

Abstract
T cell receptor (TCR) ligation induces increased diacylglycerol and Ca2+ levels in T cells, and both secondary messengers are crucial for TCR-induced nuclear factor of activated T cells (NF-AT) and NF-κB signaling pathways. One prominent calcium-dependent enzyme involved in the regulation of NF-AT and NF-κB signaling pathways is the protein phosphatase calcineurin. However, in contrast to NF-AT, which is directly dephosphorylated by calcineurin, the molecular basis of the calcium-calcineurin dependence of the TCR-induced NF-κB activity remains largely unknown. Here, we demonstrate that calcineurin regulates TCR-induced NF-κB activity by controlling the formation of a protein complex composed of Carma1, Bcl10, and Malt1 (CBM complex). For instance, increased calcium levels induced by ionomycin or thapsigargin augmented the phorbol 12-myristate 13-acetate-induced formation of the CBM complex and activation of NF-κB, whereas removal of calcium by the calcium chelator EGTA-acetoxymethyl ester (AM) attenuated both processes. Furthermore, inhibition of the calcium-dependent phosphatase calcineurin with the immunosuppressive agent cyclosporin A (CsA) or FK506 as well as siRNA-mediated knockdown of calcineurin A strongly affected the PMA + ionomycin- or anti-CD3 + CD28-induced CBM complex assembly. Mechanistically, the positive effect of calcineurin on the CBM complex formation seems to be linked to a dephosphorylation of Bcl10. For instance, Bcl10 was found to be hyperphosphorylated in Jurkat T cells upon treatment with CsA or EGTA-AM, and calcineurin dephosphorylated Bcl10 in vivo and in vitro. Furthermore, we show here that calcineurin A interacts with the CBM complex. In summary, the evidence provided here argues for a previously unanticipated role of calcineurin in CBM complex formation as a molecular basis of the inhibitory function of CsA or FK506 on TCR-induced NF-κB activity.
Keywords: Calcineurin, Immunosupressor, NF-kappaB, Phosphatase, T Cell Receptor, Bcl10, T Cells, Phosphorylation
Introduction
Upon engagement of the T cell receptor (TCR),2 phospholipase Cγ1 (PLCγ1) is induced, which in turn hydrolyzes phosphoinositol 4,5-bisphosphate, resulting in elevated levels of diacylglycerol and inositol trisphosphate. Diacylglycerol is crucial for the activation of members of the PKC family, with PKCϴ being the key PKC family member required for the activation of AP1, NF-AT, and NF-κB signaling pathways in T cells. The other product generated by phosphoinositol 4,5-bisphosphate hydrolysis, inositol trisphosphate, is an inducer of calcium influx into the cytoplasm of T cells by a process known as store-operated calcium entry. After an initial inositol trisphosphate-induced depletion of the calcium stored in the endoplasmic reticulum, CRAC channels at the plasma membrane are opened in a STIM-dependent fashion, leading to a more sustained calcium influx (1). Calcium acts as a second messenger in the T cell necessary for the activation of several signaling molecules, including the protein kinases CaMKII and PKCα as well as the protein phosphatase calcineurin, which in turn are crucial for the activation of the transcription factors NF-AT, AP1, CREB1, and NF-κB. Especially calcineurin has been demonstrated to be crucial for the activation of the AP-1, NF-AT, and NF-κB signaling pathways (1–4). In contrast to the NF-AT pathway, however, which is activated due to a direct dephosphorylation of the NF-AT transcription factors by the calcium-dependent protein phosphatase calcineurin, the molecular mechanisms by which calcineurin affects the NF-κB pathway is far less understood.
The transcription factor NF-κB is crucial for development, survival, homoeostasis, and function of T lymphocytes. One essential step in the antigen receptor-induced NF-κB signaling pathway is the formation of the CBM complex, which is composed of Carma1, Bcl10, and Malt1 (5). Whereas Bcl10 and Malt1 interact constitutively, Carma1 is recruited to this Bcl10-Malt1 complex in an inducible manner, depending on site-specific phosphorylation of Carma1. Besides PKCϴ, which seems to be the main Carma1 kinase following TCR engagement, also HPK1 and IKK2 have been reported to phosphorylate Carma1, albeit at different serine residues (6–8). Following CBM complex formation, Malt1 is ubiquitinated by TRAF6, and this Lys63-conjugated ubiquitin chain functions as an anchor for the recruitment of the IκB-kinase complex, which is subsequently activated by a still unknown mechanism (9, 10). The activated IκB-kinase complex phosphorylates IκB proteins and thereby targets these proteins for proteasomal degradation. NF-κB is subsequently liberated and translocates to the nucleus, where it supports the expression of various NF-κB target genes. Finally, TCR-induced NF-κB signaling is terminated by the phosphorylation-dependent proteasomal degradation of Carma1 and Bcl10 (5, 11, 12). The calcium-dependent phosphatase calcineurin is composed of a catalytically active A subunit (CnA) of ∼61 kDa and a regulatory B subunit (CnB) of 19 kDa. In its inactive state, the active site of calcineurin is blocked by a carboxyl-terminal localized autoinhibitory domain (1, 13). For its activation, calcineurin requires the binding of the CnB subunit as well as the calcium-calmodulin complex. The binding of the regulatory proteins causes a structural alteration of CnA and a release of the autoinhibitory domain. Immunosuppressive drugs like cyclosporin A (CsA) and tacrolimus (FK506) target calcineurin and impair the activation of key signaling pathways in lymphoid cells. For instance, pretreatment of T cells with pharmacological inhibitors specific for calcineurin, like CsA or FK506, led to a marked reduction of the TCR-induced NF-AT, JNK, and NF-κB activity (2–4). However, although calcineurin dependence of the TCR-induced NF-κB signaling pathway has been known for over a decade, the molecular mechanism by which this calcium-dependent protein phosphatase regulates NF-κB has not been clarified.
Here, we demonstrate that calcium influx is essential for the formation of the CBM complex in activated T cells. Furthermore, we provide evidence for a crucial role of the calcium-dependent protein phosphatase calcineurin for the assembly of the CBM complex and NF-κB activation. Blocking calcineurin activity with CsA or FK506 or suppression of the catalytic calcineurin A subunit by siRNA led to a drastic decrease in CBM complex formation. Moreover, calcineurin constitutively interacts with components of the CBM complex like Malt1, and inhibition of calcineurin augmented the basal Bcl10 phosphorylation in Jurkat T cells. Furthermore, basal Bcl10 phosphorylation was diminished by calcineurin coexpression in HEK293 cells. In conclusion, our data imply that calcineurin regulates the TCR-induced NF-κB signaling pathway by controlling the CBM complex assembly, most likely by the removal of inhibitory phosphate groups from the CBM complex component Bcl10.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, and siRNA-mediated Knockdown
Jurkat T cells and Jur4 cells were cultivated using RPMI supplemented with 10% FCS, glutamine, penicillin, and streptomycin. HEK293 cells were kept in DMEM supplemented with 10% fetal bovine serum, penicillin (50 units/ml), and streptomycin (50 μg/ml). Primary mouse T lymphocytes were isolated from lymph nodes and spleens of 6-week-old BL6 mice using the CD4+/CD8+ T cell isolation kit from Dynal according to the manufacturer's protocol. For the stimulation of T cells, 50 ng/ml PMA and 500 ng/ml ionomycin were used. HEK293 cells were transfected using the CaPO4 transfection method. In brief, cells were plated the day before transfection to obtain a cell density of about 50% at the day of transfection. For the transfection of one well of a 6-well plate, 1–2.5 μg of DNA was mixed with 90 μl of sterile water and 10 μl of a 2 m CaCl2 solution. After incubation for 5 min, 100 μl of 2× HeBS buffer was added, mixed, and incubated at room temperature for an additional 5 min. Subsequently, the transfection sample was added to the cells. For the suppression of calcineurin A isoforms by siRNA, Jurkat T cells were transfected with either 100 nm control siRNA or the SMARTpool siRNAs for CnAα or CnAβ as indicated, using the Nucleofection Kit V (Amaxa/Lonza). If both CnA isoforms were suppressed, a 75 nm concentration of each SMARTpool siRNA was used. The cells were subsequently incubated for 72 h prior to analysis. Jurkat T cells were transfected using the Lipofectamine LTX +PLUS reagent (Invitrogen) according to the manufacturer's protocol.
Real-time PCR Analysis
RNA from 2 × 106 cells/sample was isolated using the RNeasy Protect kit (Qiagen), and cDNA was synthesized using an avian myeloblastosis virus reverse transcriptase kit (Invitrogen). Real-time PCR analysis to determine interleukin-2 (IL2) expression levels was performed using the SYBR Green kit from Qiagen in a Lightcycler 480 system (Roche Applied Science). All measurements were performed in triplicate, and the expression of IL2 was normalized to β-actin expression. The PMA plus ionomycin (P+I)-induced increase in IL2 expression was determined using the ΔΔCp method.
Antibodies, Plasmids, and Reagents
Agonistic anti-human CD3 and anti-human CD28 antibodies were isolated from hybridoma supernatants kindly provided by Dr. Rüdiger Arnold (Deutsches Krebsforschungszentrum, Heidelberg, Germany). Goat anti-Bcl10 (sc-9560), rabbit anti-Bcl10 (sc-5611), rabbit anti-Card11 (sc-48737), rabbit anti-ERK2 (sc-154), rabbit anti-HA (sc-805), goat anti-IκBα, rabbit anti-IKKγ (sc-8330), rabbit-anti-IKK2 (sc-7607), and rabbit anti-MALT1 (sc-28246) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and rabbit anti-IκBα (44D4, catalog no. 4812) antibody was purchased from Cell Signaling. Phospho-specific antibodies for pIκBα, pCaMKII, pPKCα/βII (Thr638/641), pPKCϴ (Thr538), and pan-pPKC were purchased from Cell Signaling. Anti-FLAG M2 affinity gel (A2220) and anti-FLAG M5 antibody were purchased from Sigma. Antibodies specifically recognizing calcineurin A were obtained from BD Pharmingen (catalog no. 556350) and Stressgen (catalog no. SPA-610), and an antibody for CARD11 was from Cell Signaling (catalog no. 4435). PMA, FK506, thapsigargin, and ionomycin were purchased from Sigma-Aldrich, and EGTA-AM was from Invitrogen. CsA was obtained from Fluka. Expression vectors encoding FLAG-Bcl10WT, FLAG-Bcl10S5A, HA-Bcl10, HA-Carma1, and Myc-MALT1 were described previously (10, 14, 15) as well as the vector coding for Xpress-IKK2 (16). To create expression vectors for FLAG-CnAα or FLAG-CnAβ, the appropriate cDNA was amplified by PCR and was subsequently inserted into the BamHI and NotI sites of the pFLAG-CMV2 vector (Sigma-Aldrich). The constitutive active ΔCam mutant was inserted either into the EcoRI and BamHI sites of the pFLAG-CMV2 vector or the EcoRI and XhoI sites of a HA-pcDNA3.1 vector to generate FLAG-ΔCam and HA-ΔCam expression vectors, respectively. The inactive ΔCamH151Q mutant was generated by site-directed mutagenesis. Primer sequences are available upon request. The 3xκB luciferase reporter vector has been described previously.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting procedures were performed as described previously (17). In brief, 250–500 μg of protein extracts were mixed with 1 μg/sample of the appropriate antibody, and samples were incubated overnight at 4 °C with agitation. After incubation, 10 μl of a 50% protein G slurry was added, and the samples were further incubated for 1 h. Subsequently, the precipitates were washed extensively in TNT buffer (20 mm Tris, pH 8.0, 200 mm NaCl, 1% Triton X-100, 1 mm DTT, 50 mm NaF, 50 mm β-glycerophosphate, 50 μm leupeptin, 1 mm PMSF). The resulting immunopurified proteins were used for immunoblotting experiments. For the immunoblotting analysis, either the immunopurified protein complexes or, as indicated, 50–100 μg of a protein extract were loaded onto a standard SDS-polyacrylamide gel. SDS-PAGE and the transfer to nitrocellulose (Schleicher & Schuell) or nylon membranes (Immobilon PVDF membrane, Millipore) were performed using standard protocols. The membrane was blocked with 5% milk powder in TBS + Tween 20 prior to the incubation with the primary antibody (1:1000 in TBS + Tween 20), subsequently washed three times for 5 min each, and incubated in a TBS-Tween 20 solution containing either horseradish peroxidase-conjugated or IRDye700/800-conjugated secondary antibody (1:5000). The detection was performed using either ECL substrates from Amersham Biosciences or the Odyssey infrared scanning system (LICOR).
In Vitro Kinase Assay and in Vivo Phosphorylation Studies
For the in vitro kinase assays, the IKK complex was purified from untreated or P+I-stimulated Jurkat T cells with 1 μg of anti-NEMO antibody. Resulting immunocomplexes were washed extensively with TNT and finally with kinase assay buffer to equilibrate the samples. The kinase reaction was performed at 30 °C for 30 min after adding 10 μCi of [γ-32P]ATP and 0.5 μg of a bacterial expressed GST-IκBα (amino acids 1–53) fusion protein in kinase reaction buffer. Samples were subsequently washed extensively with TNT buffer and PBS prior to a separation by SDS-PAGE. The separated proteins were transferred to nitrocellulose membrane, and the phosphorylation was monitored by autoradiography. For the in vivo phosphorylation studies, 2 × 107 Jurkat T cells were incubated for 18 h in phosphate-free DMEM with 5% dialyzed calf serum prior to incubation with 2 mCi/ml [32P]orthophosphate for a further 6 h. For in vivo phosphorylation studies using HEK293 cells, the cells were kept in phosphate-free medium, including dialyzed FCS, for 1 h prior to the addition of [32P]orthophosphate and a further incubation for 2 h. The cells were treated as indicated and lysed in TNT, and the resulting extracts were subjected to an immunoprecipitation analysis. Precipitated proteins were separated by SDS-PAGE and transferred to nitrocellulose membrane, and the resulting membrane was used for autoradiography to monitor the phosphorylation and subsequently subjected to immunoblot analysis.
In Vitro Phosphatase Assay
For the in vitro phosphatase assay, 3 μg of purified GST-Bcl10 were subjected to a kinase assay reaction either with or without FLAG-tagged IKK2 to generate phospho-Bcl10. For this, HEK 293 cells were transiently transfected with expression vectors encoding either FLAG-IKK2 or only the FLAG epitope. The ectopically expressed proteins were purified by immunoprecipitation using anti-FLAG beads (M2, Sigma), washed extensively with TNT buffer and kinase buffer to remove contaminating proteins, and finally included in an in vitro kinase assay with soluble GST-Bcl10 following the standard kinase assay protocol as described above. After the kinase reaction, soluble GST-Bcl10 was transferred to a new Eppendorf tube, and the GST-Bcl10 proteins were pulled down using GSH-Sepharose (Sigma) and washed twice with TNT buffer and once with phosphatase assay buffer (100 mm HEPES, pH 7.4, 100 mm NaCl, 20 mm potassium acetate, 2 mm magnesium acetate, 2 mm CaCl2, 2 mm MnCl2). Finally, GSH-coupled GST-Bcl10 was subjected to a phosphatase reaction, including either only calmodulin (catalog no. 208690, Merck) or additionally recombinant calcineurin A-calcineurin B heterodimers (catalog no. BML-SE163-5000, Enzo Life Sciences). The reaction was performed for 1 h at 30 °C, and the protein-coupled beads were washed twice with PBS prior to a standard SDS-PAGE. Phospho-GST-Bcl10 was monitored by autoradiography after transfer of the proteins to a nitrocellulose membrane. To ensure equal loading of GST-Bcl10, the membrane was subjected to a standard Bcl10 immunoblot assay.
Luciferase Reporter Assay
For the reporter gene assays, a Jurkat T cell clone was used, which was stably transfected with a luciferase reporter gene under the control of a multimerized κB binding site (Jur4 cells). Cells were treated with the individual reagents as indicated, and luciferase activity was generally estimated after 6 h of treatment. Luciferase values were normalized for protein concentration (relative luciferase units/μg of protein). The experiments were done in duplicates and were repeated at least three times with similar results.
Gel Shift Analysis
For gel shift analysis (EMSA), 5 μg of nuclear proteins or whole cell extracts (DignamC extracts) from untreated or stimulated cells were incubated on ice for 20 min in a reaction containing 0.3 ng of 32P-labeled κB-specific or Oct-specific oligonucleotide, 1 μg of poly(dI:dC), and 3 μl of a binding buffer. The samples were separated on a native 5% polyacrylamide gel, and the gel was dried and subjected to autoradiography.
Determination of Relative Carma1-Bcl10 Complex Formation
To estimate the relative amount of Carma1 bound to Bcl10, autoradiographs of the appropriate immunoblots were scanned, and resulting TIFF files were analyzed by densitometry using the ImageJ software. The intensities of the Carma1 and Bcl10 signals were determined, and background intensity was subtracted. Subsequently, the ratio of Carma1 signal to Bcl10 signal was calculated, and Carma1-Bcl10 complex formation relative to the control transfection was determined. The value calculated with the control transfection was set arbitrary to 100%. A similar procedure was applied for the estimation of NF-κB activity in siRNA-transfected Jurkat T cells measured by EMSA.
RESULTS
Calcium Influx Is Crucial for the Assembly of the CBM Complex
A critical role of increased cellular calcium levels for the TCR- or phorbol ester-induced NF-κB signaling pathway has been reported previously. This augmentative effect of increased intracellular calcium levels is reflected in an augmented IκBα degradation in cells stimulated with a combination of PMA and ionomycin when compared with cells stimulated with PMA alone (Fig. 1A). Although an IκBα-degradation is detectable in both cases, the onset of IκBα degradation is much faster in cells additionally treated with ionomycin (compare lanes 2–4 with lanes 5–7). To unravel the molecular mechanism underlying the enhancing effect of calcium on PMA-induced NF-κB activity, we set out to analyze the impact of elevated cellular calcium levels on upstream signaling events. One crucial step in the TCR-induced NF-κB signaling pathway is the formation of a multisubunit protein complex consisting of Carma1, Bcl10, and Malt1 (the CBM complex). Because TCR engagement already causes calcium entry, we used the calcium ionophore ionomycin in combination with the phorbol ester PMA to dissect a potential effect of calcium on CBM complex formation. Jurkat T cells were stimulated with either ionomycin or PMA separately or in combination prior to the analysis of the CBM complex. As expected, the Bcl10-Malt1 interaction remained largely unaltered upon stimulation with either PMA or ionomycin or with a combination of both agents (Fig. 1B, top). In contrast, Carma1 recruitment to Bcl10 was moderately enhanced in Jurkat T cells stimulated with PMA alone (Fig. 1B, compare lane 1 with lanes 4 and 5). A combined treatment with PMA and the calcium ionophore ionomycin strongly augmented the Carma1 interaction with Bcl10 (Fig. 1B, lanes 2 and 3), whereas treatment of the cells with ionomycin alone had no significant effect on the Carma1-Bcl10 interaction (Fig. 1B, lanes 6 and 7). Similar effects were observed in experiments with the A3.01 T cell line (data not shown). The observed effect of PMA and/or ionomycin on the Carma1-Bcl10 interaction strongly correlated with the NF-κB activity, as monitored by the degree of IκBα degradation (Fig. 1, A and B, bottom) or by a reporter analysis (Fig. 1C) using Jurkat T cells stably transfected with a luciferase gene under the control of a multimerized NF-κB binding site (Jur4 cells). Consistently, ionomycin co-treatment also augmented Bcl10 degradation, which is a feature of activated T cells, when compared with PMA treatment alone (Fig. 1B, top, lanes 2–5). In contrast, no significant Bcl10 degradation was observed in T cells treated with ionomycin alone (Fig. 1B, top, lanes 6 and 7).
FIGURE 1.

Calcium augments the PMA-induced formation of the CBM complex. A, Jurkat T cells were stimulated with either PMA alone (50 ng/ml) or with a combination of PMA plus ionomycin for the indicated time points. Subsequently, anti-IκBα and anti-ERK2 immunoblots were performed. n.s., nonspecific. B, Bcl10 was immunoprecipitated (IP) from TNT lysates (500 μg of protein/sample) of untreated or P+I-stimulated Jurkat T cells (50 ng/ml PMA, 500 ng/ml ionomycin). The resulting immunopurified protein complexes were subjected to immunoblot analyses (IB) with the indicated antibodies. To determine the cellular levels of the CBM components, control immunoblot analyses were performed with the indicated antibodies. C, NF-κB activity upon PMA, ionomycin, or thapsigargin stimulation for 24 h was monitored by determining the luciferase activity in stably transfected Jur4 cells. All stimulations were performed in duplicate, and the mean value and S.E. (error bars) are depicted. D, anti-Bcl10 immunoprecipitation analysis using whole cell extracts from unstimulated Jurkat T cells or from Jurkat T cells pretreated with the indicated concentrations of thapsigargin (60 min) and subsequently stimulated with PMA or PMA + ionomycin for the stated times.
Because these results suggest an important role of elevated cellular calcium levels for the formation of the CBM complex, we next thought to use an alternative agent to increase cellular calcium levels in order to verify the observed effects. Thapsigargin, a sesquiterpene lactone from the plant Thapsia garganica, is known to block the Ca2+-ATPase at the endoplasmic reticulum, thus leading to increased calcium levels in the cytoplasm (18). Indeed, pretreatment of Jurkat T cells with thapsigargin (50–500 nm) augmented the PMA-induced Carma1-Bcl10 interaction drastically (Fig. 1D, compare lanes 2 and 3 with lanes 4 and 5), similar to the levels seen with PMA + ionomycin stimulation (Fig. 1D, lanes 10 and 11). Again, we observed an enhanced Bcl10 degradation (Fig. 1D) upon thapsigargin pretreatment in comparison with PMA stimulation alone. By contrast, neither a CBM complex formation nor a Bcl10 degradation was observed when cells were treated with thapsigargin alone (supplemental Fig. S1). Of note, thapsigargin pretreatment led also to an electromobility shift of the Bcl10-bound Malt1, which most likely represents the previously reported ubiquitinated Malt1 (Fig. 1D, top). Furthermore, the cumulative effect of the thapsigargin-induced calcium influx on NF-κB activity was also seen in luciferase reporter assays using Jur4 cells (Fig. 1C). Again, thapsigargin augmented the PMA-induced NF-κB activity similar to ionomycin. Although treatment with thapsigargin alone had no effect on the Carma1-Bcl10 interaction, similar to the situation upon ionomycin treatment (Fig. 1B), a slight NF-κB activation was observed in NF-κB-dependent reporter gene assays using either thapsigargin or ionomycin to increase cellular calcium levels (Fig. 1C).
Calcium Withdrawal Attenuates PMA + Ionomycin-induced CBM Complex Assembly and NF-κB Activation
To further support our findings regarding the crucial role of calcium for the CBM complex formation and NF-κB activation, we next conducted a set of reciprocal experiments by using the cell-permeable calcium chelator EGTA-AM. Pretreatment of cells with EGTA-AM (50 and 100 μm) had a profound effect on the PMA + ionomycin-induced NF-κB activation as measured by EMSA, NF-κB-dependent reporter gene assay, and IκBα degradation (Fig. 2, A and C). However, this negative effect is stimulus-specific because the EGTA-AM pretreatment only impaired the NF-κB activity upon PMA or P+I stimulation and not upon TNF-α stimulation (Fig. 2, B and C). In accordance with the results obtained with ionomycin or thapsigargin cotreatment, we observed a drastic reduction in Carma1-Bcl10 interaction by pretreating the cells with EGTA-AM prior to stimulation with PMA + ionomycin (Fig. 2D, top). Taken together, these results clearly suggest that the molecular basis of the calcium impact on the NF-κB signaling pathway is the modulation of the CBM complex formation.
FIGURE 2.

Signal-specific attenuation of NF-κB activity by the calcium chelator EGTA-AM. A, Jurkat T cells were either left untreated or were pretreated with the indicated concentrations of EGTA-AM for 30 min prior to stimulation with PMA + ionomycin as indicated. The resulting whole cell extracts were used for EMSA experiments with 32P-labeled NF-κB-specific or Oct-specific oligonucleotides. Additionally, immunoblot analyses (IB) were performed using the indicated antibodies. n.s., nonspecific. B, a similar experiment was performed using Jurkat T cells that were either left untreated or pretreated with 100 μm EGTA-AM and subsequently stimulated with PMA + ionomycin, PMA, or TNF-α, as indicated. C, Jur4 cells were either left untreated or pretreated with increasing concentrations of EGTA-AM (50 and 100 μm) for 30 min prior to a stimulation with PMA, PMA + ionomycin, or TNF-α (10 ng/ml). After 6 h, the cells were lysed, and the luciferase activity was determined. Experiments were done in duplicate, and the mean value and the S.E. (error bars) are depicted. D, Jurkat T cells were either left untreated or pretreated with 100 μm EGTA-AM for 30 min prior to the stimulation with PMA + ionomycin. Whole cell extracts were prepared and subjected to an anti-Bcl10 immunoprecipitation (IP). 50 μg of the samples were probed in control Western blot analyses with antibodies specifically recognizing IκBα or Carma1.
The Calcium-dependent Protein Phosphatase Calcineurin Is Required for CBM Complex Formation
To explore the potential role of calcineurin as mediator of the calcium effect on the NF-κB activity and especially the CBM complex formation, we first analyzed the P+I-induced NF-κB activity in Jurkat T cells alone or upon pretreatment with CsA, a well known pharmacological inhibitor of calcineurin. As described previously (3), the P+I-induced NF-κB activity was dramatically attenuated by CsA pretreatment as monitored by EMSA and IκBα degradation (Fig. 3B) or NF-κB-dependent luciferase reporter assays (Fig. 3A). Interestingly, CsA pretreatment impaired not only the NF-κB activity induced by P+I but also that induced by PMA or ionomycin alone (Fig. 3A). Furthermore, pretreatment of Jurkat T cells with CsA or FK506, another pharmacological calcineurin inhibitor, impaired the activity of the IκB-kinase complex as monitored by an in vitro kinase assay (Fig. 3C). To determine whether the observed negative effect of the calcineurin inhibitors is based on reduced CBM complex formation, we analyzed the Carma1-Bcl10 interaction in Jurkat T cells that were either stimulated with P+I alone or additionally pretreated with CsA (Fig. 3D). In accordance with the results shown in Figs. 1 and 2, we observed a marked reduction in the Carma1-Bcl10 interaction by pretreatment with CsA (Fig. 3D, compare lanes 2–4 with lanes 5–7) or with FK506 (data not shown), which was accompanied by an attenuated Bcl10 degradation in CsA-pretreated Jurkat cells (Fig. 3D). In addition, the appearance of a slower migrating Bcl10 signal was only observed with P+I-stimulated cells and not with CsA-pretreated cells (Fig. 3D, top, compare lanes 3 and 4 with lanes 6 and 7). Because this Bcl10 band shift results most likely from an IKK2-mediated Bcl10 hyperphosphorylation, which depends on a prior CBM complex assembly (14), we concluded that calcineurin is indeed crucial for the CBM complex formation. A similar effect of CsA on Carma1-Bcl10 interaction as well as Bcl10 degradation was observed in primary murine T lymphocytes (Fig. 3E). To further support our findings regarding the inhibitory effect of calcium withdrawal or calcineurin inhibition on CBM complex assembly, Jurkat T cells were pretreated with EGTA-AM or CsA prior to stimulation with agonistic anti-CD3 and anti-CD28 antibodies. Again, both inhibitors led to a distinct reduction in signal-induced Carma1-Bcl10 interaction (Fig. 3F). To determine whether calcineurin inhibition by CsA would impair other signaling pathways, including calcium-dependent signaling pathways, in PMA + ionomycin-stimulated Jurkat T cells, the activation status of different PKCs as well as CaMKII was determined by using phospho-specific antibodies for the activated isoforms of these kinases (Fig. 4). As expected, CsA pretreatment caused a significant decrease in IκBα phosphorylation (Fig. 4A, compare lanes 3 and 4 with lanes 7 and 8). Moreover, CsA pretreatment also caused a moderate reduction in p65 phosphorylation at Ser536 (Fig. 4C). However, the phosphorylation pattern of CaMKII showed only a slight reduction after CsA pretreatment (Fig. 4B) (data not shown). In contrast, we detected a moderate increase in PKCϴ phosphorylation, whereas the phosphorylation levels measured by pPKCα and pan-phospho-PKC antibodies showed, if anything, only a weak increase and no reduction. Collectively, the differences seen in the phosphorylation patterns of the different PKC isoforms or of CaMKII cannot explain the reduced CBM complex formation observed in CsA-pretreated cells.
FIGURE 3.

Negative effect of cyclosporin A and FK506 on TCR-induced NF-κB activity and CBM complex formation. A, Jur4 cells were pretreated with CsA (200 ng/ml, 1 h) and stimulated with P+I, PMA, or ionomycin (6 h) prior to estimation of luciferase activity. The mean value and S.E. (error bars) of two independently performed experiments are depicted. B, EMSA experiments using NF-κB- or Oct-specific probes in combination with whole cell extracts from untreated, CsA-pretreated (200 ng/ml), or P+I-stimulated Jurkat T cells (top). The same extracts were used for immunoblot analyses (IB) with antibodies recognizing IκBα, phospho-p65 (Ser536), and ERK2 (bottom). C, the IKK complex was immunopurified from Jurkat T cells pretreated with CsA or FK506 prior to P+I stimulation using an anti-NEMO antibody, and the kinase activity (KA) was estimated. Phosphorylation was determined by autoradiography. IκBα degradation was determined by an additional immunoblot. D, anti-Bcl10 immunoprecipitation analysis (IP) (top) of whole cell extracts from Jurkat T cells pretreated with CsA (200 ng/ml) for 1 h. Selected samples were additionally stimulated with P+I as indicated. A fraction of the whole cell extracts (50 μg/sample) was subjected to control immunoblot analyses (bottom). E, a similar anti-Bcl10 immunoprecipitation analysis as described in D was performed using primary murine T cells. F, Jurkat T cells were pretreated with CsA (200 ng/ml) or EGTA-AM (100 μm) for 1 h prior to the stimulation with agonistic anti-CD3 (1 μg/ml, plate-bound) and anti-CD28 (5 μg/ml, soluble). Subsequently, an anti-Bcl10 immunoprecipitation analysis was performed (top). To monitor the cellular expression levels of the analyzed proteins, additional immunoblot analyses were performed (bottom).
FIGURE 4.
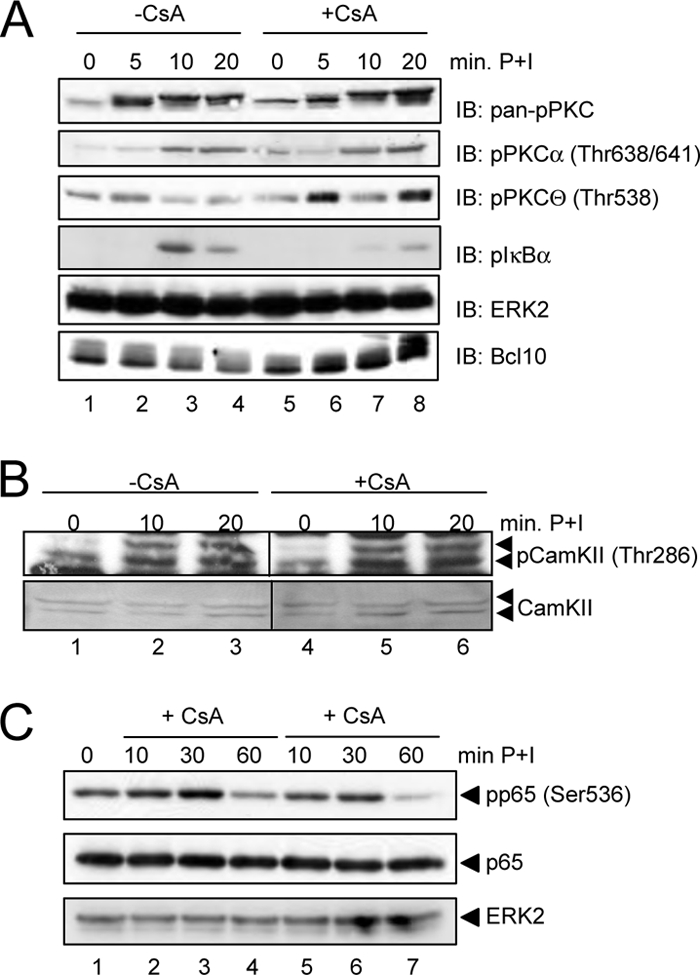
Analysis of the CsA effect on different signaling pathways. A, Jurkat T cells were either left untreated or pretreated with CsA (200 ng/ml) prior to stimulation with PMA + ionomycin. Resulting whole cell extracts were subjected to immunoblot analysis (IB) with the indicated phospho-specific antibodies to monitor the activation of PKC proteins, especially PKCϴ and PKCα, and phospho-IκBα. B, to monitor the activation status of CaMKII, cytoplasmic extracts also used in Fig. 3B were analyzed with anti-phospho-CaMKII and anti-CaMKII antibodies. C, cytoplasmic extracts from Jurkat T cells either left untreated or pretreated with CsA for 30 min prior to P+I stimulation were analyzed with the phospho-p65-specific antibody.
Reduced NF-κB Activation and CBM Complex Formation by siRNA-mediated Suppression of Calcineurin A
To ensure that the negative effects of CsA and FK506 on Carma1-Bcl10 interaction as well as NF-κB activation depend on their inhibitory effect on calcineurin, siRNAs specific for the two isoforms of the catalytic subunit CnA, CnAα and CnAβ, were used to suppress their expression in Jurkat T cells. Both isoforms are expressed in Jurkat T cells with nearly identical levels (Fig. 5A, lanes 1 and 2), and only the suppression of both catalytically active calcineurin isoforms led to a marked attenuation of the PMA + ionomycin-induced Carma1-Bcl10 interaction (Fig. 5A, compare lanes 1 and 2 with lanes 7 and 8; for a quantification, see Fig. 5A, right). Consistent with the results seen in the Carma1-Bcl10 interaction analysis, the NF-κB activity as monitored by EMSA was only reduced when both CnA isoforms were suppressed in combination (Fig. 5B). A similar effect was observed after siRNA-mediated suppression of both CnA isoforms in Jurkat T cells stimulated with agonistic anti-CD3 + CD28 antibodies (supplemental Fig. S2). IL2 expression is regulated by NF-AT, NF-κB, and AP1 transcription factors and is highly sensitive to CsA treatment (19). To estimate the degree of calcineurin inhibition by siRNA-mediated suppression of the CnA isoforms, quantitative PCR analysis (Fig. 5C, top) of untreated and P+I-stimulated Jurkat T cells transiently transfected with the different CnA-specific siRNAs were performed. Again, the combined suppression of both CnA isoforms had the strongest effect on P+I-induced IL2 expression, whereas the suppression of the individual CnA isoforms led to a gradual decrease in IL2 expression. Notably, parallel immunoblot analysis revealed that P+I stimulation induced calcineurin A expression (Fig. 5C, bottom), which suggests that a remaining calcineurin activity even in siRNA-transfected cells might be possible. However, in general, the effect of siRNA-mediated CnA knockdown on the IL2 expression reflected the degree of NF-κB and CBM complex inhibition.
FIGURE 5.

siRNA-mediated suppression of CnAα and CnAβ attenuates P+I-induced NF-κB activity. A, anti-Bcl10 immunoprecipitation analyses (IP) were performed using whole cell extracts from untreated or P+I-stimulated Jurkat T cells that were transfected with different siRNAs. Control immunoblot analyses (IB) were performed to determine the cellular levels of the indicated proteins. Two independent experiments were used for the quantification of the relative P+I-induced Carma1-Bcl10 interaction (bottom). Calculation was performed on the basis of the signal intensity measured by densitometry. B, Jurkat T cells transfected with the indicated siRNA were either left untreated or stimulated with PMA + ionomycin. Resulting whole cell extracts were subjected to EMSA experiments using NF-κB- or Oct-specific probes (top). To monitor the siRNA-mediated knockdown of CnA isoforms, additional immunoblot analyses were performed using the indicated antibodies (bottom). n.s., nonspecific. Relative NF-κB binding activity was determined by normalizing the background subtracted signals of the NF-κB-specific bands to the intensity of the Oct-specific signals. C, to estimate the functionality of the siRNA-mediated suppression of CnA isoforms, the IL2 expression in untreated or P+I-stimulated (8 h) Jurkat cells, which were transfected with the stated siRNAs, was determined by quantitative PCR analysis (top). All samples were measured in triplicate and normalized for β-actin expression. Protein extractions of transfected cells were done in parallel, and control immunoblots were performed with the indicated antibodies (bottom). Error bars, S.E.
Inhibition of Calcineurin by CsA or EGTA-AM Causes Increased Basal Phosphorylation of Bcl10
Having established a role of calcineurin for the PMA + ionomycin- or anti-CD3 + CD28-induced assembly of the CBM complex, we next aimed to identify the molecular mechanism(s) by which calcineurin affects the CBM complex formation. Because the CBM complex formation is highly regulated by the phosphorylation of Carma1 and Bcl10, an in vivo phosphorylation study was performed by labeling Jurkat T cells with [32P[orthophosphate prior to a treatment with either CsA or EGTA-AM alone or in conjunction with PMA + ionomycin stimulation (Fig. 6) to gain further insight into the effect of calcium-calcineurin on the phosphorylation status of Carma1, Malt1, and Bcl10. Subsequently, endogenous Bcl10, Malt1, or Carma1 were isolated by immunoprecipitation with the appropriate antibodies. If calcineurin affects the CBM complex formation by rendering the phosphorylation status of one of these CBM complex components, treatment with the calcineurin inhibitor CsA should increase the phosphorylation of the targeted protein. Indeed, pretreatment of Jurkat T cells with either CsA or EGTA-AM distinctively increased the basal Bcl10 phosphorylation (Fig. 6, top, lanes 1–3). By contrast, the PMA + ionomycin-induced increase of Bcl10 phosphorylation was not dramatically affected by CsA or EGTA-AM pretreatment (Fig. 6, top, lanes 4–6). Furthermore, neither CsA nor EGTA-AM pretreatment led to an increase in basal Carma1 phosphorylation; instead, a slight decrease was observed (Fig. 6, bottom, lanes 1–3). In cells stimulated with PMA + ionomycin, CsA and EGTA-AM either had no effect or led to a mild reduction in Carma1 phosphorylation (Fig. 6, bottom, lanes 4–6). In addition, the phosphorylation status of Malt1 remained unchanged upon treatment with CsA or EGTA-AM irrespective of PMA + ionomycin stimulation (data not shown).
FIGURE 6.
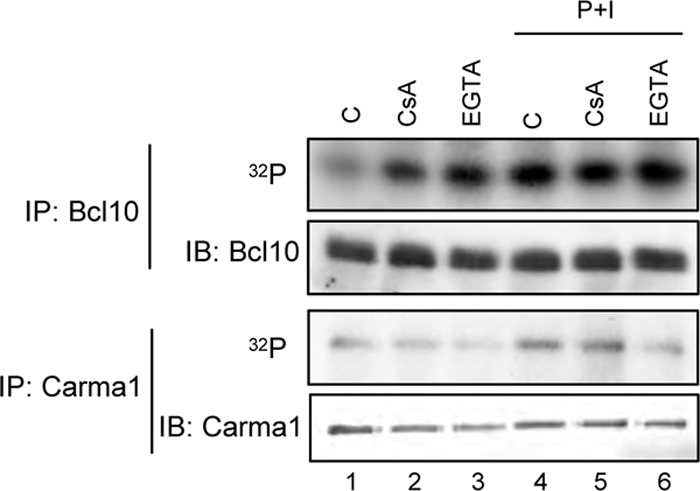
Altered phosphorylation status of Bcl10 upon EGTA-AM and CsA treatment in P+I-stimulated Jurkat T cells. Bcl10 or Carma1 proteins were immunoprecipitated (IP) using whole cell extracts from radiolabeled Jurkat T cells (100 μCi/ml [32P]orthophosphate, 6 h). Cells were either left untreated or treated with CsA (200 ng/ml) or EGTA-AM (100 μm) during the last 1 h of radiolabeling prior to a PMA + ionomycin stimulation for 10 min (lanes 4–6). IB, immunoblot.
The increased basal Bcl10 phosphorylation in Jurkat T cells upon CsA or EGTA-AM treatment points to the possibility that Bcl10 might be a substrate for the phosphatase activity of calcineurin. Bcl10 has been shown to be a target of different kinases, like CaMKII or IKK2. Important, however, is the notion that all described Bcl10 phosphorylations exert a negative effect on CBM complex formation and NF-κB activity (14, 20). To further analyze the potential role of calcineurin for Bcl10-dephosphorylation, Bcl10 was ectopically expressed in HEK293 cells either alone or in combination with IKK2 or calcineurin Aα. As expected, the coexpression of IKK2 led to a distinct increase in one slowly migrating Bcl10-specific signal (Fig. 7A, compare lanes 2 and 4, signal III), which resembles a IKK2-mediated Bcl10 phosphorylation (14, 20). This slower migrating signal, however, was reduced upon coexpression of calcineurin Aα (Fig. 7A, lanes 3 and 5), again arguing for a role of calcineurin in Bcl10 dephosphorylation. To prove that this slower migrating Bcl10 signal indeed represents a Bcl10 phosphoisoform, two different experimental strategies were employed. First, another in vivo phosphorylation analysis was performed using HEK293 cells cotransfected with FLAG-Bcl10 together with either a constitutive active ΔCam mutant or an inactive ΔCamH151Q mutant alone or in conjunction with Xpress-IKK2 prior to a labeling of the cells with [32P]orthophosphate. As shown in Fig. 7B, the coexpression of ΔCamWT, but not of ΔCamH151Q, led to a distinct reduction of the basal Bcl10 phosphorylation in HEK293 cells (Fig. 7B, lanes 2–4, reduced signals II and III). Coexpression of IKK2, on the other hand, augmented the Bcl10 phosphorylation, especially signal III. However, the addition of ΔCamWT caused again a decrease in the IKK2-mediated Bcl10 phosphorylation (Fig. 7B, lanes 5 and 6). As a second experimental setup to determine whether signal III represents a phosphoisoform of Bcl10, immunopurified exogenously expressed Bcl10 was treated with shrimp alkaline phosphatase, and the Bcl10 pattern was subsequently analyzed by immunoblot. As expected, coexpression of IKK2 further enhanced the appearance of the slowest migrating Bcl10 signal, whereas the coexpression of the constitutive active ΔCam mutant reduced this signal (Fig. 7C, lanes 6–8 and 10–12). Moreover, treatment of the immunopurified Bcl10 with shrimp alkaline phosphatase led to a strong reduction of the slower Bcl10 signal (signal III), further supporting the notion that this slower migrating Bcl10 signal represents a Bcl10 phosphoisoform (Fig. 6C, lanes 1–4).
FIGURE 7.

Calcineurin affects the phosphorylation status but not the stability of Bcl10 in HEK293 cells. A, immunoblot analyses using whole cell extracts from transiently transfected HEK293 cells with antibodies recognizing IKK2 (top), calcineurin A (middle), or Bcl10 (bottom). B, FLAG-Bcl10 was ectopically expressed in HEK293 cells either alone or in conjunction with HA-ΔCamWT, a catalytically inactive ΔCam mutant (ΔCamH151Q) or Xpress-IKK2, as indicated. Transfected cells were radiolabeled (100 μCi/ml [32P]orthophosphate, 2 h), and resulting whole cell extracts were subjected to anti-FLAG immunoprecipitation experiment. Phosphorylation was determined by autoradiography following an anti-Bcl10 immunoblot to ensure the successful precipitation. C, FLAG-Bcl10 was immunoprecipitated (IP) from transiently transfected HEK293 cells, and immunocomplexes were incubated at 37 °C for 1 h with shrimp alkaline phosphatase (SAP; 1 unit, lanes 1–4). Subsequently, the immunocomplexes were subjected to anti-Bcl10 immunoblot analysis (IB). Expression levels of the FLAG-Bcl10 (top, lanes 13–16), HA-ΔCam (middle), or Xpress-IKK2 (bottom) were determined by immunoblot analyses. D, Jurkat T cells were transiently transfected with expression vectors for FLAG-Bcl10WT, FLAG-Bcl10S5A, and FLAG-ΔCam, as indicated. The cells were either left untreated or were stimulated with P+I for 3 h prior to immunoblot analysis with the indicated antibodies. E, HEK293 cells transiently transfected with expression vectors for FLAG-Bcl10, HA-ΔCam, or Xpress-IKK2 were either left untreated or treated with cycloheximide (50 ng/ml) for different times prior to immunoblot analyses with antibodies for Bcl01, IKK2, or HA epitope. As a positive control for cycloheximide efficacy, c-Myc levels were analyzed.
The opposite effect of calcineurin and IKK2 on the phosphorylation pattern of Bcl10 implied that known IKK2 target sites in Bcl10 might be dephosphorylated by calcineurin. To prove this hypothesis and to test whether calcineurin affects the basal Bcl10 phosphorylation also in T cells, Jurkat T cells were transiently transfected with expression vectors for FLAG-Bcl10WT or a mutant of Bcl10 with serine to alanine substitutions at positions Ser134, Ser136, Ser138, Ser141, and Ser144, which are known IKK2 target sites (FLAG-Bcl10S5A). Subsequently, the cells were either left untreated or stimulated with P+I for 3 h to induce Bcl10 degradation. Inactivation of the five serine residues in Bcl10 caused a loss of two specific Bcl10 signals, which correspond to phosphoisoforms of Bcl10 (signals II and III, Fig. 7D, lanes 1 and 5). As expected, coexpression of ΔCam reduced the intensity of signal III and partially of signal II (compare lanes 1 and 2 with lanes 3 and 4). Interestingly, only signal I was decreased upon P+I stimulation irrespective of whether it was caused by Bcl10WT or Bcl10S5A. Together, these results suggest that calcineurin causes a dephosphorylation of Bcl10 also in Jurkat T cells, and the central serine residues at the positions Ser134, Ser136, Ser138, Ser141, and Ser144 are likely to be targets of the calcineurin-mediated Bcl10 dephosphorylation.
Phosphorylation of Bcl10, especially at Ser138, has been linked to a signal-induced degradation of Bcl10. Because pretreatment with CsA led to a partial stabilization of Bcl10 during PMA + ionomycin stimulation (Fig. 3D), we next analyzed whether IKK2-mediated phosphorylation or calcineurin-mediated dephosphorylation would affect the stability of Bcl10, especially the Bcl10 phosphoisoform (signal III). HEK293 cells were transfected with expression vectors either for FLAG-Bcl10 alone (Fig. 7E, lanes 1–4), together with ΔCam (lanes 5–8), with Xpress-IKK2 (lanes 9–12), or with a combination of ΔCam and Xpress-IKK2 (lanes 13–16). To monitor the stability of the ectopically expressed Bcl10 protein, the cells were either left untreated or were treated with cycloheximide for different times. As expected, coexpression of IKK2 or ΔCam altered the phosphorylation pattern of Bcl10. Whereas signal III was increased upon IKK2 coexpression, it was diminished in all ΔCam-expressing cells. Bcl10 appears to be a relatively stable protein in HEK293 cells because its levels remained stable upon 8 h of cycloheximide treatment, whereas endogenous c-Myc protein or HA-ΔCam showed a shorter half-life (Fig. 7E, lanes 1–8). Importantly, neither coexpression of IKK2 nor coexpression of ΔCam had a marked effect on the stability of exogenously expressed Bcl10, especially signal III. Thus, these results suggest that the stability of Bcl10 is not directly regulated by IKK2-mediated Bcl10 hyperphosphorylation or calcineurin-mediated Bcl10 dephosphorylation. To further determine whether the reduced phosphorylation of Bcl10 upon calcineurin coexpression in intact cells is indeed caused by a direct dephosphorylation of Bcl10 by calcineurin, we performed an in vitro phosphatase experiment using recombinant proteins (Fig. 8). GST-Bcl10 fusion protein was phosphorylated by IKK2 in vitro, and phospho-GST-Bcl10 was purified and subsequently subjected to an in vitro assay including either calmodulin alone or a combination of calmodulin with increasing amounts of the calcineurin A-calcineurin B heterodimer. Consistent with our previous results, we observed also in this in vitro phosphatase assay a distinct reduction of Bcl10 phosphorylation by the addition of active calcineurin (Fig. 8, compare lanes 1, 3, and 5). Taken together, these data imply that calcineurin can act as a Bcl10 phosphatase and open up the possibility that one of the molecular mechanisms by which calcineurin positively affects the CBM complex assembly is the removal of negative acting phosphate groups from Bcl10.
FIGURE 8.
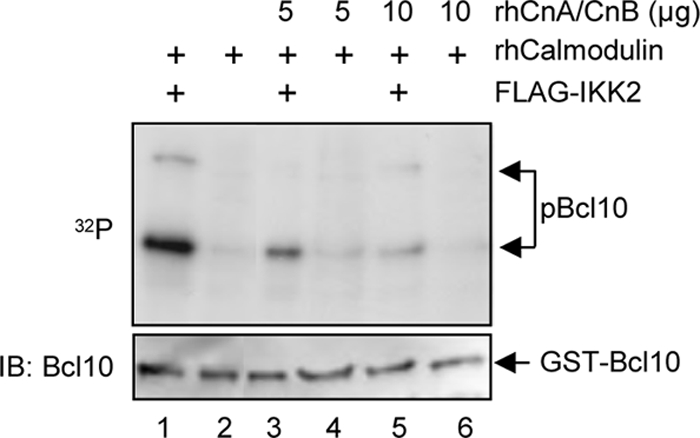
Calcineurin is a Bcl10 phosphatase in vitro. For the in vitro phosphatase assay, bacterial expressed GST-Bcl10 fusion protein was coupled to GSH-agarose prior to a kinase assay with FLAG-IKK2. The phosphorylated GST-Bcl10 proteins were subsequently washed twice with TNT buffer and once with phosphatase reaction buffer. The purified phosphorylated GST-Bcl10 protein was then subjected to an in vitro phosphatase assay by adding either recombinant calmodulin alone (150 units/sample, lanes 1 and 2) or in conjunction with increasing amounts of recombinant calcineurin A-calcineurin B (lanes 3–6). The samples were separated by SDS-PAGE and transferred to nitrocellulose membrane, and the resulting membrane was subsequently exposed to x-ray films to determine the phosphorylation status of Bcl10 (32P). To control for equal GST-Bcl10 loading, the membrane was additionally subjected to an anti-Bcl10 immunoblot analysis (IB) (bottom).
Calcineurin A Interacts with the CBM Complex
Having identified calcineurin as potential Bcl10 phosphatase, we next aimed to determine whether calcineurin might interact with components of the CBM complex. To this end, interaction studies were performed upon ectopical expression of CnAα, CnAβ, or the constitutive active deletion mutant of calcineurin Aα (ΔCam) in conjunction either with Carma, Malt1, or Bcl10 in HEK293 cells (Fig. 9, A–C). Indeed, a CnA-Malt1 protein-protein interaction was observed with both calcineurin A isoforms, CnAα and CnAβ, whereas Carma1 was only capable of a CnAα interaction and did not form a complex with CnAβ (Fig. 9, A and B). In contrast to Malt1 and Carma1, an interaction of calcineurin A with Bcl10 was not detectable upon ectopical coexpression in HEK293 cells (Fig. 9C), possibly due to a low stability of this enzyme-substrate complex. However, similar to the results shown in Fig. 7, coexpression of calcineurin Aα and more pronounced ΔCam led again to a marked decrease in the slower migrating phospho-Bcl10 signal (signal III). To further analyze the interaction of calcineurin with the CBM complex, an additional interaction study was performed by immunoprecipitating endogenous Malt1 from untreated Jurkat T cells or from Jurkat T cells stimulated with PMA + ionomycin. Here, we observed a constitutive interaction of calcineurin A with Malt1 (Fig. 9D, lanes 1–3). These results consistently suggest that CnA interacts physically with the CBM complex.
FIGURE 9.

Calcineurin A interacts with the components of the CBM complex. A–C, to determine whether calcineurin A interacts with the CBM complex anti-FLAG immunoprecipitations (IP) were performed with the whole cell extracts from transiently transfected HEK293 cells, and interaction of Carma1 (A), Malt1 (B), or Bcl10 (C) with the different CnA proteins was monitored with the appropriate antibodies (top). To ensure the expression of the ectopically expressed proteins, a fraction of the whole cell extracts (10%) was analyzed by additional immunoblot experiments (IB) (bottom). *, IgH. D, analysis of the calcineurin interaction with the CBM complex in Jurkat T cells. Immunoprecipitation experiments (500 μg/sample) were performed with an anti-Malt1 antibody, and immunoblot analyses (40 μg/sample, lanes 1–4) used whole cell extracts from untreated or P+I-stimulated Jurkat T cells. NRS, normal rabbit serum.
DISCUSSION
A multitude of signal transduction events are initiated upon engagement of the antigen receptors in T and B cells. One critical event during this process is the activation of the phospholipase Cγ1, which in turn hydrolyzes phosphoinositol 4,5-bisphosphate to diacylglycerol and inositol trisphosphate, leading to the activation of the Ras and PKC pathways on the one hand and to a calcium influx on the other. This calcium influx is known as store-operated calcium entry and depends on the calcium sensors of the STIM family and the CRAC calcium channels (1). Several signaling pathways, including the NF-AT, AP1, and NF-κB pathways, need both signaling branches (the diacylglycerol-induced and the calcium-dependent signals) for their full activity. One essential calcium-dependent signaling protein is the protein phosphatase calcineurin, which has been shown to be crucial for the TCR-induced activation of NF-AT as well as NF-κB (1, 3, 4). However, in contrast to the NF-AT pathway, the molecular mechanism underlying the calcium-calcineurin dependence of the NF-κB pathway is less well understood. Therefore, we set out to determine the molecular mechanisms by which this calcium-dependent phosphatase affects the NF-κB signaling pathway. One essential step in the NF-κB signaling pathway initiated upon antigen-receptor triggering is the assembly of a multiprotein complex consisting of Carma1, Bcl10, and Malt1, also known as the CBM complex (21). Here, we provide evidence that calcium influx is crucial for the proper formation of the CBM complex. For instance, we show that an additional calcium-influx, either induced by ionomycin or by thapsigargin treatment, augmented the PMA-induced Carma1-Bcl10 interaction distinctively (Fig. 1). By contrast, a pretreatment with the cell-permeable calcium chelator EGTA-AM attenuated the PMA- and P+I-induced NF-κB activity. In addition, the CBM complex formation but had no effect on the TNF-α-induced NF-κB activation (Fig. 2), thus further demonstrating the necessity of elevated cellular calcium levels for the formation of the CBM complex.
An active calcineurin is composed of a catalytically active A subunit, a regulatory B subunit, and a calcium-calmodulin complex (13). Inhibition of calcineurin with the pharmaceutical inhibitor CsA or FK506 impairs the IKK activation and thus the IκBα degradation specifically induced by TCR engagement but not upon TNF-α stimulation (Fig. 3, A–C) (3, 4). Here, we demonstrate that the basis for the negative effect of these pharmaceutical calcineurin inhibitors is the attenuation of the CBM complex assembly. Pretreatment of Jurkat T cells as well as primary murine T cells with CsA strongly reduces the Bcl10-Carma1 interaction by P+I stimulation or by stimulation with agonistic anti-CD3 + CD28 antibodies (Fig. 3, D–F). Importantly, a similar reduction in Bcl10-Carma1 interaction was achieved by the combined transfection of siRNAs for CnAα and CnAβ but only very moderately by transfection of the individual CnA siRNAs (Fig. 5A). However, the degree of inhibition in the Carma1-Bcl10 interaction as well as NF-κB activation was always more pronounced by pretreatment with CsA than by siRNA-mediated suppression of CnA. The reason for the lower efficacy of the siRNA-mediated CnA suppression is not completely clear. However, one can speculate that the inhibition by CsA might be more efficient because CsA impairs the activity of all CnA isoforms, whereas a residual CnA expression remained in case of the siRNA-mediated knockdown experiments, which would allow a remaining calcineurin activity (Fig. 5C). Alternatively, the third isoform of CnA, CnAγ, might also contribute to the regulation of the CBM complex formation by calcineurin. Expression of CnAγ is thought to be restricted to the testis. However, data mining revealed that this calcineurin A isoform is also expressed in T and B lymphocytes at the RNA level (GeneATLAS).
The formation of the CBM complex appears to be highly regulated by post-translational modifications, involving the phosphorylation of Carma1 and Bcl10. Carma1 phosphorylation upon TCR ligation, which can be mediated by PKCϴ, HPK1, IKK2, CaMKII, and casein kinase I, is required for CBM complex formation, and thus Carma1 phosphorylation represents mostly a positive signal (7, 8, 22). In contrast, the phosphorylation of Bcl10 has been linked to negative functions (14, 23, 24). For instance, the phosphorylation of the five serine residues (Ser134, Ser136, Ser138, Ser141, and Ser144) in the carboxyl-terminal part of Bcl10 by IKK2 has been linked to a remodeling of the Bcl10-Malt1 heterodimer, leading to resolution of CBM complex assembly (14). Phosphorylation of Bcl10 at these carboxyl-terminal serine residues does not influence the Carma1-Bcl10 interaction directly but affects CBM complex function by altering the structure of the Malt1-Bcl10 heterodimer. The disturbed Malt1-Bcl10 interaction in turn causes a reduced recruitment of Carma1 as part of a negative feedback mechanism to terminate the TCR-induced NF-κB pathway. A similar mechanism might also underlie the positive calcineurin effect shown in the present study. Although Malt1-Bcl10 interaction remained unchanged upon calcineurin inhibition by CsA or by specific siRNAs (Figs. 3 and 5), calcineurin might dephosphorylate Bcl10, causing an alteration of the Malt1-Bcl10 heterodimer structure and thus promoting an efficient Carma1 recruitment. Interestingly, the role of basal Bcl10 phosphorylation in unstimulated T cells or during the early phase of TCR-induced signaling has not been fully elucidated. However, a basal Bcl10 phosphorylation can be observed in Jurkat T cells as well as HEK293 cells (Figs. 6 and 7) (20). The hypothesis that calcineurin might affect the CBM complex formation by removing one or more negatively acting phosphate groups from the Bcl10 protein is supported by the increased basal Bcl10 phosphorylation in CsA- or EGTA-treated Jurkat T cells (Fig. 6) (which suggests that calcineurin dephosphorylates Bcl10 in vivo). In addition, the decrease in Bcl10 phosphorylation in HEK293 and Jurkat T cells upon coexpression of calcineurin A or more pronounced upon coexpression of the constitutive active mutant ΔCam further supports our hypothesis. Moreover, we observed a protein-protein interaction of the calcineurin A subunit or a constitutive active deletion mutant of calcineurin Aα (ΔCam) with Malt1 or Carma1. In addition, calcineurin A forms a protein complex with the CBM complex in Jurkat T cells as demonstrated by the Malt1-calcineurin A interaction (Fig. 9D). The reason for the lack of Bcl10-CnA interaction with ectopically expressed proteins remains unclear. However, one explanation could be that Malt1 is required for a stable Bcl10-CnA interaction. Alternatively, the recently published Bcl10-calmodulin interaction might also contribute to the recruitment of calcineurin to Bcl10 (25). Experiments are under way in our laboratory to clarify this question.
Taken together, the evidence provided in this study clearly demonstrates that calcium influx is crucial for the formation of the CBM complex during TCR-induced NF-κB activation. Furthermore, our data strongly suggest that calcineurin is one of the calcium-dependent signaling proteins involved in the assembly of the CBM complex (Fig. 10). However, calcineurin might not be the only calcium-dependent enzyme required for this process. Blocking CaMKII with the specific inhibitor KN93, for instance, had a profound effect on the PMA- and TCR-induced NF-κB activity (26). Furthermore, PKCα has also been demonstrated to affect the antigen receptor-induced NF-κB activation in T cells. However, PKCα seems to modulate the TCR-induced NF-κB activity indirectly by affecting PKCϴ, the central regulator of CBM complex assembly (27). CaMKII, on the other hand, has been identified as both a Bcl10 and a Carma1 kinase and could participate in the control of CBM complex assembly by phosphorylating Carma1 at Ser109 (23, 28). The reduced Carma1 phosphorylation upon EGTA-AM pretreatment would support the hypothesis of an additional calcium-dependent but CsA-insensitive effect on Carma1 phosphorylation. However, although PKCα or CaMKII might contribute to the overall calcium dependence of the TCR-induced NF-κB activation, they seem not to be negatively affected by CsA (Fig. 4). From these results, especially the calineurin-mediated dephosphorylation of Bcl10 in vitro (Fig. 8), we concluded that the positive effect of calcineurin is rather direct and does not involve the activation of other calcium-dependent signaling pathways. Although we provide evidence that CBM complex formation might be affected by the calcineurin-mediated dephosphorylation of Bcl10, further work is needed to clarify the exact role of calcineurin for the TCR-induced NF-κB signaling pathway. For instance, it will be necessary to identify the serine residues in Bcl10 that are dephosphorylated by calcineurin. To this end, it seems likely that one or more of the five already identified carboxyl-terminal IKK2 phosphorylation sites are targeted by calcineurin because calcineurin coexpression reverted the IKK2-mediated Bcl10 hyperphosphorylation (Fig. 7, A–C). Furthermore, the Bcl10S5A mutant is not affected by calcineurin (Fig. 7). Another question pertains to the kinases responsible for basal Bcl10 phosphorylation. IKK2 and CaMKII have been identified as Bcl10 kinases, which are responsible for Bcl10 phosphorylation upon cell activation. However, whether the basal Bcl10 phosphorylation is also conducted by these kinases or conducted by still unknown kinases needs to be clarified in future experiments. The answers to these questions will not only provide a better understanding of the regulation of the TCR-induced NF-κB signaling pathway but might also lead to the development of more specific inhibitors for the different CnA isoforms in order to avoid the toxicity of the currently used immunosuppressors CsA and FK506.
FIGURE 10.

Hypothetical model of the calcineurin effect on CBM complex assembly. In resting T cells, Carma1 is hypophosphorylated and Bcl10, included in a heterodimer with Malt1, displays a low basal phosphorylation (gray boxes). Furthermore, calcineurin and PKCϴ activity remains low in unstimulated T cells. Engagement of T cell receptor, however, induces PLCγ1-dependent Ca2+ influx and diacylglycerol (DAG)-mediated PKCϴ activation (white box, center). Activated PKCQ phosphorylates Carma1, and activated calcineurin dephosphorylates Bcl10. Hypophosphorylated Bcl10 and phospho-Carma1 are a prerequisite for the efficient formation of the CBM complex. As a negative feedback mechanism, Bcl10 gets rephosphorylated or even hyperphosphorylated, for example by IKK2 or by CaMKII. The CBM complex becomes inactive, and Bcl10 is subsequently degraded.
Supplementary Material
Acknowledgments
We thank Dr. Edward Felder (University of Ulm) for technical support and Drs. Bernd Baumann and Thomas Wirth for plasmids and Jur4 cells. We thank Dr. Rüdiger Arnold (Deutsches Krebsforschungszentrum) for the kind gift of hybridoma cell lines expressing agonistic anti-CD3 and anti-CD28 antibodies.
This work was supported by a Juniorprofessor program grant of the Ministry of Science and Culture of the State of Baden-Württemberg (to M. W. K.) and by Deutsche Forschungsgemeinschaft Grant MA 2367/3-2 (to R. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- TCR
- T cell receptor
- NF-κB
- nuclear factor κB
- NF-AT
- nuclear factor of activated T cells
- IKK
- IκB kinase
- NEMO
- NF-κB essential modulator
- CsA
- cyclosporin A
- CnA and CnB
- calcineurin A and B, respectively
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- AM
- acetoxymethyl ester
- PMA
- phorbol 12-myristate 13-acetate
- P+I
- PMA plus ionomycin.
REFERENCES
- 1. Oh-hora M., Rao A. (2008) Curr. Opin. Immunol. 20, 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Werlen G., Jacinto E., Xia Y., Karin M. (1998) EMBO J. 17, 3101–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marienfeld R., Neumann M., Chuvpilo S., Escher C., Kneitz B., Avots A., Schimpl A., Serfling E. (1997) Eur. J. Immunol. 27, 1601–1609 [DOI] [PubMed] [Google Scholar]
- 4. Trushin S. A., Pennington K. N., Algeciras-Schimnich A., Paya C. V. (1999) J. Biol. Chem. 274, 22923–22931 [DOI] [PubMed] [Google Scholar]
- 5. Thome M., Weil R. (2007) Trends Immunol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 6. Sommer K., Guo B., Pomerantz J. L., Bandaranayake A. D., Moreno-García M. E., Ovechkina Y. L., Rawlings D. J. (2005) Immunity 23, 561–574 [DOI] [PubMed] [Google Scholar]
- 7. Shinohara H., Maeda S., Watarai H., Kurosaki T. (2007) J. Exp. Med. 204, 3285–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brenner D., Brechmann M., Röhling S., Tapernoux M., Mock T., Winter D., Lehmann W. D., Kiefer F., Thome M., Krammer P. H., Arnold R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14508–14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bidère N., Snow A. L., Sakai K., Zheng L., Lenardo M. J. (2006) Curr. Biol. 16, 1666–1671 [DOI] [PubMed] [Google Scholar]
- 10. Oeckinghaus A., Wegener E., Welteke V., Ferch U., Arslan S. C., Ruland J., Scheidereit C., Krappmann D. (2007) EMBO J. 26, 4634–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hinz M., Scheidereit C. (2007) Sci. STKE 2007, pe19. [DOI] [PubMed] [Google Scholar]
- 12. Moreno-García M. E., Sommer K., Shinohara H., Bandaranayake A. D., Kurosaki T., Rawlings D. J. (2010) Mol. Cell. Biol. 30, 922–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shibasaki F., Hallin U., Uchino H. (2002) J. Biochem. 131, 1–15 [DOI] [PubMed] [Google Scholar]
- 14. Wegener E., Oeckinghaus A., Papadopoulou N., Lavitas L., Schmidt-Supprian M., Ferch U., Mak T. W., Ruland J., Heissmeyer V., Krappmann D. (2006) Mol. Cell 23, 13–23 [DOI] [PubMed] [Google Scholar]
- 15. Welteke V., Eitelhuber A., Düwel M., Schweitzer K., Naumann M., Krappmann D. (2009) EMBO Rep. 10, 642–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palkowitsch L., Leidner J., Ghosh S., Marienfeld R. B. (2008) J. Biol. Chem. 283, 76–86 [DOI] [PubMed] [Google Scholar]
- 17. Marienfeld R., May M. J., Berberich I., Serfling E., Ghosh S., Neumann M. (2003) J. Biol. Chem. 278, 19852–19860 [DOI] [PubMed] [Google Scholar]
- 18. Inesi G., Sagara Y. (1992) Arch Biochem. Biophys. 298, 313–317 [DOI] [PubMed] [Google Scholar]
- 19. Serfling E., Avots A., Neumann M. (1995) Biochim. Biophys. Acta 1263, 181–200 [DOI] [PubMed] [Google Scholar]
- 20. Zeng H., Di L., Fu G., Chen Y., Gao X., Xu L., Lin X., Wen R. (2007) Mol. Cell. Biol. 27, 5235–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulze-Luehrmann J., Ghosh S. (2006) Immunity 25, 701–715 [DOI] [PubMed] [Google Scholar]
- 22. Moreno-García M. E., Sommer K., Haftmann C., Sontheimer C., Andrews S. F., Rawlings D. J. (2009) J. Immunol. 183, 7362–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishiguro K., Ando T., Goto H., Xavier R. (2007) Mol. Immunol. 44, 2095–2100 [DOI] [PubMed] [Google Scholar]
- 24. Scharschmidt E., Wegener E., Heissmeyer V., Rao A., Krappmann D. (2004) Mol. Cell. Biol. 24, 3860–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edin S., Oruganti S. R., Grundström C., Grundström T. (2010) Mol. Immunol. 47, 2057–2064 [DOI] [PubMed] [Google Scholar]
- 26. Hughes K., Edin S., Antonsson A., Grundström T. (2001) J. Biol. Chem. 276, 36008–36013 [DOI] [PubMed] [Google Scholar]
- 27. Trushin S. A., Pennington K. N., Carmona E. M., Asin S., Savoy D. N., Billadeau D. D., Paya C. V. (2003) Mol. Cell. Biol. 23, 7068–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishiguro K., Green T., Rapley J., Wachtel H., Giallourakis C., Landry A., Cao Z., Lu N., Takafumi A., Goto H., Daly M. J., Xavier R. J. (2006) Mol. Cell. Biol. 26, 5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.