The discovery of the Rab32/38-specific binding protein Varp4 (VPS9-ankyrin-repeat protein; official symbol in the National Center for Biotechnology Information is Ankrd27) has recently been reported (18, 19), and as its name indicates, Varp protein contains an N-terminal VPS9 (vacuolar protein sorting 9) domain and C-terminal tandem ankyrin repeat domains (named ANKR1 and ANKR2). The Varp VPS9 domain possesses Rab21-GEF (guanine nucleotide exchange factor) activity (20), and the ANKR1 domain functions as a specific GTP-Rab32/38-binding site (18, 19). Even more recently, VAMP7/TI-VAMP (vesicle-associated membrane protein) has been shown to interact with the region between the ANKR1 domain and the ANKR2 domain and to regulate the neurite outgrowth of hippocampal neurons (21), but whether the Varp·VAMP7 complex is involved in melanogenesis has never been demonstrated. Because knockdown of Varp or overexpression of the ANKR1 domain, i.e. Rab32/38-binding site, in cultured melanocytes causes the disappearance of Tyrp1 signals from peripheral melanosomes (18), Varp is likely to be involved in trafficking of Tyrp1 in concert with Rab32/38. However, the functional interaction between Rab32/38 and Varp (or VAMP7 and Varp) and the involvement of Rab21-GEF activity in Tyrp1 trafficking are not fully understood, and the structural basis of the molecular recognition of Rab32/38 by the Varp ANKR1 domain, including the residue(s) that are critical for Rab32/38 (or ANKR1) recognition, has never been elucidated.
In this study, we identified critical residues for the Rab32/38-Varp interaction in Rab32/38 (or Varp) by performing Ala-based site-directed mutagenesis. A switch II mutant of Rab38, which carries a Val-to-Ala mutation at amino acid position 78 (named Rab38(V78A)), completely lost its Varp binding activity without any change in its intrinsic GTPase activity or subcellular localization. We showed that whereas wild-type Rab38 is able to restore the peripheral melanosomal localization of Tyrp1 in Rab32/38-deficient melanocytes, the Rab38(V78A) mutant is incapable of rescuing Tyrp1-trafficking deficiency and that ANKR1 point mutants, Varp(Q509A) and Varp(Y550A), are also unable to rescue Tyrp1 deficiency in Varp-deficient melanocytes. We then investigated two Varp mutants carrying a point mutation in the VPS9 domain and a Varp deletion mutant lacking the VAMP7-binding site (named ΔVID) and showed that VAMP7 binding to Varp is required for Tyrp1 trafficking, but that Rab21-GEF activity is not. We discuss the structure-function relationships of Varp in Tyrp1 trafficking to melanosomes in melanocytes on the basis of these findings.
EXPERIMENTAL PROCEDURES
Materials
Horseradish peroxidase (HRP)-conjugated anti-FLAG tag (M2) mouse monoclonal antibody was obtained from Sigma. HRP-conjugated anti-T7 tag mouse monoclonal antibody and anti-T7 tag antibody-conjugated agarose were purchased from Merck. Anti-Tyrp1 mouse monoclonal antibody (Ta99) was from ID Labs (Ontario, Canada). Anti-Tyrp1 goat polyclonal antibody and anti-actin goat polyclonal antibody were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-VAMP7 mouse monoclonal antibody (SYBL1; ab36195) and anti-DsRed rabbit polyclonal antibody were from Abcam K. K. (Tokyo, Japan) and Clontech, respectively. Anti-red fluorescent protein rabbit polyclonal antibody and anti-green fluorescent protein (GFP) rabbit polyclonal antibody were from MBL (Nagoya, Japan). Alexa 488/594/633-conjugated anti-mouse IgG goat antibody was from Invitrogen. Anti-Rab21 rabbit polyclonal antibody was raised against glutathione S-transferase (GST)-Rab21 and affinity-purified by exposure to antigen-bound Affi-Gel 10 beads (Bio-Rad) as described previously (22). All other reagents used in this study were analytical grade or the highest grade commercially available.
Site-directed Mutagenesis and Plasmid Construction
Mutant mouse Rab32 expression plasmids carrying a Gly-to-Ala mutation at amino acid position 87 (G87A), N88A, or V92A and mutant mouse Rab38 expression plasmids carrying a V19A, G73A, N74A, or V78A were produced by two-step PCR techniques essentially as described previously (23). In brief, PCRs were performed to generate two DNA fragments having overlapping ends into which specific alterations were introduced, e.g. Rab32-G87A-5′ primer, 5′-AGGAACGGTTTGCCAACATG-3′; and Rab32-G87A-3′ primer, 5′-CATGTTGGCAAACCGTTCCT-3′ (substituted nucleotides are underlined). After purification of the two DNA fragments, they were combined to generate the fusion product by a second PCR. The sequences of other mutagenic oligonucleotides used in this study are summarized in supplemental Table 1. The resulting mutant Rab32/38 cDNAs were subcloned into the pEF-FLAG tag expression vector (24), pGBD-C1 vector (25), pEGFP-C1 vector (Clontech), and pGEX-4T-3 vector (GE Healthcare). Mutant mouse Varp expression plasmids carrying a D310A, Y350A, Q509A, C544A, K546A, Y550A, R557A, W575A, or Y577A mutation or carrying a deletion of amino acid residues 641–707 (i.e. ΔVID) (21) were similarly produced by the method described above (23). The sequences of the mutagenic oligonucleotides for the Varp mutants used are also summarized in supplemental Table 1. To obtain the full-length mutant Varps, each point-mutated Varp fragment (e.g. amino acid residues 442–590) was amplified by two-step PCRs (e.g. the first PCR with Varp-ANKR1-N and Varp-ApaI-3′ primers and the second PCR with Varp-HindIII and Varp-ApaI-3′ primer) and then substituted for the wild-type fragment by using appropriate restriction enzyme sites (e.g. HindIII and artificial ApaI sites). A small interfering RNA (siRNA)-resistant Varp mutant (named VarpSR) was produced by a similar method. Deletion mutants of Varp (individual ANK repeat mutants and ANKR1 mutants lacking a single ANK repeat; see Fig. 4A for details) were also produced by using conventional PCR or two-step PCR techniques and specific oligonucleotides (see supplemental Table 1) as described previously (23, 26). The resulting mutant Varp cDNAs were subcloned into the pEF-T7 tag expression vector (24), pGAD-C1 vector (25), pAct2 vector (Clontech), and/or pmStrawberry (pmStr)-C1 vector (Clontech). The cDNA sequence of all of the mutants described above was verified by DNA sequencing. The cDNA of mouse VAMP7 was amplified from melanocyte cDNAs by PCR with the following oligonucleotides (24): 5′-GGATCCGGTCAGATTGAAGTCATGGCCATT-3′ (VAMP7-5′ primer, sense) and 5′-AGAAACTGGCATTTTCTGAGAGTAG-3′ (VAMP7-3′ primer, antisense). The open reading frame of VAMP7 was then amplified by using the following oligonucleotides with a BamHI site (underlined) or a stop codon (boldface): 5′-GGATCCATGGCCATTCTTTTT-3′ (VAMP7 Met primer, sense) and 5′-TTATTTCTTCACACAGTTTGGCCAT-3′ (VAMP7 stop primer, antisense). pEGFP-C1-VAMP7 and an siRNA-resistant VAMP7 mutant (named VAMP7SR; supplemental Table 1) were similarly produced by the method described above. pSilencer 2.1-U6 neo (Ambion, Austin, TX)-VAMP7 site 1 (st1 target site, 5′-GTGTTTTGGCTGCACAACT-3′) and pSilencer-VAMP7 site 2 (st2 target site, 5′-GAGCCTAGACAAAGTGATG-3′) were prepared as described previously (13). Other expression plasmids, including pEF-FLAG-Rab32/38, pGEX-4T-3-Rab32/38, pEGFP-C1-Rab32/38, pEF-T7-Varp, pmStr-C1-Varp, pSilencer-Varp, and pSilencer-Rab21, were prepared as described elsewhere (18, 27). siRNA against Rab32 (Oligo 32-1) was synthesized as described previously (17).
FIGURE 4.

All four ANK repeats are required for the Rab32/38 binding activity of the Varp ANKR1 domain. A, schematic representation of the truncated mutants of the ANKR1 domain of Varp used in this study. ANKR1-1 contains amino acid residues 462–494; ANKR1-2 contains amino acid residues 495–527; ANKR1-3 contains amino acid residues 528–560; and ANKR1-4 contains amino acid residues 561–596. ANKR1-Δ1 lacks amino acid residues 462–494 (Δ462–494); ANKR1-Δ2 lacks amino acid residues 495–527 (Δ495–527); ANKR1-Δ3 lacks amino acid residues 528–560 (Δ528–560); and ANKR1-Δ4 lacks amino acid residues 561–596 (Δ561–596). B, yeast two-hybrid assays revealed that all four ANK repeats in the ANKR1 are required for Rab32/38 binding. Yeast cells containing pAct2 plasmid expressing each Varp-ANKR1 mutant and pGBD plasmid expressing Rab32 mutant (left panels) or Rab38 mutant (right panels) were streaked on SC-LW (top panels) and SC-AHLW (bottom panels) and incubated at 30 °C. Note that none of the ANKR1 deletion mutants of Varp grew on SC-AHLW (i.e. selection medium) but that they grew normally on SC-LW.
Co-immunoprecipitation Assays in COS-7 Cells
T7-tagged Varp and FLAG-tagged Rab32/38 were transiently expressed in COS-7 cells, and their associations were evaluated in the presence of 0.5 mm GTPγS by co-immunoprecipitation assay with anti-T7 tag (or anti-FLAG tag) antibody-conjugated agarose beads as described previously (24, 28). Proteins bound to the beads were analyzed by 10% SDS-PAGE followed by immunoblotting with HRP-conjugated anti-T7 tag antibody (1:10,000 dilution) and HRP-conjugated anti-FLAG tag antibody (1:10,000 dilution). Immunoreactive bands were visualized by enhanced chemiluminescence.
Yeast Two-hybrid Assays
Yeast two-hybrid assays were performed by using pGBD-C1-Rab lacking the C-terminal geranylgeranylation site and pGAD-C1-Varp or pAct2-Varp (wild-type or its mutants) or by using pGBD-C1-VAMP7 and pAct2-Varp (wild-type or its mutants) as described previously (18, 29). The yeast strain, medium, culture conditions, and transformation protocol used are as described in Ref. 25.
Immunofluorescence Analysis
The immortal mouse melanocyte cell lines melan-a, derived from a black mouse, and melan-cht, derived from a chocolate mouse, both a generous gift of Dorothy C. Bennett, St. George's Hospital Medical School, London, UK, were cultured on glass-bottom dishes (35-mm dish; MatTek, Ashland, MA) as described previously (17, 30, 31). Transfection of pmStr-C1-Varp, pEGFP-C1-Rab32/38, and/or a control vector (pmStr-C1 or pEGFP-C1 alone) into melan-a cells was achieved by using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. Co-transfection of pEGFP-C1-Rab38 with siRab32 into melan-cht cells was achieved by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Two days after transfection, cells were fixed in 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and stained with anti-Tyrp1 mouse monoclonal antibody (1:100 dilution). Tyrp1 signals were visualized with anti-mouse Alexa Fluor 488/594/633 IgG and then examined for fluorescence with a confocal fluorescence microscope (Fluoview FV500; Olympus, Tokyo, Japan) as described previously (31). The images were processed with Adobe Photoshop software (CS4). To quantitatively measure Tyrp1 signals, the images of the enhanced green fluorescent protein (EGFP)-Rab38-expressing cells and/or the mStr (monomeric Strawberry)-Varp-expressing cells were captured at random (n >30 from three independent dishes) with the confocal microscope, and fluorescent signals of Tyrp1 were quantified with MetaMorph software (Molecular Devices, Sunnyvale, CA).
GTP Binding Assay and Measurement of Intrinsic GTPase Activity
GST-Rab32 (wild-type and V92A) and GST-Rab38 (wild-type and V78A) were expressed in Escherichia coli JM109 and purified by standard protocols. A 200 pmol amount of purified GST-Rab32/38 protein was incubated for 10 min at 30 °C with 6.7 pmol of [α-32P]GTP (Muromachi Yakuhin Kaisha Ltd., Tokyo, Japan) and 800 pmol of cold GTP (Sigma) in 50 mm Tris-HCl (pH 8.0), 50 mm NaCl, 2.5 mm EDTA, and 0.5 mg/ml bovine serum albumin. After the addition of MgCl2 (final concentration 10 mm), the mixture was passed through a PD-10 column (GE Healthcare) filled with Sephadex G-25 (GE Healthcare), and the 3.0–3.5-ml elution fractions were collected (32). Bound GTP was quantified with an FLA-3000 fluorescent and radioisotope imaging analyzer (FujiFilm, Tokyo, Japan) and an imaging plate. The intrinsic GTPase activity of Rab32/38 (∼2 pmol) was measured by incubating at 30 °C for 60, 120, or 180 min. The reaction was halted by addition of an equal volume of stop buffer (20 mm EDTA and 0.4% SDS) and incubation for 5 min at 70 °C. A 2-μl sample was dropped on a TLC plate (Merck) and developed in 0.5 m LiCl and 1 m formic acid. The amounts of GTP and GDP were determined with a FLA-3000 fluorescent and radioisotope imaging analyzer and an imaging plate.
Sequence Analysis
Sequence alignment of the switch II region of mouse or human Rabs or of each ANK repeat in the ANKR1 domain of Varp was performed by GENETYX-MAC (version 15.0.1; GENETYX Corp., Tokyo, Japan). Varp sequences from various species were obtained from the public data base.
RESULTS
Identification of Critical Residues in Rab32/38 for Varp Binding by Site-directed Mutagenesis
To identify the critical residues for Varp binding in Rab32/38, we first compared the amino acid sequences of 60 mammalian Rab isoforms. We especially focused on the switch II region of Rab32/38, because previous mutational and/or structural analyses of the Rab3·rabphilin-Rab-binding domain (RBD) and the Rab27·Slac2-a-synaptotagmin-like protein homology domain (SHD) had indicated that several amino acid residues in the switch II region of Rab3/27 are critical for recognition by its effector (33–37). We found that three amino acids, the Gly at amino acid position 87 (Gly-87), Asn-88, and Val-92 of Rab32 (or Gly-73, Asn-74, and Val-78 of Rab38), are only conserved in the switch II region of Rab32 and Rab38 alone and not of any of the other 58 Rabs (asterisks in supplemental Fig. S1 and Fig. 1A). Each of these three amino acids of Rab32/38 was replaced by Ala by site-directed mutagenesis, and the resulting Rab32/38 mutants were subjected to Varp binding assays by performing yeast two-hybrid assays as described previously (18). As shown in Fig. 1B, neither the Rab32(V92A) mutant nor the Rab38(V78A) mutant bound Varp, whereas the other mutants seemed to bind Varp normally. Because a G19A mutation of Rab38 had been reported in chocolate mice (15), we also investigated the effect of the G19A mutation on Varp binding. However, Rab38(G19A) bound Varp, the same as the wild-type protein (Fig. 1B, bottom panels), suggesting that the G19A mutation affects Rab38 functions other than effector binding (e.g. lipid modification (17)). The defective Varp binding activity of the Rab32(V92A) mutant and Rab38(V78A) mutant was confirmed by co-immunoprecipitation assays in COS-7 cells (Fig. 1C). Consistent with the results of the yeast two-hybrid assays described above, neither Rab32(V92A) nor Rab38(V78A) interacted with Varp (lanes 2 and 4 in the middle panel of Fig. 1C), in contrast to the wild-type proteins (lanes 1 and 3).
FIGURE 1.
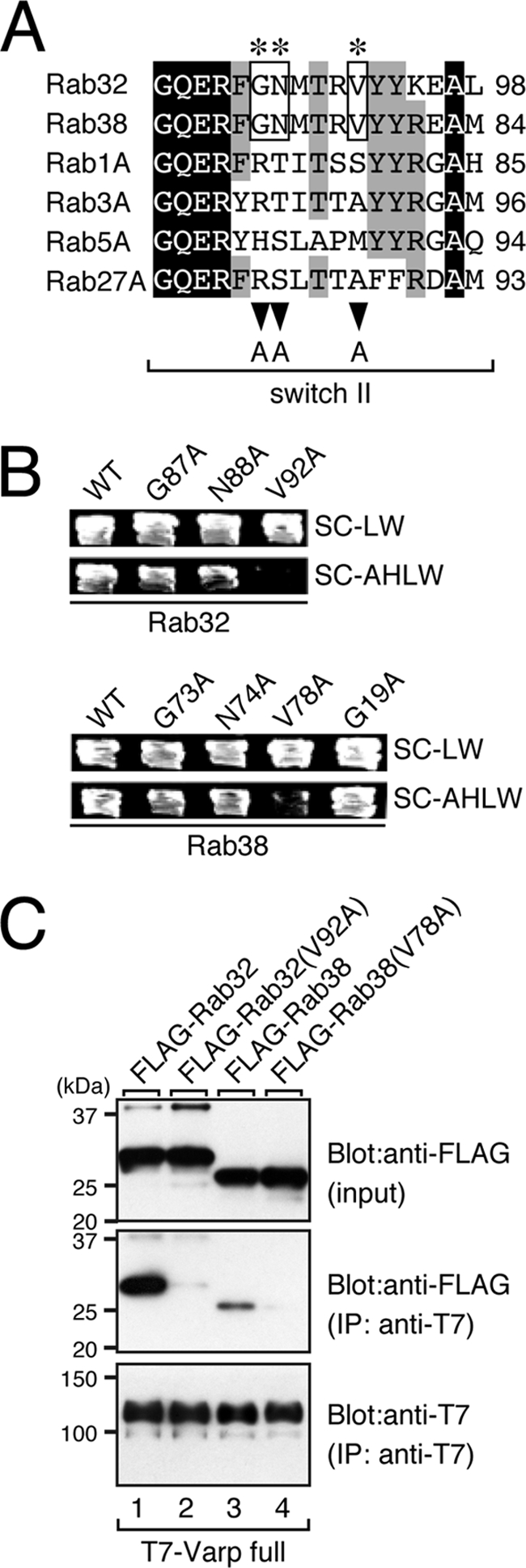
Identification of critical residues responsible for Varp binding in the switch II region of Rab32/38 by site-directed mutagenesis. A, sequence alignment of the switch II regions of mouse Rab1A, Rab3A, Rab5A, Rab27A, Rab32, and Rab38. Identical residues and residues conserved in more than five of the sequences are shown against a black background and a gray background, respectively. The asterisks indicate the positions of three highly conserved amino acids (boxed) in the switch II region of Rab32 and Rab38 that were the focus of the Ala-based site-directed mutagenesis (see also supplemental Fig. S1). B, yeast two-hybrid assays revealed that the Val-92 of Rab32 and Val-78 of Rab38 in the switch II region are critical for Varp binding. Yeast cells containing the pAct2 plasmid expressing Varp and pGBD plasmid expressing the Rab32 mutant or Rab38 mutant were streaked on SC-LW (top panels) and SC-AHLW (selection medium; bottom panels) and incubated at 30 °C. Note that the V92A mutation of Rab32 and V78A mutation of Rab38 completely abrogated Varp binding activity, whereas other mutants, including the Rab38(G19V) mutant found in chocolate mice (15), had virtually no effect on Varp binding. C, T7-tagged Varp-full and FLAG-tagged Rab32/38 or their VA mutants were co-expressed in COS-7 cells, and their associations were analyzed in the presence of 0.5 mm GTPγS by co-immunoprecipitation assays with anti-FLAG tag antibody-conjugated agarose beads as described previously (24). Co-immunoprecipitated FLAG-tagged Rab32/38 (or their VA mutants) (middle panel) and immunoprecipitated (IP) T7-tagged Varp-full (bottom panel) were detected with HRP-conjugated anti-FLAG tag antibody and HRP-conjugated anti-T7 tag antibody, respectively. Input means 1/70 volume of the reaction mixture used for immunoprecipitation (top panel). The positions of the molecular mass markers (in kilodaltons) are shown on the left. Note that neither Rab32(V92A) nor Rab38(V78A) bound Varp-full, consistent with the results of the yeast two-hybrid assays shown in B.
We next investigated whether the V92A mutation of Rab32 (or V78A mutation of Rab38) affected other properties of Rab32 (or Rab38), e.g. its GTP binding activity or intrinsic GTPase activity. To do so, we expressed and purified wild-type and mutant recombinant proteins with a GST tag from bacteria. Because both the Rab32(V92A) mutant and Rab38(V78A) mutant were easily obtained free of contamination by degradation products, the same as the wild-type proteins (Fig. 2A), these mutations were unlikely to affect protein folding or stability. We found that both mutants exhibit normal GTP binding activity (Fig. 2B) and intrinsic GTPase activity (Fig. 2C), although the intrinsic GTPase activity of Rab32/38 was very weak. Moreover, the subcellular localization of the EGFP-tagged Rab32(V92A) mutant and Rab38(V78A) mutant seemed to be the same as that of the wild-type proteins (Fig. 2D, top panels). Actually, co-localization of Rab32/38 mutants with Tyrp1 was not significantly different from the co-localization of the wild-type Rab32/38 protein (supplemental Fig. S2A). Furthermore, neither mutant affected the Tyrp1 distribution (Fig. 2D, bottom panels), suggesting that these mutants are unlikely to function as a dominant negative mutant in Tyrp1 trafficking. These results allowed us to conclude that the V92A mutation of Rab32 (or V78A mutation of Rab38) specifically impairs Varp binding ability.
FIGURE 2.

Characterization of the Rab32(V92A) mutant and Rab38(V78A) mutant. A, purified GST-Rab32/38 used in this study. Approximately 5 μg each of purified GST-Rab32(WT) (lane 2), GST-Rab32(V92A) (lane 3), GST-Rab38(WT) (lane 4), and GST-Rab38(V78A) (lane 5) was subjected to 10% SDS-PAGE followed by staining with Coomassie Brilliant Blue R-250. Note that the GST-Rab32/38 proteins were purified with little contamination by degradation products. The positions of the molecular mass markers (in kilodaltons) are shown on the left (lane 1). B, GTP binding activity of Rab32/38 mutants. a.u., arbitrary units. C, intrinsic GTPase activity of Rab32/38 mutants. GTP hydrolysis by Rab32/38 was measured, and the results are expressed as the amount of the GTP-bound form of Rab32/38 after the reaction as a percentage of the amount before the reaction. The bars represent the means ± S.D. of three determinations. Note that the GTP binding activity and intrinsic GTPase activity of Rab32(V92A) (or Rab38(V78A)) were the same as those of the wild-type protein. D, normal distribution of Rab32/38 mutants in melanocytes. Melan-a cells transiently expressing EGFP-Rab32(WT), EGFP-Rab32(V92A), EGFP-Rab38(WT), or EGFP-Rab38(V78A) (green, top panels) were immunostained with anti-Tyrp1 mouse monoclonal antibody (red, bottom panels). No differences in the distribution of EGFP-Rab32/38 or Tyrp1 were observed between the wild-type-expressing and mutant protein-expressing cells (see also supplemental Fig. S2A). Scale bars, 20 μm.
Rab38(V78A) Mutant Is Incapable of Restoring Tyrp1 Trafficking in Melan-cht Cells
If the Varp binding ability of Rab38 is actually essential for Tyrp1 trafficking in vivo, the Rab38(V78A) mutant described above should not restore Tyrp1 trafficking deficiency in Rab38-defective melanocytes from chocolate mice. Although the immortal melanocyte cell line melan-cht derived from a chocolate mouse has recently been established, because the melan-cht cells exhibited normal Tyrp1 distribution as a result of the compensatory effect of Rab32 (17), we used Rab32-knockdown melan-cht cells (i.e. Rab32/38-deficient melanocytes) in the rescue experiments. Consistent with a previous report (17), Rab32-deficient melan-cht cells exhibited reduced Tyrp1 signals and perinuclear clustering of Tyrp1 (Fig. 3A, top row of panels). Re-expression of Rab38 with the EGFP tag completely restored strong peripheral Tyrp1 signals on melanosomes (Fig. 3A, 2nd row of panels from the top), whereas expression of Rab38(V78A), which specifically lacks Varp binding activity, failed to rescue the reduced Tyrp1 signals or perinuclear clustering of Tyrp1 in Rab32-deficient melan-cht cells (Fig. 3A, bottom row of panels). The absence of any rescue effect by Rab38(V78A) must not be attributable to a low level of expression of the mutant protein, because EGFP fluorescence indicated that a sufficient amount of mutant protein was expressed in the melan-cht cells (data not shown), and both the wild-type and V78A mutant form of Rab38 were expressed equally in melan-a cells (supplemental Fig. S3A). It should be noted that Rab38(G73A) and Rab38(N74A), both of which retained Varp binding ability, were capable of restoring peripheral Tyrp1 signals on melanosomes (Fig. 3, A, 3rd and 4th rows of panels from the top, and B).
FIGURE 3.

Re-expression of Rab38, but not the Varp-binding-deficient Rab38(V78A) mutant, restored the peripheral melanosomal distribution of Tyrp1 in Rab32-deficient melan-cht cells (i.e. Rab32/38-deficient melanocytes). A, Rab38-defective melan-cht cells were co-transfected with siRab32 and pEGFP-C1 or pEGFP-C1-Rab38 (WT, G73A, N74A, or V78A), and the cells were then immunostained with anti-Tyrp1 mouse monoclonal antibody. Note that reduced and perinuclear Tyrp1 distributions are evident in the Rab32/38-deficient cells (top rows of panels), whereas re-expression of Rab38 (2nd row of panels from the top), but not the Varp-binding-deficient Rab38(V78A) mutant (bottom row of panels), in Rab32/38-deficient cells restored strong peripheral distribution of Tyrp1. Scale bars, 20 μm. B, quantification of Tyrp1 signals shown in A. The bars represent the means ± S.E. of data from three independent dishes (n >30). **, p < 0.01 (Student's unpaired t test) in comparison with the mock control; NS, not significant. a.u., arbitrary units.
Identification of Critical Residues in Varp for Rab32/38 Binding by Performing Site-directed Mutagenesis
In the next set of experiments, we attempted to identify critical residues in the Varp ANKR1 domain for Rab32/38 binding (18). Two additional ANK repeat domain-containing proteins, ORP1L (38) and centaurin β2/ACAP2 (39), have recently been shown to bind Rab7 and Rab35, respectively, but nothing is known about the structural basis of the Rab recognition by their ANK repeat domains. Because ORP1L (38) and centaurin β2 (39) contain three ankyrin repeats, we first investigated how many ANK repeats are required for recognition of Rab32/38 by the Varp ANKR1 domain and which ANK repeats are most important for Rab32/38 recognition. We did so by performing systematic deletion analyses (Fig. 4A). The results of yeast two-hybrid assays showed that, in contrast to the wild-type Varp ANKR1 domain, none of the ANK repeats (i.e. ANKR1-1–4) or ANKR1 mutants lacking any of the ANK repeats (i.e. ANKR1-Δ1 to Δ4) bound Rab32 or Rab38, thereby indicating that all four ANK repeats in ANKR1 are required for specific recognition of Rab32/38. However, we cannot completely rule out the possibility that deletion of individual ANK repeats may destabilize or alter the overall structure of the ANKR1 domain, which indirectly causes loss of the Rab32/38 binding activity of ANKR1-Δ1–4 mutants.
To identify the residues in each ANK repeat in the ANKR1 domain that are critical for Rab32/38 binding, we next attempted to select candidate residues responsible for Rab binding based on the following criteria: (i) amino acid residues not conserved in the ANKR2 domain (i.e. exclusion of the consensus sequence of the ANK repeat domains) and (ii) amino acid residues conserved in a variety of species (from silkworm to mammals) (Fig. 5A). Based on these criteria, we selected seven candidate residues, Gln-509, Cys-544, Lys-546, Tyr-550, Arg-557, Trp-575, and Tyr-577 (asterisks in Fig. 5A), none of which are conserved in ORP1L or centaurin β2, and each of the candidate amino acids was replaced by Ala by site-directed mutagenesis (arrowheads in Fig. 5A). The resulting Varp mutants were subjected to a Varp binding assay by using yeast two-hybrid assays as described previously (18). As shown in Fig. 5B, neither Varp(Q509A) nor Varp(Y550A) bound Rab32 or Rab38, whereas the Varp(C544A), Varp(K546A), and Varp(R557A) mutants retained the ability to bind Rab32/38. The Varp(W575A) mutant and Varp(Y577A) mutant showed reduced binding activity toward Rab32/38, because yeast cells expressing either of these mutants were able to grow on the selection medium; however, their growth rate was clearly slower than that of cells expressing wild-type Varp (Fig. 5B). The lack of Rab32/38 binding activity by the Varp(Q509A) mutant and Varp(Y550A) mutant was confirmed by two independent approaches. In the first approach, neither the Varp(Q509A) mutant nor the Varp(Y550A) mutant with a monomeric Strawberry tag co-localized with EGFP-Rab32/38 in melan-a cells, whereas other Varp mutants, including the Varp(W575A) mutant and Varp(Y577A) mutant, clearly co-localized with EGFP-Rab32/38 (arrowheads in Fig. 6 and supplemental Fig. S4). In the second approach, neither the Varp(Q509A) mutant nor the Varp(Y550A) mutant was co-purified with Rab32/38 in co-immunoprecipitation assays (lanes 3 and 4 in the middle panels of Fig. 5C), whereas the Varp(R557A) mutant bound Rab32/38 normally, the same as the wild-type protein did (lanes 2 and 5 in the middle panels of Fig. 5C). On the basis of these results, we decided to use the Varp(Q509A) mutant and Varp(Y550A) mutant as Rab32/38-binding-deficient mutants in the subsequent analysis and the Varp(R557A) mutant as a negative control.
FIGURE 5.

Identification of the critical residues responsible for Rab32/38 binding in the ANKR1 domain of Varp by site-directed mutagenesis. A, sequence alignment of the ANKR1 domain of human (Hs, Homo sapiens), mouse (Mm, Mus musculus), chick (Gg, Gallus gallus), zebra fish (Dr, Danio rerio), sea urchin (Sp, Strongylocentrotus purpuratus), and silkworm (Bm, Bombyx mori) Varp. Residues conserved in more than five of the sequences are shown against a black background. The asterisks indicate the positions of the seven highly conserved amino acids, Gln-509, Cys-544, Lys-546, Tyr-550, Arg-557, Trp-575, and Tyr-577, in the ANKR1 domain that were the focus of the Ala-based site-directed mutagenesis. B, yeast two-hybrid assays revealed that the Gln-509 and Tyr-550 of Varp are critical for Rab32/38 binding. Yeast cells containing pGAD plasmid expressing Varp and pGBD plasmid expressing Rab32 (left panels) or Rab38 (right panels) were streaked on SC-LW (top panels) and SC-AHLW (selection medium; bottom panels) and incubated at 30 °C. Note that the Q509A, Y550A, W575A, and Y577A mutations dramatically reduced Rab32/38 binding activity, although the former two mutations reduced Rab32/38 binding activity more severely than the latter two mutations, based on the growth rate of the yeast cells and the results of the immunofluorescence analysis (see Fig. 6 and supplemental Fig. S4A). By contrast, the C544A, K546A, and R557A mutations had little or no effect on Rab32/38 binding. C, FLAG-tagged Rab32/38 and T7-tagged Varp-full or their mutants were co-expressed in COS-7 cells, and their associations were analyzed in the presence of 0.5 mm GTPγS by co-immunoprecipitation assays with anti-FLAG tag antibody-conjugated agarose beads as described previously (24). Co-immunoprecipitated T7-tagged Varp-full (or their mutants) (middle panels) and immunoprecipitated FLAG-tagged Rab32/38 (bottom panels) were detected with HRP-conjugated anti-FLAG tag antibody and HRP-conjugated anti-T7 tag antibody, respectively. Input means 1/70 volume of the reaction mixture used for immunoprecipitation (IP) (top panels). The positions of the molecular mass markers (in kilodaltons) are shown on the left. Note that neither Varp(Q509A) nor Varp(Y550A) bound Rab32/38 (lanes 3 and 4 in the middle panel), whereas Varp(R557A) normally bound Rab32/38 (lane 5 in the middle panel), consistent with the results of the yeast two-hybrid assays shown in B.
FIGURE 6.

Co-localization between Rab38 and Varp mutants. Melan-a cells transiently expressing EGFP-Rab38 together with mStr-Varp(WT) (top row of panels), mStr-Varp(Q509A) (2nd row of panels from the top), mStr-Varp(Y550A) (3rd row of panels from the top), or mStr-Varp(R557A) (bottom row of panels) are shown. Note that Rab32/38-binding-deficient mutants of Varp were mostly distributed in the cytosol and that they were not co-localized with EGFP-Rab38 (insets in the 2nd and 3rd rows of panels). By contrast, there was clear co-localization between Rab38 and Varp(WT) or Varp(R557A), which retained Rab32/38 binding ability (arrowheads in the insets). The insets are magnified views of the boxed areas. Scale bars, 20 μm.
Overexpression of Varp in Melanocytes Caused a Reduction in Tyrp1 Signals
We also attempted to perform rescue experiments by expressing Rab32/38-binding-deficient Varp mutants in Varp knockdown cells. To do so, we first prepared an siRNA-resistant Varp mutant (named VarpSR) by replacing six nucleotides that did not result in any amino acid substitutions. As anticipated, expression of the VarpSR mutant in COS-7 cells was resistant to Varp short hairpin RNA (shRNA), whereas expression of wild-type Varp was clearly suppressed by the Varp shRNA (supplemental Fig. S5A). To our surprise, however, expression of the VarpSR mutant alone in melan-a cells caused a dramatic reduction in Tyrp1 signals (supplemental Fig. S5B). This effect should not be attributable to the six nucleotide substitutions, because expression of wild-type Varp (i.e. no SR mutation) alone also caused a dramatic reduction in Tyrp1 signals (Fig. 7A, left panels). Interestingly, expression of the Rab32/38-binding-deficient Varp(Q509A) mutant or Varp(Y550A) mutant had no effect on the Tyrp1 signals, whereas expression of Varp(R557A), which retains Rab32/38-binding ability (Fig. 5, B and C), caused a dramatic reduction in Tyrp1 signals (Fig. 7, A and B). Because co-expression of mStr-Varp with EGFP-Rab32/38 had no effect on the Tyrp1 signals or their distribution (18), an excess of Varp molecules that are free of Rab32/38 may negatively regulate Tyrp1 trafficking in living cells.
FIGURE 7.

Expression of Varp alone caused a dramatic reduction in Tyrp1 signals in melan-a cells. A, melan-a cells transiently expressing Varp(WT), Varp(Q509A), Varp(Y550A), or Varp(R557A) (red, 2nd row of panels from the top) were immunostained with anti-Tyrp1 mouse monoclonal antibody (green, top row of panels). Note the clearly reduced Tyrp1 signals in the Varp(WT)- or Varp(R557A)-expressing cells (the cells have been outlined in white), whereas the Rab32/38-binding-deficient Varp mutants, i.e. Varp(Q509A) and Varp(Y550A), had no effect on Tyrp1 staining. Scale bars, 20 μm. B, quantification of Tyrp1 signals shown in A. The bars represent the means ± S.E. of data from three independent dishes (n >30). **, p < 0.01 (Student's unpaired t test) in comparison with the mock control. a.u., arbitrary units.
To overcome this problem, we attempted to simultaneously transfect three plasmids, pEGFP-C1-Rab38, pmStr-C1-VarpSR, and pSilencer-Varp, into melan-a cells. Although knockdown of Varp in melan-a cells caused a dramatic reduction in Tyrp1 signals (Fig. 8A, top row of panels), re-expression of mStr-VarpSR together with EGFP-Rab38 in Varp-deficient cells restored peripheral Tyrp1 distribution (Fig. 8A, 2nd row of panels from the top). By contrast, however, re-expression of the Rab32/38-binding-deficient mStr-VarpSR(Q509A) mutant or VarpSR(Y550A) mutant in Varp-deficient melan-a cells did not rescue Varp deficiency, even in the presence of EGFP-Rab38 (Fig. 8A, 3rd and 4th rows of panels from the top). It should be noted that re-expression of mStr-VarpSR(R557A), which retains Rab32/38-binding ability, together with EGFP-Rab38 restored peripheral Tyrp1 distribution in Varp-deficient cells (Fig. 8A, bottom row of panels).
FIGURE 8.

Re-expression of Varp, but not the Rab32/38-binding-deficient Varp(Q509A) or Varp(Y550A) mutant, restored the peripheral distribution of Tyrp1 in Varp-deficient melan-a cells. A, melan-a cells were co-transfected with shVarp, pEGFP-C1-Rab38, and pmStr-C1 vectors (pmStr-C1 or pmStr-C1-VarpSR (WT, Q509A, Y550A, or R557A)) and then immunostained with anti-Tyrp1 mouse monoclonal antibody. Note the clear disappearance of Tyrp1 signals in Varp-deficient melan-a cells (top row of panels), whereas re-expression of wild-type VarpSR (2nd row of panels from the top) and the Rab32/38-binding VarpSR(R557A) mutant (bottom row of panels), but not the Rab32/38-binding-deficient VarpSR(Q509A) mutant or VarpSR(Y550A) mutant (3rd and 4th rows of panels from the top; the cells have been outlined in white), restored strong peripheral distribution of Tyrp1 in Varp-deficient melan-a cells. Scale bars, 20 μm. B, quantification of Tyrp1 signals shown in A. The bars represent the means ± S.E. of data from three independent dishes (n >50). **, p < 0.01 (Student's unpaired t test) in comparison with the wild-type Varp; a.u., arbitrary units.
Effect of VPS9 Point Mutants and the ΔVID Mutant of Varp on Tyrp1 Trafficking
In addition to the Rab32/38 effector domain, i.e. the ANKR1 domain, Varp contains a VPS9 Rab21-GEF domain (20) and a VAMP7-interaction domain between the ANKR1 domain and ANKR2 domain (21); however, the functional involvement of Rab21-GEF activity (or Rab21 itself) and VAMP7 binding activity (or VAMP7 itself) in Tyrp1 trafficking to melanosomes has never been investigated. Therefore, we finally attempted to investigate their involvement in Tyrp1 trafficking by the knockdown-rescue approach as described above. Because recent structural studies (40, 41) have already identified several critical residues for guanine nucleotide exchange factor activity of the VPS9 domain (Fig. 9A, arrowheads), we mutated two conserved residues in the VPS9 domain (Asp-310 and Tyr-350) to Ala by site-directed mutagenesis (named Varp(D310A) and Varp(Y350A)). As anticipated, both VPS9 point mutants completely abrogated GDP-Rab21 binding activity as assessed by yeast two-hybrid assays (Fig. 9B, top row in the SC-AHLW panel) without any change in GTP-Rab32/38 binding activity or VAMP7 binding activity (Fig. 9B, bottom three rows in the SC-AHLW panel). If the Rab21-GEF activity of the Varp VPS9 domain is also essential for Tyrp1 trafficking, these two VPS9 point mutants should not support Tyrp1 trafficking in Varp-deficient cells. To our surprise, however, both the VarpSR(D310A) mutant and VarpSR(Y350A) mutant completely restored the peripheral melanosomal distribution of Tyrp1 in Varp-deficient cells (Fig. 9, C and D). Although Rab21 was endogenously expressed in melan-a cells (supplemental Fig. S6B), and it has been proposed that endosomal trafficking regulates melanosomal biogenesis (2), knockdown of endogenous Rab21 by specific shRNA had no effect on either Tyrp1 distribution or Tyrp1 signals (Fig. 9E). These results allowed us to conclude that the Rab21-GEF activity of Varp is not essential for Tyrp1 trafficking in melanocytes.
FIGURE 9.

Rab21-GEF activity is not required for peripheral melanosomal distribution of Tyrp1 in melanocytes. A, schematic representation of VPS9 point mutants, Varp(D310A) and Varp(Y350A), used in this study. The arrowheads indicate the positions of two highly conserved amino acids (D310A and Y350A) in the VPS9 domain (40, 41) that were the focus of the Ala-based site-directed mutagenesis. B, yeast two-hybrid assays revealed that the Asp-310 and Tyr-350 of Varp are critical for GDP-Rab21 binding. Yeast cells containing pGAD (or pAct2) plasmid expressing VarpSR and pGBD plasmid expressing the constitutive negative form of Rab21 (top rows), the constitutive active form of Rab32 (2nd rows from the top), the constitutive active form of Rab38 (3rd rows from the top), or VAMP7 (bottom rows) were streaked on SC-LW (top panels) and SC-AHLW (selection medium; bottom panels) and incubated at 30 °C. Note that the D310A mutation and Y350A mutation abrogated Rab21 binding activity, whereas both the VarpSR(D310A) mutant and VarpSR(Y350A) mutant exhibited normal Rab32/38 binding activity and VAMP7 binding activity. C, re-expression of VarpSR VPS9 point mutants in Varp-deficient melan-a cells fully restored the peripheral distribution of Tyrp1. Melan-a cells were co-transfected with shVarp, pEGFP-C1-Rab38, and pmStr-C1 vectors (pmStr-C1 or pmStr-C1-VarpSR (WT, D310A, or Y350A)) and then immunostained with anti-Tyrp1 mouse monoclonal antibody. The insets are magnified views of the boxed areas and show co-localization between EGFP-Rab38 and mStr-Varp mutants (yellow signals). Scale bars, 20 μm. D, quantification of the Tyrp1 signals shown in C. The bars represent the means ± S.E. of data from three independent dishes (n >50). **, p < 0.01 (Student's unpaired t test) in comparison with the wild-type Varp; NS, not significant. a.u., arbitrary units. E, knockdown of Rab21 by specific shRNA had no effect on the Tyrp1 signals or their distribution. Melan-a cells were co-transfected with shRab21 and pEGFP-C1 vector and then immunostained with anti-Tyrp1 mouse monoclonal antibody. Scale bar, 20 μm. The efficiency of Rab21 siRNA is shown in supplemental Fig. S6.
We then evaluated the importance of the interaction between Varp and VAMP7 in Tyrp1 trafficking by using a Varp-ΔVID mutant, which lacks the VAMP7 interaction domain (Fig. 10A) (21). The Varp-ΔVID mutant completely lacked VAMP7 binding activity, consistent with a previous report (21), but it was capable of binding GDP-Rab21 and GTP-Rab32/38 (Fig. 10B), although the Rab32/38 binding activity of Varp-ΔVID appeared to be reduced based on the growth rate of the yeast cells. The results of the immunofluorescence analysis, however, showed that the Varp-ΔVID mutant co-localized with EGFP-Rab38 (Fig. 10C, inset), indicating that the Varp-ΔVID mutant retained the ability to bind Rab38 in cultured cells. The rescue experiment clearly showed that the VarpSR-ΔVID mutant is incapable of restoring the peripheral melanosomal distribution of Tyrp1 in Varp-deficient cells (Fig. 10, C and D). Moreover, we discovered that VAMP7 is endogenously expressed in cultured melanocytes (supplemental Fig. S7B) and that its knockdown by specific shRNA phenocopies Varp deficiency, i.e. the disappearance of Tyrp1 signals, especially from peripheral melanosomes (Fig. 10E). This knockdown phenotype should not be attributable to the off-target effect of the shVAMP7, because the same phenotype was observed with two independent shRNAs for VAMP7 (supplemental Fig. S7A and data not shown) and the disappearance of Tyrp1 signals in VAMP7-deficient melanocytes was completely rescued by re-expression of VAMP7SR (Fig. 10E). These results indicated that the Varp-VAMP7 interaction is essential for the peripheral melanosomal distribution of Tyrp1 in melanocytes.
FIGURE 10.

Interaction between Varp and VAMP7 is necessary for the peripheral melanosomal distribution of Tyrp1 in melanocytes. A, schematic representation of the Varp-ΔVID mutant (i.e. deletion of 641–707 amino acids), which lacks VAMP7 binding activity (21), used in this study. B, VarpSR-ΔVID mutant lacks VAMP7 binding activity alone and retains binding capacity for Rab21, Rab32, and Rab38. Yeast cells containing pGAD (or pAct2) plasmid expressing VarpSR and pGBD plasmid expressing the constitutive negative form of Rab21 (top row of panels), the constitutive active form of Rab32 (2nd row of panels from the top), the constitutive active form of Rab38 (3rd row of panels from the top), or VAMP7 (bottom row of panels) were streaked on SC-LW (left panels) and SC-AHLW (selection medium; right panels) and incubated at 30 °C. C, Re-expression of VarpSR-ΔVID mutant in Varp-deficient melan-a cells did not restore the peripheral distribution of Tyrp1. Melan-a cells were co-transfected with shVarp, pEGFP-C1-Rab38, and pmStr-C1 vectors (pmStr-C1 or pmStr-C1-VarpSR (WT or ΔVID)) and then immunostained with anti-Tyrp1 mouse monoclonal antibody. The inset is a magnified view of the boxed area and shows co-localization between Rab38 and Varp-ΔVID (yellow signals). Scale bars, 20 μm. D, quantification of the Tyrp1 signals shown in C. The bars represent the means ± S.E. of data from three independent dishes (n >50). **, p < 0.01 (Student's unpaired t test). a.u., arbitrary units. E, knockdown of VAMP7 by specific shRNA dramatically reduced the Tyrp1 signals. Melan-a cells were co-transfected with shVAMP7 and pEGFP-C1 vectors (pEGFP-C1 or pEGFP-C1-VAMP7SR) and then immunostained with anti-Tyrp1 mouse monoclonal antibody. The inset is a magnified view of the boxed area and shows co-localization between Tyrp1 and VAMP7 (yellow signals). Scale bars, 20 μm. The efficiency of VAMP7 shRNA is shown in supplemental Fig. S7.
DISCUSSION
We have previously shown that the Rab32/38-specific binding protein Varp is expressed in cultured melanocytes and that its knockdown by a specific shRNA caused a dramatic reduction in Tyrp1 signals (18), suggesting that a Varp·Rab32/38 complex regulates the transport of Tyrp1 to melanosomes, but whether interaction between Varp and Rab32/38 in melanocytes is essential for Tyrp1 trafficking remained unclear. In this study, we identified the residues of Rab32/38 (or Varp) that are critical for the formation of the Varp·Rab32/38 complex by performing Ala-based site-directed mutagenesis (Figs. 1 and 5), and we evaluated the functional significance of the Varp-Rab32/38 interaction in Tyrp1 trafficking in melanocytes (Figs. 3 and 8) by the knockdown-rescue approach. The results showed that neither Varp-binding-deficient Rab mutants (e.g. Rab38(V78A)) nor Rab32/38-binding-deficient Varp mutants (i.e. Varp(Q509A) and Varp(Y550A)) mediated the peripheral distribution of Tyrp1, whereas other Rab38 point mutants that possessed Varp-binding ability (e.g. Rab38(G73A) and Rab38(N74A)) and Varp point mutants with Rab32/38-binding ability (e.g. Varp(R557A)) behaved the same way as the respective wild-type protein in terms of the Tyrp1 distribution in melanocytes (Figs. 3 and 8). These findings enabled us to conclude that the interaction between Rab32/38 and Varp in melanocytes is essential for the transport of Tyrp1 and for its peripheral distribution. We also demonstrated by the same knockdown-rescue approach that the VAMP7 binding activity of Varp, but not its Rab21-GEF activity, is essential for the peripheral melanosomal distribution of Tyrp1 in melanocytes (Figs. 9 and 10). The requirement for the Rab21-GEF activity of Varp may be cell type-specific (neurons versus melanocytes) or cargo-specific (endosomal trafficking in neurites versus Tyrp1-containing vesicle trafficking), whereas VAMP7 binding activity is required for both neurite outgrowth and Tyrp1 transport. Because VAMP7 functions as a v-SNARE (42) and its partner t-SNARE syntaxin-3 has been shown to be localized on melanosomes (43), it is tempting to speculate that the VAMP7-syntaxin-3 interaction is involved in the fusion of Tyrp1-containing vesicles with melanosomes. Another suspected function of VAMP7 is as a bridge between the Varp·Rab32/38 complex and the adaptor complex AP-3, which is required for tyrosinase transport (44), because the Longin domain of VAMP7 interacts with AP-3 (45). Further work will be necessary to determine the mechanism(s) by which the Varp·VAMP7 complex regulates Tyrp1 transport at the molecular level.
Another important but unexpected finding in this study was that the level of expression of Varp in melanocytes is a key factor in the quality control of Tyrp1 in melanocytes. We found that either knockdown of Varp or overexpression of Varp alone in melanocytes caused a dramatic reduction in Tyrp1 signals (Fig. 7 and supplemental Fig. S5B). Interestingly, however, co-expression of Varp with Rab38 in melanocytes had no effect on Tyrp1 distribution or signals, suggesting that Varp free of Rab32/38 negatively regulates Tyrp1 trafficking, possibly by a proteasome-mediated degradation of Tyrp1 (18) by unknown mechanisms, because the proteasome inhibitor MG132 greatly attenuated the reduction in Tyrp1 signals induced by overexpression of Varp (data not shown). As it has recently been reported that tyrosinase is degraded by endoplasmic reticulum-associated degradation (46–48) and that Rab32/38 is also involved in tyrosinase trafficking (17), a similar degradation mechanism may operate for Tyrp1. It would therefore be interesting to determine whether compounds that promote the endoplasmic reticulum-associated degradation of tyrosinase (49) affect the function of Varp. An attempt to determine whether Varp is also involved in the trafficking of other melanogenic enzymes, including tyrosinase, and their quality control is now under way in our laboratory.
In summary, we performed a structure-function analysis of Varp by site-directed mutagenesis in combination with the knockdown-rescue approach in cultured melanocytes. Our findings clearly indicated that Varp functions as a Rab32/38 effector in melanocytes and regulates Tyrp1 trafficking to melanosomes and that VAMP7 binding activity, but not Rab21-GEF activity, is also required for this process. The results further indicate that Varp is a key regulator of the quality control of Tyrp1 in melanocytes.
Supplementary Material
Acknowledgments
We thank Dr. Dorothy C. Bennett (St. George's Hospital Medical School, London, UK) for kindly donating melan-a and melan-cht cells, Megumi Aizawa for technical assistance, and members of the Fukuda laboratory for valuable discussions.
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, and Technology of Japan (to N. O. and M. F.), by a grant from the Global COE Program (Basic and Translational Research Center for Global Brain Science) from the Ministry of Education, Culture, Sports, and Technology of Japan (to N. O. and M. F.), and by a grant from the Kato Memorial Bioscience Foundation (to M. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. S1–S7.
- Varp
- VPS9-ankyrin-repeat protein
- ANK
- ankyrin
- EGFP
- enhanced green fluorescent protein
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate.
REFERENCES
- 1. Marks M. S., Seabra M. C. (2001) Nat. Rev. Mol. Cell Biol. 2, 738–748 [DOI] [PubMed] [Google Scholar]
- 2. Raposo G., Marks M. S. (2007) Nat. Rev. Mol. Cell Biol. 8, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tomita Y., Suzuki T. (2004) Am. J. Med. Genet. 131C, 75–81 [DOI] [PubMed] [Google Scholar]
- 4. Pfeffer S. R. (2001) Trends Cell Biol. 11, 487–491 [DOI] [PubMed] [Google Scholar]
- 5. Fukuda M. (2008) Cell. Mol. Life Sci. 65, 2801–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stenmark H. (2009) Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 7. Hume A. N., Collinson L. M., Rapak A., Gomes A. Q., Hopkins C. R., Seabra M. C. (2001) J. Cell Biol. 152, 795–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bahadoran P., Aberdam E., Mantoux F., Buscà R., Bille K., Yalman N., de Saint-Basile G., Casaroli-Marano R., Ortonne J. P., Ballotti R. (2001) J. Cell Biol. 152, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu X., Rao K., Bowers M. B., Copeland N. G., Jenkins N. A., Hammer J. A., 3rd (2001) J. Cell Sci. 114, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 10. Fukuda M., Kuroda T. S., Mikoshiba K. (2002) J. Biol. Chem. 277, 12432–12436 [DOI] [PubMed] [Google Scholar]
- 11. Wu X. S., Rao K., Zhang H., Wang F., Sellers J. R., Matesic L. E., Copeland N. G., Jenkins N. A., Hammer J. A., 3rd (2002) Nat. Cell Biol. 4, 271–278 [DOI] [PubMed] [Google Scholar]
- 12. Strom M., Hume A. N., Tarafder A. K., Barkagianni E., Seabra M. C. (2002) J. Biol. Chem. 277, 25423–25430 [DOI] [PubMed] [Google Scholar]
- 13. Kuroda T. S., Fukuda M. (2004) Nat. Cell Biol. 6, 1195–1203 [DOI] [PubMed] [Google Scholar]
- 14. Van Gele M., Dynoodt P., Lambert J. (2009) Pigment Cell Melanoma Res. 22, 268–282 [DOI] [PubMed] [Google Scholar]
- 15. Loftus S. K., Larson D. M., Baxter L. L., Antonellis A., Chen Y., Wu X., Jiang Y., Bittner M., Hammer J. A., 3rd, Pavan W. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 4471–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park M., Serpinskaya A. S., Papalopulu N., Gelfand V. I. (2007) Curr. Biol. 17, 2030–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wasmeier C., Romao M., Plowright L., Bennett D. C., Raposo G., Seabra M. C. (2006) J. Cell Biol. 175, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tamura K., Ohbayashi N., Maruta Y., Kanno E., Itoh T., Fukuda M. (2009) Mol. Biol. Cell 20, 2900–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang F., Zhang H., Zhang X., Wang Y., Ren F., Zhang X., Zhai Y., Chang Z. (2008) Biochem. Biophys. Res. Commun. 372, 162–167 [DOI] [PubMed] [Google Scholar]
- 20. Zhang X., He X., Fu X. Y., Chang Z. (2006) J. Cell Sci. 119, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 21. Burgo A., Sotirakis E., Simmler M. C., Verraes A., Chamot C., Simpson J. C., Lanzetti L., Proux-Gillardeaux V., Galli T. (2009) EMBO Rep. 10, 1117–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukuda M., Mikoshiba K. (1999) J. Biol. Chem. 274, 31428–31434 [DOI] [PubMed] [Google Scholar]
- 23. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989) Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 24. Fukuda M., Kanno E., Mikoshiba K. (1999) J. Biol. Chem. 274, 31421–31427 [DOI] [PubMed] [Google Scholar]
- 25. James P., Halladay J., Craig E. A. (1996) Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukuda M., Kojima T., Aruga J., Niinobe M., Mikoshiba K. (1995) J. Biol. Chem. 270, 26523–26527 [DOI] [PubMed] [Google Scholar]
- 27. Itoh T., Fujita N., Kanno E., Yamamoto A., Yoshimori T., Fukuda M. (2008) Mol. Biol. Cell 19, 2916–2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fukuda M., Kanno E. (2005) Methods Enzymol. 403, 445–457 [DOI] [PubMed] [Google Scholar]
- 29. Fukuda M., Kanno E., Ishibashi K., Itoh T. (2008) Mol. Cell. Proteomics 7, 1031–1042 [DOI] [PubMed] [Google Scholar]
- 30. Bennett D. C., Cooper P. J., Hart I. R. (1987) Int. J. Cancer 39, 414–418 [DOI] [PubMed] [Google Scholar]
- 31. Kuroda T. S., Ariga H., Fukuda M. (2003) Mol. Cell. Biol. 23, 5245–5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Itoh T., Satoh M., Kanno E., Fukuda M. (2006) Genes Cells 11, 1023–1037 [DOI] [PubMed] [Google Scholar]
- 33. Ostermeier C., Brunger A. T. (1999) Cell 96, 363–374 [DOI] [PubMed] [Google Scholar]
- 34. Fukuda M. (2002) J. Biol. Chem. 277, 40118–40124 [DOI] [PubMed] [Google Scholar]
- 35. Fukuda M. (2003) J. Biol. Chem. 278, 15373–15380 [DOI] [PubMed] [Google Scholar]
- 36. Kukimoto-Niino M., Sakamoto A., Kanno E., Hanawa-Suetsugu K., Terada T., Shirouzu M., Fukuda M., Yokoyama S. (2008) Structure 16, 1478–1490 [DOI] [PubMed] [Google Scholar]
- 37. Itzen A., Goody R. S. (2008) Structure 16, 1437–1439 [DOI] [PubMed] [Google Scholar]
- 38. Johansson M., Rocha N., Zwart W., Jordens I., Janssen L., Kuijl C., Olkkonen V. M., Neefjes J. (2007) J. Cell Biol. 176, 459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanno E., Ishibashi K., Kobayashi H., Matsui T., Ohbayashi N., Fukuda M. (2010) Traffic 11, 491–507 [DOI] [PubMed] [Google Scholar]
- 40. Delprato A., Merithew E., Lambright D. G. (2004) Cell 118, 607–617 [DOI] [PubMed] [Google Scholar]
- 41. Delprato A., Lambright D. G. (2007) Nat. Struct. Mol. Biol. 14, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chaineau M., Danglot L., Galli T. (2009) FEBS Lett. 583, 3817–3826 [DOI] [PubMed] [Google Scholar]
- 43. Chi A., Valencia J. C., Hu Z. Z., Watabe H., Yamaguchi H., Mangini N. J., Huang H., Canfield V. A., Cheng K. C., Yang F., Abe R., Yamagishi S., Shabanowitz J., Hearing V. J., Wu C., Appella E., Hunt D. F. (2006) J. Proteome Res. 5, 3135–3144 [DOI] [PubMed] [Google Scholar]
- 44. Höning S., Sandoval I. V., von Figura K. (1998) EMBO J. 17, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinez-Arca S., Rudge R., Vacca M., Raposo G., Camonis J., Proux-Gillardeaux V., Daviet L., Formstecher E., Hamburger A., Filippini F., D'Esposito M., Galli T. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9011–9016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watabe H., Valencia J. C., Yasumoto K., Kushimoto T., Ando H., Muller J., Vieira W. D., Mizoguchi M., Appella E., Hearing V. J. (2004) J. Biol. Chem. 279, 7971–7981 [DOI] [PubMed] [Google Scholar]
- 47. Ando H., Watabe H., Valencia J. C., Yasumoto K., Furumura M., Funasaka Y., Oka M., Ichihashi M., Hearing V. J. (2004) J. Biol. Chem. 279, 15427–15433 [DOI] [PubMed] [Google Scholar]
- 48. Kageyama A., Oka M., Okada T., Nakamura S., Ueyama T., Saito N., Hearing V. J., Ichihashi M., Nishigori C. (2004) J. Biol. Chem. 279, 27774–27780 [DOI] [PubMed] [Google Scholar]
- 49. Ando H., Ichihashi M., Hearing V. J. (2009) Int. J. Mol. Sci. 10, 4428–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.