

Abstract
Homodimerization is an essential step for membrane type 1 matrix metalloproteinase (MT1-MMP) to activate proMMP-2 and to degrade collagen on the cell surface. To uncover the molecular basis of the hemopexin (Hpx) domain-driven dimerization of MT1-MMP, a crystal structure of the Hpx domain was solved at 1.7 Å resolution. Two interactions were identified as potential biological dimer interfaces in the crystal structure, and mutagenesis studies revealed that the biological dimer possesses a symmetrical interaction where blades II and III of molecule A interact with blades III and II of molecule B. The mutations of amino acids involved in the interaction weakened the dimer interaction of Hpx domains in solution, and incorporation of these mutations into the full-length enzyme significantly inhibited dimer-dependent functions on the cell surface, including proMMP-2 activation, collagen degradation, and invasion into the three-dimensional collagen matrix, whereas dimer-independent functions, including gelatin film degradation and two-dimensional cell migration, were not affected. These results shed light on the structural basis of MT1-MMP dimerization that is crucial to promote cellular invasion.
Keywords: Cell Migration, Cell Surface Enzymes, Extracellular Matrix, Matrix Metalloproteinase, Protease
Introduction
Degradation of extracellular matrix by proteinases is a crucial step during cell migration in tissue because the extracellular matrix is a physical barrier (1). Membrane type 1 matrix metalloproteinase (MT1-MMP or MMP-14)7 plays an essential role in this process (1, 2). MT1-MMP belongs to the matrix metalloproteinase (MMP) family, containing shared domain structures of a signal peptide, a propeptide, a catalytic domain, a hinge (linker-1), and a hemopexin (Hpx) domain (2–4). As a membrane proteinase, it also has a stalk region (linker-2), a transmembrane domain, and a short cytoplasmic tail of 20 amino acids at its C terminus. MT1-MMP cleaves a wide variety of extracellular matrix components, such as collagens I, II, and III, laminins 1 and 5, aggrecan core protein, fibrin and fibronectin, vitronectin, and lumican (5, 6). MT1-MMP also activates other MMPs, such as proMMP-2 (7) and proMMP-13 (8), expanding its proteolytic repertoire on the cell surface. It also cleaves several cell surface proteins, such as CD44 (9), transglutaminase (10), low density lipoprotein receptor-related protein (11), αv integrin (12, 13), and syndecan-1 (14). All of these proteolytic events result in modification of the cellular microenvironment, which enhances cell migration in tissues (15).
Among these proteolytic activities of MT1-MMP, activation of proMMP-2 on the cell surface is considered to be important, especially during in vivo growth of well differentiated cancer cells, where degradation of own basement membrane is a limiting factor to expand their tumor mass (16). MT1-MMP is unable to degrade type IV collagen, a major component of the basement membrane (6), whereas activated MMP-2 does (17). In the activation process, MT1-MMP forms a complex with TIMP-2, which then acts as a receptor for proMMP-2, resulting in a trimolecular MT1-MMP·TIMP-2·proMMP-2 complex (18). To cleave the propeptide of proMMP-2, an additional TIMP-2·free MT1-MMP needs to be positioned in close proximity to this trimolecular complex, and this is achieved by the formation of a homodimeric complex of MT1-MMP (19, 20). Disruption of this dimer formation effectively inhibited proMMP-2 activation on the cell surface (19, 20).
Another important activity of MT1-MMP is collagen degradation. For MT1-MMP to cleave collagen on the cell surface, the enzyme is also required to be in a dimeric state (21). Again, disruption of dimerization of MT1-MMP effectively inhibited its collagenolytic activity (21, 22) and MT1-MMP-dependent cellular invasion into collagen matrix (23).
Homodimerization of MT1-MMP has emerged as an important mechanism to regulate two major activities of MT1-MMP on the cell surface: proMMP-2 activation (19, 20, 22, 24) and collagen degradation (21, 22). Previously, we identified two regions of MT1-MMP that are involved in the formation of a dimerization interface: the Hpx domain and the transmembrane (TM) domain (22). By analyzing domain deletion/substitution mutants, we found that the Hpx domain-dependent dimer plays a key role in collagen degradation on the cell surface and that the TM domain-dependent dimer plays a key role in proMMP-2 activation. In the full-length enzyme, a dimer assembles through both interfaces, and interfering with Hpx-dependent dimer formation by co-expression of either soluble or membrane-anchored Hpx domain also inhibits the TM-dependent dimerization, probably due to steric hindrance (22). This suggests that there may be a cross-talk between the two domains to determine the dimerization status of the MT1-MMP molecules on the cell surface and that the Hpx domain-dependent dimer is the major determinant for dimerization of full-length MT1-MMP. However, the structural nature of the MT1-MMP dimer interface is not known at present.
In this report, we determined the crystal structure of the Hpx domain of MT1-MMP (Hpx-14), which showed symmetrical and asymmetrical dimer interactions with respect to an adjoining molecule in the crystal. Mutagenesis studies revealed that the symmetrical interaction of Hpx-14 is the dimer interface of full-length MT1-MMP. This interaction was also found to be crucial for the expression of biological activities of full-length MT1-MMP on the cell surface.
MATERIALS AND METHODS
Cell Culture and Transfection
COS7 cells, HT1080 cells, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM; BioWhittaker) supplemented with 10% fetal bovine serum and penicillin/streptomycin (BioWhittaker). Cells were transfected with expression plasmids using FuGene 6TM (Roche Applied Science) for COS7 and HT1080 cells and Lipofectamine 2000 (Invitrogen) for HeLa cells according to the manufacturer's instructions.
Construction of MT1-MMP Hpx Domain (Hpx-14) Expression Plasmids for Escherichia coli and Mammalian Cells
For crystallization of the Hpx-14, we amplified a sequence coding for Asn317–Gly511 by PCR, attaching Met at the N terminus and His6 at the C terminus using MT1-MMP cDNA as a template, and subcloned into the pET3a E. coli expression vector (Novagen). The generated cDNA was confirmed by DNA sequencing. For mammalian expression of the soluble Hpx domain construct, we created the catalytic domain (Tyr112–Pro312) and transmembrane/cytoplasmic domain (Ala536–Val582) deletion mutant of MT1-MMP (MT1FΔCatTM) using the PCR extension method as described previously with FLAG-tagged MT1-MMP (MT1F) as a template (21). In MT1F, a FLAG tag was inserted after the last residue of the propeptide, Arg111 (25). A chimeric protein construct of MT1F-NGFR was made as described previously (19). The mutations to disrupt symmetrical interaction (D385K/T412A/Y436F (DTY/KAF)) and asymmetrical interaction (R339A/W340A/D391K (RWD/AAK)) were introduced into the soluble Hpx domain created by the PCR extension method using MT1FΔCatTM as a template. These mutations were also introduced in MT1F with the PCR extension method using MT1F as a template. Furthermore DTY/AAF, DTY/DAF, and DTY/DTF mutations were introduced in both soluble Hpx domain constructs and full-length MT1F by the PCR extension method using MT1FΔCatTM, Hpx-DTY/KAF, MT1F, and MT1F-DTY/KAF as templates. All of the constructs were confirmed by DNA sequencing and subcloned into the pSG5 mammalian expression vector (Stratagene).
Protein Purification
The Hpx-14 for crystallization was expressed in the E. coli strain BL21pLysS as inclusion bodies, purified using a nickel-chelating column, and refolded as described previously (19). Protein folding was confirmed by a shift of electrophoretic mobility under reducing and non-reducing conditions in SDS-PAGE (25 and 21 kDa, respectively) due to the presence of one disulfide bond.
The Hpx-14 and its mutants were also purified from culture media of COS7 cells (six 100-mm dishes) transiently transfected with the expression plasmid. Conditioned media were applied to anti-FLAG antibody-conjugated beads (Sigma-Aldrich) and eluted with the FLAG peptide of DYKDDDDK (200 μg/ml) in 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 10 mm CaCl2, 0.02% NaN3 (TNC buffer) with 0.05% Brij35. The samples were dialyzed against TNC buffer with 0.05% Brij35 to remove FLAG peptide.
Sedimentation Velocity Experiment
Sedimentation velocity and sedimentation equilibrium runs were carried out on an analytical ultracentrifuge (model E; Beckman Coulter instruments, Inc., Fullerton, CA) equipped with electronic speed and temperature control and a photoelectric scanner. The buffer used in all of the sedimentation experiments contained 20 mm Tris-HCl (pH 7.5). The density of this buffer was measured with a pycnometer at 20 °C. For a second series of equilibrium runs at 4 °C, 20,000 rpm were employed with detection at 280 and 260 nm at high protein concentrations (model XL-I; Beckman Coulter instruments, Inc., Fullerton, CA). Samples were prepared at different concentrations (0.25, 0.5, and 1 mg/ml) in 50 mm HEPES (pH 7.5), 150 mm NaCl, 10 mm CaCl2 buffer. Centrifugation was performed at 20,000 rpm at different temperatures (20 and 4 °C) for 48–72 h until equilibrium was achieved. For data analysis, the software Ultrascan 9.0 (B. Demeler, University of Texas Health Science Center, San Antonio, TX) and Origin were used. No temperature-dependent differences were observed. Mass spectrometry after the equilibrium runs proved that no significant degradation of the protein had occurred. Analysis regarding the dimer content (5–20%) was performed with Origin (OriginLab, Northampton, MA) according to a model, which assumed equilibrium conditions for the monomer-dimer formation.
Small Angle X-ray Scattering
The small angle x-ray scattering patterns were collected at the EMBL beamline X33 (Deutsches Elektronen Synchrotron, Hamburg) using the MAR345 Image plate detector (26). Solutions of the Hpx-14 at a protein concentration between 1 and 2.3 mg/ml were recorded at a sample-to-detector distance of 2.7 meters covering the range of momentum transfer 0.10 < s < 5.0 nm−1 (s = 4π sin(θ)/λ, where 2θ is the scattering angle and λ = 0.15 nm is the wavelength of the radiation). To check for radiation damage during the scattering experiments, two successive 2-min exposures of the solutes were compared, and no changes were detected. The data were processed using standard procedures by the programs PRIMUS (27) and GNOM (28) to compute the radius of gyration (Rg) and maximum dimension (Dmax). The molecular mass (MM) of the solute was estimated by calibration against a reference solution of bovine serum albumin. The overall parameters and scattering patterns from the atomic models of monomeric and dimeric Hpx-14 were calculated by the program CRYSOL (29). The volume fractions of monomers and dimers in the mixture were computed by the program OLIGOMER (30), minimizing the discrepancy (1) between the linear combination of the intensities of the two components and the experimental data I(s) from the mixture, where N is the number of experimental points, c is a scaling factor, and Icalc(s) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively.
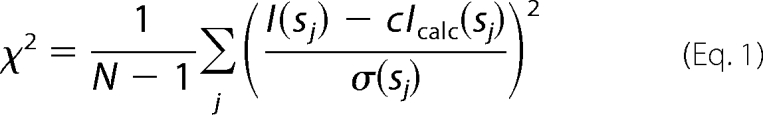 |
Crystallization, Data Collection, and Structure Determination
The Hpx domain of MT1-MMP was crystallized using the sitting drop vapor diffusion technique. 1 μl of protein at a concentration of 5.0 mg/ml in 20 mm Tris-HCl (pH 7.5) was mixed with 1 μl of reservoir buffer, 2.1 m malic acid, pH 6.0, 40% (v/v) acetone. The crystals appeared after a few days at room temperature. For data collection, crystals were transferred into cryobuffer (20% perfluoropolyether PFO-X125/03) and were cryocooled under a stream of nitrogen gas at 100 K. Data were collected using synchrotron radiation at beamline BW6 of the Deutsches Elektronen Synchrotron (Hamburg, Germany), employing a Mar Research CCD detector at a wavelength of 1.050 Å. The diffraction data were processed and scaled by the programs DENZO/SCALEPACK. The crystal diffracted to 1.7 Å, belonging to the trigonal space group P3121 containing one molecule in the asymmetric unit with a Matthews coefficient of Vm = 2.5 Å3/Da, corresponding to a solvent content of 50.4%.
Molecular replacement was performed with the program PHASER (31) using a monomer of the Hpx domain of MMP-9 (Protein Data Bank (PDB) code 1ITV). For electron density map calculations, a rigid body and positional refinement was performed, followed by simulated annealing and energy minimization using CNS (32). Model building was performed using the program MAIN (33). Subsequent cycles of rebuilding and refinement finally resulted in a model with a crystallographic R value of 18.5% and an Rfree of 20.7%. Stereo chemical coordinate validation was done using MOLPROBITY (34), showing an excellent stereochemistry for the model (Table 1). All of the structure model illustrations were generated by PyMOL (DeLano Scientific LLC).
TABLE 1.
Data collection and refinement statistics for Hpx-14
| Parameter | Value |
|---|---|
| Data collection | |
| Crystal content | MT1-MMP Hpx domain (Hpx-14) |
| Space group | P3121 |
| Cell constants (Å) | a = b = 77.21, c = 66.82, a = b = 90°, g = 120° |
| Resolution range (Å)a | 20.0–1.70 (1.76–1.70) |
| Unique reflections | 25,677 (2388) |
| Completeness (%) | 99.7 (98.5) |
| Multiplicity | 9.5 (8.0) |
| I/σ | 37.5 (4.6) |
| Rlinearb | 0.05 (0.35) |
| Refinement | |
| Resolution range (Å)a | 20.0–1.70 (1.76–1.70) |
| Completeness (%) | 99.5 (98.6) |
| Reflection used for refinement | 25,607 (2388) |
| Reflections in working set | 24,310 (2262) |
| Reflections in test set (5%) | 1297 (126) |
| Rcryst (%)c | 18.5 (25.8) |
| Rfree (%)d | 20.7 (28.6) |
| Root mean square deviationd bond lengths (Å) | 0.006 |
| Root mean square deviationd bond angles (degrees) | 1.40 |
| Protein molecules per asymmetric unit | 1 |
| Residue number (B-factors (Å2)) | 196 (16.6) |
| Water molecules (B-factors (Å2)) | 235 (28.0) |
| Chlorine ion (B-factors (Å2)) | 1 (22.9) |
| Calcium ion (B-factors (Å2)) | 1 (21.8) |
| Ramachandran plote | |
| Favored | 190 (97.9%) |
| Allowed | 4 (2.1%) |
| Outliers | 0 (0%) |
| PDB accession code | 3C7X |
a Values in parentheses are for the highest shell.
b Rlinear = Σh,k,l|I(h,k,l) − 〈I(h,k,l)〉|/Σh,k,lI(h,k,l).
c Rcryst = Σh,k,l|Fo(h,k,l) − Fc(h,k,l)|/Σh,k,lFo(h,k,l).
d Rfree is the R-value calculated with 5% of reflections not used in refinement.
e According to the MOLPROBITY server.
Gel Permeation Column Chromatography
Mammalian expressed MT1-MMP Hpx domain (Hpx-14) was subjected to gel filtration on a Superdex 75 or Superdex 200 column (Amersham Biosciences) connected to an Äkta FPLC (Amersham Biosciences). The column was equilibrated with TNC buffer with 0.05% Brij35 and calibrated with molecular mass markers including aprotinin (6.5 kDa), ribonuclease A (13.7 kDa), carbonic anhydrase (29 kDa), and conalbumin (75 kDa).
Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE)
BN-PAGE was carried out according to Swamy et al. (35). The Hpx samples were immunoprecipitated from the media of COS7 cells (three 100-mm dishes each) transiently transfected with expression plasmids for wild-type and mutant Hpx constructs using anti-FLAG beads (Sigma) and eluted with FLAG peptide (200 μg/ml) in the BN-PAGE sample buffer. The samples were then dialyzed against BN-PAGE sample buffer overnight to remove FLAG peptide and subjected to BN-PAGE followed by Western blot using anti-FLAG M2 antibody. The molecular weight marker for native gel used in this experiment was purchased from Invitrogen.
Phagokinetic Track Motility Assay
The phagokinetic track motility assay was performed as previously reported (9, 36–38). HT1080 cells were transiently transfected with MT1-MMP and EGFP plasmids and cultured on colloidal gold-coated glass coverslips. After 24 h, cells were fixed, and transfected cells were identified by EGFP signals. Images of tracks of transfected cells were captured under bright field microscopy, and the areas of the motility tracks were measured using ImageJ software. 50 cells/treatment group were measured.
In Situ Gelatin Degradation Assay and Indirect Immunofluorescence Staining
Coverslips were coated with Alexa568-conjugated gelatin (F-gelatin) prepared with an Alexa568-labeling kit (Invitrogen) as described previously (19). Transfected COS7 cells were cultured in the chamber slides for 1.5 h. Cells were then fixed with 3% paraformaldehyde in TBS and subjected to indirect immunofluorescent staining. After blocking with 5% goat serum and 3% bovine serum albumin in TBS for 1 h at room temperature, cells were incubated with an anti-FLAG M1 antibody (5 μg/ml) at room temperature for 2 h without permeabilizing cells. 1 mm CaCl2 was included throughout the procedure of washing and incubation for the staining with the anti-FLAG M1 antibody. Alexa488-conjugated goat anti-mouse IgG (Invitrogen) was used to visualize the antigen signal. Because anti-FLAG M1 antibody can only recognize the FLAG tag at the N terminus of the molecule (25), only active forms of the enzyme can be stained with this procedure. The signals were analyzed by an Ultraview confocal microscope (PerkinElmer Life Sciences) with a ×60 objective lens.
In Situ Collagen Degradation Assay
The experiments were done as described previously (21). 6-well culture plates were coated with a thin layer of chilled neutralized PureColTM collagen (Inamed Biomaterials, Fremont, CA) at 2.7 mg/ml in 1× RPMI medium (typically 100 μl/well) and incubated for 60 min at 37 °C for fibril formation, and COS7 cells (4 × 105/well) were then seeded on the film. 18 h later, cells were transfected with the expression plasmids in the growth medium (10% FBS/DMEM) using FUGENE6TM according to the manufacturer's instructions. The following day, culture medium was changed to serum-free DMEM, and cells were cultured for a further 3 days at 37 °C. The remaining collagen film was exposed by removing cells using repeated treatment with PBS containing 0.5 mg/ml trypsin and 1 mm EDTA. The collagen film was then fixed with 3% paraformaldehyde for 20 min at room temperature. Collagen was visualized by staining with Coomassie Brilliant Blue R-250, and the images were captured by a CCD camera-equipped microscope (Nikon TE-2000) with a ×20 objective lens. Degraded areas were visualized as a white, unstained, non-collagen-containing zone. In this assay, stained collagen was trypsin-resistant, suggesting that it was intact fibrillar collagen.
Microcarrier Bead Invasion Assay
The microcarrier bead invasion assay was carried out as described previously (23). HeLa cells were attached to Cytodex 3TM microcarrier beads (Sigma-Aldrich) by incubating trypsinized cells with sterile beads at 37 °C overnight with gentle agitation. Cells were then transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction. 24 h later, the beads and attached cells were suspended in a 2 mg/ml type I collagen gel of Cellmatrix type 1-A (Nitta Gelatin) and cultured for 40 h in the DMEM supplemented with 10% FBS and antibiotics. At the end of the culture period, images were taken using the ×10 objective lens on a CCD camera-equipped microscope (Nikon TE-2000). The distance cells had migrated from the bead surface was measured using Volocity software (Improvision, PerkinElmer Life Sciences).
RESULTS
Analysis of MT1-MMP Hpx Domain in Solution
The Hpx-14 expressed in E. coli and folded from the inclusion bodies had an apparent molecular mass on SDS-PAGE of 25 kDa under reducing conditions and 21 kDa under non-reducing conditions, indicating that the folded protein contains a disulfide bond as expected (Fig. 1A). Similar results were observed with the Hpx domain of MMP-1 (Hpx-1) (Fig. 1A). Gel filtration of Hpx-14 indicated that the molecular mass of Hpx-14 was around 32 kDa in solution, whereas Hpx-1 eluted around 17 kDa (Fig. 1B). The sedimentation coefficient of Hpx-14 determined by analytical ultracentrifugation was 2.74 S (Fig. 1C), and the molecular mass estimated was 40 kDa, whereas the molecular mass of Hpx-1 was around 21 kDa. The molecular mass of Hpx-14 was also determined by small angle x-ray scattering analysis (Fig. 1D). The overall parameters of the solute (Rg = 2.2 ± 0.1 nm, MM = 35 ± 5 kDa) are between those of the Hpx-14 monomer (Rg = 1.7 nm, MM = 23 kDa) and dimer (Rg = 2.5 nm, MM = 46 kDa). The maximum size of the dissolved particles (Dmax = 8 ± 1 nm) is close to that of the dimeric Hpx-14 (8 nm). These results suggest that the solute represents an equilibrium mixture of monomers and dimers. Using the computed scattering from the high resolution models, it was predicted that the Hpx-14 in this preparation was a mixture of 62% of monomers and 38% of dimers. Both predicted scattering from the monomer and the dimer failed to fit the experimental data (discrepancy χ = 2.9 and 3.3, respectively), and a mixture of 62% monomer and 38% dimer fits the best (discrepancy χ = 1.2). These data confirmed that Hpx-14 indeed forms a dimer in solution and potentially that the dimeric state is dynamic.
FIGURE 1.

Detection of Hpx domain dimer. A, purified E. coli-expressed and refolded Hpx domains of MT1-MMP (Hpx-14) and MMP-1 (Hpx-1) were subjected to SDS-PAGE analysis in reducing (R) and non-reducing (NR) conditions. The bands were visualized by staining with Coomassie Brilliant Blue R-250. Note that both Hpx domains shifted molecular mass between reducing and non-reducing conditions. B, purified Hpx-14 and Hpx-1 (1 μm) were subjected to analytical gel filtration of Superdex 75. Note that although Hpx-14 and Hpx-1 exhibited similar molecular mass around 21 kDa on SDS-PAGE under non-reducing conditions, Hpx-14 was eluted around 32 kDa and Hpx-1 around 17 kDa. C, purified Hpx-14 was subjected to sedimentation velocity experiments with analytical ultracentrifugation. Plot of the sedimentation coefficient S from several scans for the Hpx-14. S = 2.74 indicates that it has a molecular mass around 40 kDa. D, small angle x-ray scattering patterns from hemopexin14. Dots with error bars, experimental data; smooth curves, computed curves; blue dashed line and green dashed line, scattering from the crystallographic models of monomeric and dimeric hemopexin14, respectively, calculated by CRYSOL (29); solid red line, scattering from the best fit mixture of monomers and dimers computed by OLIGOMER (27).
Crystal Structure of Hpx-14
To analyze the dimer interface, we solved the crystal structure of Hpx-14 (PDB code 3C7X). The crystals of Hpx-14 belong to space group P3121, containing one molecule in the asymmetric unit. The final model was refined at a resolution of 1.7 Å with an Rfree of 20.7% (see Table 1). Hpx-14 belongs to the topological family of β-propeller domains, which encompasses proteins with 4–8 blades of β-strands. All Hpx domains of the MMP family consist of a 4-fold propeller and show the overall shape of a disc or shallow cylinder. The structure of Hpx-14 has four structurally homologous repeats of 35–45 residues, each repeat forming a blade or β-leaflet, around a central pseudo-4-fold axis. This axis exhibits a central funnel-like tunnel, which traverses the molecule and in this way connects both flat disc surfaces (39, 40) (Fig. 2A). Accordingly, the fourth blade of this β-propeller domain packs against the first one. Each blade (I–IV) of Hpx-14 is made up of a three-stranded anti-parallel β-sheet (strands β1–β3) (Fig. 2). The outermost strand is a combination of very short β-strands interrupted by a bulge, which are folded against β3. The blades are arranged in such a way that the sequentially and structurally first inner strands β1 within each blade are oriented with radially directed hydrogen bonds to the following strands that run along the shaft of the propeller. The blades accumulate a twist from one strand to its outer neighbor, such that the outermost strand is nearly perpendicular to the innermost. The resulting toroidal domain structure is tethered by a disulfide bridge between the two terminal blades. One sodium ion binds to the exit side of the channel of the carbonyl oxygen atoms of Asp416 (2.3 Å; blade III), Asn369 (2.4 Å; blade II), Thr370-Og (2.7 Å; blade II), and Arg464 (2.6 Å; blade IV), and with one of the surrounding water molecules, Wat56 (2.5 Å). Deeper in the channel, a well defined density was observed, which could be interpreted as a chloride ion. It is situated next to NH- of Ala371 (3.1 Å; blade II) and the water Wat19 molecule (2.9 Å), whereas its charge may be compensated by the nearby sodium ion.
FIGURE 2.

MT1-MMP Hpx domain structure. A, stereo representation of the Hpx-14 structure. View along “entry side” of the channel through the disc-like propeller domain. The four blades are labeled I–IV, β-strands are labeled β1–β5, and α-helices are labeled α1–α4. The sodium ion is shown as an orange sphere, and chlorine is shown as a blue sphere. B, structure overlay of Hpx-14 (green) with Hpx-1 (orange), Hpx-2 (yellow), Hpx-9 (pink), and Hpx-13 (blue). The Hpx-14 structure has extra loops in the outermost strand of blades I and IV (left). Also, Hpx-14 has a much shorter loop in the back of the molecule in blade III (right). PDB codes of structural data of Hpx-1, Hpx-2, Hpx-9, and Hpx-13 used in this figure are 1SU3, 1GEN, 1ITV, and 1HPX, respectively.
In Fig. 2B, the structure of Hpx-14 is superimposed on already known structures of the Hpx-1 (orange; PDB code 1FBL) (41), the Hpx domain of MMP-2 (Hpx-2; yellow; PDB code 1GEN) (42, 43), MMP-9 (Hpx-9; pink; PDB code 1ITV) (44), and MMP-13 (Hpx-13; blue; PDB code 1PEX) (45). Although the overall Hpx structures match very well, MT1-MMP contains an additional bulge in the outer strand of blade 1 in place of the β4 strand (Fig. 2B, left), an additional bulge in the outer strand of blade IV in place of the β4 strand (Fig. 2B, left), and a shorter loop in blade III in the back of the molecule (Fig. 2B, right). These differences make folding of Hpx-14 unique among the known four-bladed β-propeller domains.
Potential Dimerization Interfaces in the Crystal Structure
Two different potential dimeric forms of Hpx-14 were observed in the crystal. The first interaction constitutes a symmetrical dimer. The interface between blades II/III of molecule A and III/II of molecule B is generated by the crystallographic symmetry operation (2-fold axis), which relates the two molecules by a 180° rotation around an axis running tilted by ∼30° to the propeller axes. The residues involved in formation of this interface are shown in Fig. 3. Two major interaction sites were found. The first one involves Asp385, Lys386, and Lys434. Asp385 forms a salt bridge with Lys386 (blade II) within molecule A, forming an interface to interact with Lys434 (blade III) in molecule B. The second one is the direct hydrogen bond between Thr412 (molecule A, blade III) and the Tyr436 (molecule B, blade III) with its Oγ (2.67 Å). Because this interface is based on a 2-fold symmetry, these interactions are contributed in the same way by both molecules, A and B, forming close contacts. A number of water molecules are found in the interface and participate in additional interactions of the hydrogen bond network. Analysis of this mode of interaction on the Protein Interaction Calculation (PIC) server with default values indicated that it has two direct contacts of Thr412 Oγ (A)-Tyr436 Oη (B) and Tyr436 Oη (A)-Thr412 Oγ (B) and four water-mediated contacts of Asp385 Oδ1/Oδ2 (A)-Lys434 Nζ (B), Lys434 Nζ (A)-Asp385 Oδ1/Oδ2 (B), Lys404 Nζ (A)-Tyr449 carbonyl O (B), and Tyr449 carbonyl O (A)-Lys404 Nζ (B), supporting our analyses. Analysis of this interface on the PISA server provided an interface area of 344.9 Å2 for both molecule A and B. When surface complementarity values were calculated on the class PPI server that classifies homoprotein-protein interfaces structurally, we obtained a value of 0.073, close to the 0.1 that is considered as a shape of a complementary pair.
FIGURE 3.

Symmetrical dimer interactions. A, symmetrical interaction of molecule A (mol A) and B (mol B) are shown. The left panel shows a characteristic view of this form of dimer, and the right panel displays a 90° rotated view. B, wall-eye stereo view of the interface of the symmetrical interactions. Polar contacts between the residues and molecules are shown by dotted lines. Red spheres represent the water molecules bound to the protein. C, surface representation of the symmetrical Hpx-14 dimer interface. The residues involved in the interaction (Asp385, Thr412, and Tyr436) are colored in red and annotated. D, each blade is colored differently to show orientation of the molecules. Blue, blade I; green, blade II; yellow, blade III; red, blade IV.
The second mode of interaction results in an asymmetrical dimer (Fig. 4). These contacts between blades I/II of molecule C and I/II/III/IV of molecule D also have a mixed character of interaction (Fig. 4). Most notably, side chains of Arg339 and Trp340 in molecule C form a bulge and fit into a concave space of molecule D. The side chain of Arg339 (C) seems to interact with Met422 (D) that locates the deepest site of the concave space, whereas the side chain Trp340-Nϵ (C) forms a salt bridge to Glu472 (D). Secondarily, Asp391 (D) forms an indirect water-mediated salt bridge with Lys386 (C). Again, many water molecules were observed here and contribute to indirect interactions of a hydrogen-bonded network as well. Analysis of this mode of interaction on the PIC server identified two direct contacts of Glu472 Oϵ1 (C)-Trp340 Nϵ (D) and Glu332 Oϵ2 (C)-Arg362 Nϵ (D) and two water-mediated contacts of Glu373 Oϵ2 (C)-Arg339 Nη1/Nη2 (D) and Asp391 Oδ2 (C)-Lys386 Nζ (D), supporting our analyses. PISA server analysis provided an interface area of 528.4 for molecule C and 552.1 for molecule D. However, when surface complementarity values were calculated at the class PPI server, we obtained a value of 0.033, which is much lower than the 0.073 of the symmetrical interaction. Other modes of interaction were also carefully investigated, but they arise from crystal packing.
FIGURE 4.

Asymmetrical dimer interactions. A, asymmetrical interaction of molecule C (mol C) and D (mol D). The left panel shows a characteristic view of this form of dimer, and the right panel displays a 90° rotated view. B, wall-eye stereo view of the interface of the symmetrical interactions. Polar contacts between the residues and molecules are shown by dotted lines. Red spheres represent the water molecules bound to the protein. C, surface representation of the asymmetrical Hpx-14 dimer interface. The residues involved in the interaction (Arg339, Trp340, Asp391, and Lys386) are colored in red and annotated.
Symmetrical Dimerization Provides a Dimer Interface for MT1-MMP
To examine which mode of dimerization is related to the biological dimer, residues involved in the two types of interactions were subjected to mutagenesis. For the symmetrical dimer, disrupting interactions involving Asp385, Thr412, and Tyr436 were expected to have an impact on dimer formation as described above. Because Asp385 is located on an exposed loop (blade II), it was mutated to Lys to cause repulsion with Lys386. To disrupt hydrogen bonds between Thr412 (A) and Tyr436 (B), Thr412 was mutated to Ala. Because Tyr436 is packed between Pro411 and Pro450, it was mutated to Phe to create a minimal impact to the local environment, resulting in a total three-amino acid replacement (Hpx-DTY/KAF). For the asymmetrical dimer, mutations disrupting interactions involving Arg339, Trp340, and Asp391 were expected to have an impact as described above. Because the Arg339 and Trp340 side chains form a bulge, they were mutated to Ala to diminish the interface. In addition, Asp391 was mutated to Lys to cause repulsion against Lys386 in the other partner, resulting in a three-amino acid replacement as well (Hpx-RWD/AAK).
These Hpx-14 mutants and the wild-type Hpx-14 were expressed in COS7 cells as soluble proteins and purified by an anti-FLAG affinity column (Fig. 5A). We chose a mammalian system to ensure that mutant proteins are folded correctly. The two purified Hpx-14 mutants behaved identically to the wild type on SDS-PAGE under reducing and non-reducing conditions, confirming that they contain one disulfide bond. When these Hpx domains were analyzed by gel filtration on Superdex 75, they eluted around 40 kDa, suggesting that they are all forming dimers (Fig. 5B). However, the peak of Hpx-DTY/KAF elution was clearly asymmetric with a noticeable tail to lower molecular masses, suggesting that the dimeric interaction of this mutant was weaker, resulting in partial dissociation. The elution pattern of Hpx-RWD/AAK was very similar to that of the wild-type Hpx-14, suggesting that the RWD/AAK mutation had no effect on Hpx-14 dimerization. These results suggest that asymmetric dimerization is the result of crystal packing.
FIGURE 5.

Biochemical analysis of mutant Hpx-14. A, mammalian expressed purified Hpx domains (DTY, D396K/T412A/Y436F; RWD, R339A/W340A/D391K) were subjected to SDS-PAGE analysis under reducing (Red) and non-reducing (Non-Red) conditions. The bands were visualized by Coomassie Brilliant Blue R-250. Note that all three Hpx-14 proteins exhibit similar molecular mass in both reducing (23.3 kDa) and non-reducing conditions (20 kDa). B, purified Hpx-14 proteins were subjected to analytical gel filtration. Fractions of 0.5 ml were collected, and each fraction was TCA-precipitated and analyzed by Western blotting using an anti-FLAG M2 antibody (top). The distribution of the total intensity of the band in different fractions was plotted (bottom). The peak of Hpx-DTY was noticeably tailed to lower molecular mass, whereas both WT and RWD mutant Hpx domains was not. C, purified Hpx14 proteins were subjected to BN-PAGE. Note that Hpx-DTY migrated at 30 and 19 kDa mass, suggesting that it is partially dissociated.
To elaborate on this finding, we subjected these Hpx-14 proteins to BN-PAGE (Fig. 5C). BN-PAGE allows proteins to be electrophoresed according to their molecular weight in their native conformation (35, 46). On BN-PAGE, Hpx-WT migrated around 30 kDa, Hpx-DTY/KAF primarily around 19 kDa, and Hpx-RWD/AAK around 41 kDa (Fig. 5C). The data confirmed the gel filtration finding that DTY/KAF mutations failed to form a stable dimer. The RWD/AAK mutations did not dissociate the dimeric complex but seemed to increase the apparent molecular mass.
To further examine the effect of these mutations on dimeric complex formation, we carried out two additional assays. We have previously reported that a FLAG-tagged MT1-MMP/nerve growth factor receptor chimera mutant (MT1F-NGFR) can be used to monitor ectodomain-dependent dimerization by detecting tyrosine phosphorylation at its cytoplasmic domains (19), and its co-expression with soluble Hpx-14 inhibited the dimerization of MT1F-NGFR by competing in the Hpx-driven dimerization (21). Therefore, we utilized this system to test the ability of soluble Hpx-14 mutants to block the dimerization of MT1F-NGFR. If the mutation abraded the ability of Hpx14 to dimerize, such a mutant could not interfere with the dimerization. As shown in Fig. 6B, when an excess amount of the wild-type Hpx-14 (Hpx-WT) was co-expressed, the level of phosphotyrosine was reduced by 35%. Similarly, expression of Hpx-RWD/AAK inhibited phosphorylation by 22%, which is about 63% efficiency compared with the wild-type Hpx-14. However, when Hpx-DTY/KAF was co-expressed, there was no reduction in the phosphotyrosine level of MT1F-NGFR, suggesting that the DTY/KAF mutation affected Hpx-14 dimerization. The expression levels of these Hpx-14 variants and the wild type in these cells were equivalent (Fig. 6B).
FIGURE 6.

Effect of mutations on the dimer formation of MT1-MMP on the cell surface. A, schematic representation of mutant constructs used in the experiments. Pro, propeptide; FLAG, FLAG tag (DYKDDDDK); Cat, catalytic domain; L1, linker 1 (hinge); Hpx, hemopexin domain; NGFR, nerve growth factor receptor derived sequences; TM, transmembrane domain; Zn, catalytic zinc ion. B, COS7 cells were transfected with expression plasmids for mutant constructs as indicated. Cells were lysed and subjected to Western blotting analyses using anti-phosphotyrosine antibody (PY, top) and anti-FLAG M2 antibody (FLAG, bottom). Relative intensities of phosphotyrosine bands were measured by ImageJ, taking the band intensity of anti-FLAG M2 antibody as a control, and indicated under the phosphotyrosine blot. Note that overexpression of Hpx-DTY/KAF failed to inhibit dimer formation of MT1F-NGFR detected by anti-phosphotyrosine. C, COS7 cells expressing FLAG-tagged Hpx-14 constructs with or without expression of non-tagged full-length MT1-MMP were fixed and subjected to cell surface immunolocalization of soluble Hpx-14 using anti-FLAG M2 antibody and full-length MT1-MMP using anti-MT1-MMP hinge region antibody. Note that Hpx-WT and Hpx-RWD/AAK localized on the cell surface of MT1-MMP-expressing cells, whereas Hpx-DTY/KAF failed to do so. None of the Hpx mutants localized on the cell surface without co-expression of full-length MT1-MMP (right).
Next, we examined the ability of Hpx-14 mutants to associate with a full-length MT1-MMP on the cell surface by immunofluorescent staining. Again, this interaction is due to the complex formation of soluble Hpx domain with the Hpx domain in full-length MT1-MMP molecule expressed on the cell surface (21). When soluble FLAG-tagged wild-type Hpx-14 was co-expressed with non-tagged wild-type full-length MT1-MMP, soluble Hpx-14 was detected on the cell surface as expected (Fig. 6C). Similarly, Hpx-RWD/AAK was retained on the cell surface in an MT1-MMP expression-dependent manner, but Hpx-DTY/KAF was not detected on the cell surface. These data again suggest the importance of Asp385, Thr412, and Tyr436 for Hpx-14 dimerization. Together, these data corroborate that the symmetrical interaction is the biological dimer and that the asymmetrical interaction is an artifact due to the crystal packing.
Disruption of Dimerization of Full-length MT1-MMP on the Cell Surface by Hpx Mutation and Its Effects on Biological Activities
To investigate whether the symmetrical dimerization of Hpx-14 is crucial for biological activities of MT1-MMP, the Hpx domain mutations were introduced in the context of full-length MT1-MMP and their activities on proMMP-2 activation, cell migration promotion, gelatin film degradation, collagen film degradation, and three-dimensional collagen matrix invasion were examined. As shown in Fig. 7A, wild-type MT1F and MT1F-RWD/AAK expressed in COS7 cells efficiently activated exogenously added proMMP-2, converting it to the active 62-kDa form, although the level of activation by MT1F-RWD/AAK was slightly lower than that of MT1F. MT1F-DTY/KAF, however, did not activate proMMP-2 efficiently, leaving the majority of proMMP-2 as a pro form. Overexpression of MT1F, MT1F-DTY/KAF, and MT1F-RWD/AAK in HT1080 cells significantly enhanced cell migration to a similar extent compared with mock-transfected cells (Fig. 7B). Cell surface levels of wild-type MT1F and mutants and their localization on the cell surface were indistinguishable, and their abilities to degrade gelatin film were similar (Fig. 7C). The collagen film degradation was, however, affected differently by mutations. Cells expressing the DTY/KAF mutant exhibited little collagenolytic activity, whereas those with the RWD/AAK mutant exhibited levels that were significantly higher but lower than those of wild-type MT1F (Fig. 7D). The three-dimensional collagen matrix invasion was measured using HeLa cells on microcarrier beads (23). Because HeLa cells do not produce endogenous MT1-MMP, mock cells showed no invasion activity into collagen matrix. When wild-type MT1F was transfected, a significant number of the cells started to invade into collagen, but MT1F-DTY/KAF-expressing cells invaded collagen much less efficiently. On the other hand, MT1F-RWD/AAK-expressing cells were more invasive than MT1F-DTY/KAF-expressing cells but less invasive than cells transfected with wild-type MT1F (Fig. 7E).
FIGURE 7.

Effect of mutations in the Hpx domain on the biological functions of MT1-MMP. A, COS7 cells were transfected with the expression plasmids for MT1F, MT1F-DTY/KAF, and MT1F-RWD/AAK or empty vector (Mock). Cells were then reacted with purified proMMP-2 in the serum-free medium for 14 h. Media were subjected to zymography analysis (top) and cell lysates for Western blotting using anti-FLAG M2 antibody (bottom). B, HT1080 cells were transfected with expression plasmids for MT1F, MT1F-DTY/KAF, and MT1F-RWD/AAK together with pEGFP. After 24 h, cells were subjected to a phagokinetic motility assay as described under “Materials and Methods.” In each transfection group, migrated areas of GFP-positive cells were measured. Top, representative pictures of each transfected group; bottom, data with mean ± S.E. (error bars) (n = 50). *, p < 0.001. C, transfected COS7 cells were cultured on Alexa568-labeled gelatin-coated coverslips for 3 h. Cells were then fixed and stained with anti-FLAG M1 antibody without permeabilization. Mock cells were also stained with Alexa 647 phalloidin to visualize cells. Note that MT1F and its mutant-expressing cells degraded gelatin, creating non-fluorescent areas detected by dark zone. D, COS7 cells expressing MT1F and mutants were subjected to collagen film degradation assay. MT1F-DTY/KAF-expressing cells failed to degrade collagen film efficiently. E, HeLa cells expressing MT1F and the mutants were subjected to microcarrier bead invasion assay as described under “Materials and Methods.” Representative images for each transfection group are shown in the left panels, and data with means ± S.E. are shown in the right panels (n = 267). ***, p < 0.0001.
The DTY/KAF mutation disrupts two critical interactions at the interface: interactions between Asp385/Lys386 (molecule A) and Lys434 (molecule B) and a hydrogen bond between Thr412 (molecule A) and Tyr436 (molecule B). To examine the significance of each interaction further, we created three more mutants: DTY/AAF, DTY/DAF, and DTY/DTF. The DTY/AAF mutation was less drastic compared with the DTY/KAF mutation because Asp385 was changed to Ala. The DTY/DAF mutation was created to examine whether the disruption of only the hydrogen bond between Thr412 and Tyr436 would be sufficient to disrupt dimerization. The DTY/DTF mutant was the most minimal mutation (Y436F) and removed only the hydroxyl group of Tyr436. These mutations were incorporated into both the soluble Hpx domain (Fig. 8A) and full-length MT1F constructs (Fig. 8C). Co-expression of each of the soluble Hpx mutants with non-tagged full-length MT1-MMP (MT1-WT) indicated that DTY/AAF did not show any association with the cell surface, but both DTY/DAF and DTY/DTF mutants showed some cell surface association, although the levels of cell surface binding were much lower than that of wild-type Hpx-14 (Fig. 8B). Introduction of the DTY/AAF mutation into full-length MT1F showed significantly lower levels of proMMP-2 activation than that of wild-type MT1F, and it was the same as the DTY/KAF mutation, suggesting that mutation of Asp385 to Ala has the same effect as mutation to lysine. However, DTY/DAF and DTY/DTF mutants activated proMMP-2 to a level similar to that of wild-type MT1F, suggesting that interactions involving Asp385 are important for proMMP-2 activation to take place. In the case of collagen degradation (Fig. 8E), again DTY/KAF and DTY/AAF mutants showed significantly lower levels of collagenolytic activity compared with wild-type MT1F. Interestingly, DTY/DAF and DTY/DTF also showed significantly lower levels of collagenolytic activity than wild-type MT1F, although the level of activity was slightly higher than that of the DTY/KAF or DTY/AAF mutant, suggesting that collagenolytic activity is more sensitive to the mode of dimerization than proMMP-2 activation.
FIGURE 8.

Effect of mutations in symmetrical interface on biological function of MT1-MMP on the cell surface. A, schematic representation of mutants at symmetrical interface in soluble Hpx domain constructs used in B. B, COS7 cells expressing FLAG-tagged Hpx-14 constructs with or without expression of non-tagged full-length MT1-MMP were fixed and subjected to cell surface immunolocalization of soluble Hpx-14 using anti-FLAG M2 antibody and full-length MT1-MMP using an anti-MT1-MMP hinge region antibody. Note that Hpx-DTY/DAF and Hpx-DTY/DTF associated with the cell surface of MT1-WT-expressing cells weakly, whereas Hpx-DTY/KAF and Hpx-DTY/AAF did not associate at all. C, schematic representation of mutants at the symmetrical interface in full-length MT1F constructs used in D and E. D, COS7 cells were transfected with the expression plasmids for MT1F mutants as indicated or empty vector (Mock). Cells were then reacted with purified proMMP-2 in the serum-free medium for 8 h. Media were subjected to zymography analysis (top) and cell lysates for Western blotting using anti-FLAG M2 antibody (bottom). E, COS7 cells expressing MT1F and mutants were subjected to collagen film degradation assay as described under “Materials and Methods.”
DISCUSSION
Dimerization of MT1-MMP on the cell surface has emerged as an important mechanism to regulate two major activities of the enzyme, namely proMMP-2 activation and collagen degradation. This process is primarily driven by Hpx-14 dimerization, but the dimer interface was not known. In this study, we have addressed this question by solving the crystal structure of Hpx-14. The overall structure of Hpx-14 is similar to the Hpx domains of other MMPs, including MMP-1, MMP-2, MMP-9, and MMP-13, that consist of a four-bladed β-propeller (39). The structural overlay of Hpx-14 with Hpx-1, -2, -9, and -13 revealed that the Hpx-14 structure has distinguishing features. Each of the four propeller blades consists of three anti-parallel β-sheets, and the outermost strand is a combination of two very short β-sheets interrupted by a bulge, whereas blades I and IV at least in other MMPs have four anti-parallel β-sheets without a bulge (39). It has been shown that Hpx-9 forms a homodimer through the outermost strand of β-sheet (β4) in blade IV (44). In the case of Hpx-14, β4 is interrupted by a bulge, which may explain the different mode of homodimeric Hpx interaction from that of Hpx-9. In addition, the loop in the blade III outer surface in Hpx-14 is much shorter than those in Hpx-1, -2, -9, and -13, which commonly have more extended loops. This region is involved in the symmetrical dimer interaction of Hpx-14, and this shorter loop may facilitate dimerization of Hpx-14. Among all of the Hpxs, only MMP-9 possesses an intact β4 strand in blade II. The variation in Hpx domains among MMPs may facilitate interaction with specific binding partners and fulfill their distinct roles and functions.
We found two potential dimer interaction surfaces for the Hpx-14 in the crystal structure, consisting of a symmetrical and an asymmetrical dimer. Mutagenesis studies identified the symmetric interaction as fundamental for MT1-MMP to fulfill its biological function. The triple mutation designed to disrupt the symmetrical interaction (DTY/KAF) weakened the physical interaction of Hpx-14, and this was sufficient to significantly affect the dimer-dependent functions of the enzyme on the cell surface. Based on these observations, we have tentatively modeled a full-length MT1-MMP dimer on the cell surface (Fig. 9). The symmetrical interface is characterized by a 2-fold rotation based on the crystallographic symmetry, which allows both enzyme units to be orientated for dimerization through the transmembrane domain of the two molecules A and B. This model incorporates our previous finding that the enzyme forms dimers through both the Hpx and the transmembrane domains. The hinge region (linker-1) of MT1-MMP with 34 amino acids is longer than those of other MMPs, except that of MMP-9 with 64 amino acids, and is also O-glycosylated (47). Rosenblum et al. (48) recently reported that O-glycosylation of proMMP-9 at the linker makes this region flexible and can be extended as much as 85 Å in distance, providing enhanced dynamics of catalytic and Hpx domains. Therefore, we expect the linker-1 of MT1-MMP to be also flexible and extended, which makes it difficult to predict how the catalytic domain will be arranged relative to the Hpx domains on the cell surface.
FIGURE 9.

Potential molecular arrangement of full-length MT1-MMP on the cell surface. Based on the symmetrical interaction, other domains of MT1-MMP molecules are put together to form a full-length model. Because the structures of linker-1, linker-2, TM domain, and cytoplasmic domain are not known, we illustrated these regions schematically. Linker-1 has four O-glycosylation sites, which would extend the structure. Cys574 in the cytoplasmic domain is palmitoylated, which would create a loop structure. The relative positioning of the two catalytic domains cannot be predicted at this time because the structure of the O-glycosylated linker-1 is not known and is expected to be dynamic but should allow activation of proMMP-2 on the cell surface by forming a proMMP-2·TIMP-2·(MT1-MMP)2 complex.
We and others have previously reported that the Hpx domain of MT1-MMP is dispensable for proMMP-2 activation on the cell surface by analyzing Hpx domain-deleted mutants (22, 49). The present study, however, showed that the MT1F-DTY/KAF and MT1F-DTY/AAF mutants, which only form a weak dimer through the Hpx domain, are poor activators of proMMP-2. One possible explanation for this discrepancy is that the presence of the Hpx domain that cannot form a correct orientation may restrict the arrangement of the two catalytic domains, resulting in inefficient proMMP-2 activation. In the case of the complete Hpx deletion, both correct and restricted orientation of the Hpx domain is absent, and the catalytic domain arrangement may be more flexible, which allows efficient proMMP-2 activation. This hypothesis is supported by Cao et al. (50), who have shown that a MT1-MMP mutant with its Hpx domain replaced by that of MMP-2 could not efficiently activate proMMP-2 on the cell surface. Because the Hpx domain derived from MMP-2 does not form a homodimer, this may present a situation similar to that of MT1F-DTY/KAF or MT1F-DTY/AAF. We have previously reported that expression of the constitutively active form of Rac1 enhanced the Hpx-dependent dimerization and proMMP-2 activation by full-length MT1-MMP on the cell surface (19). Ingvarsen et al. (24) also reported that their divalent monoclonal anti-MT1-MMP Hpx domain antibody enforced the Hpx dimerization of MT1-MMP on the cell surface, resulting in enhanced proMMP-2 activation. Therefore, although the Hpx domain is dispensable, it acts as a device that controls proMMP-2 activation by forming a dimer in full-length enzymes on the cell surface.
Other dimerization-dependent activities of MT1-MMP, cell surface collagenolytic activity (21) and three-dimensional collagen matrix invasion activity measured by microcarrier beads, were also inhibited by the DTY/KAF mutations. These results indicate that the symmetrical dimer interface involving residues Asp385, Lys386, Thr412, and Tyr436 in blade II and blade III are the sites where these two Hpx-14 domains interact on the cell surface. Gelatin film degradation, cell surface localization, and cell migration promotion activity measured by a phagokinetic track motility assay were not affected by these mutations. Cell migration detected by this assay is CD44 shedding-dependent (9), which indicates that interaction with and shedding of CD44 do not require dimerization of the MT1-MMP Hpx domain.
Our data showed that disrupting the interaction involving Asp385 is crucial to maintain stable dimer interaction (Fig. 8B). This interaction is also significant to proMMP-2 activation activity (Fig. 8D). However, disruption of hydrogen bonds between Thr412 and Tyr436 is sufficient for reducing collagenolytic activity of MT1-MMP (Fig. 8E). These data indicate that collagen degradation requires a stricter dimer interaction than proMMP-2 activation. This may be due to the requirement of correct orientation of both of the catalytic domains, and two Hpx domains are essential for effective interaction with the insoluble substrate and recognition of the cleavage site. These observations not only indicate that the symmetrical interaction through blade II/III and blade III/II is the biological dimer interface but also suggest that slight changes in the mode of dimerization of Hpx-14 affect biological activities of MT1-MMP differently.
The asymmetrical interaction does not mirror a biological dimer interface because the RWD/AAK mutations did not dissociate the dimer of the Hpx-14 domain. Interestingly, the mutation seemed to affect the apparent molecular size of the dimer on BN-PAGE. Hpx-WT was around 30 kDa, whereas Hpx-RWD was found to be 40 kDa. Because BN-PAGE can separate proteins in their native conformation, the higher apparent mass may suggest that the Stokes radius of the Hpx-RWD/AAK dimer is larger, but no differences were detected upon gel filtration (Fig. 4), suggesting that the higher molecular mass detected in BN-PAGE might have been caused by the experimental condition in BN-PAGE for this particular mutant. Interestingly, incorporation of the RWD/AAK mutations into full-length MT1-MMP notably reduced the collagen film degradation activity, although the effect of the RWD/AAK mutations was much less than that of the DTY/KAF mutations. Because the positions of Arg339, Trp340, and Asp391 mutated in the RWD/AAK mutant are distant from the symmetrical dimer interface and these mutations are unlikely to affect the symmetrical dimer interface, one possibility is that the RWD/AAK mutations might have affected the interaction with collagen. Another possibility is that the RWD/AAK mutation somehow modified the dynamics of the Hpx domain, which might be necessary for collagenolytic activity (51). Nevertheless, these data highlight that the Hpx domain plays an important role in expressing MT1-MMP activity on the cell surface.
Our study elucidated the biological dimer interface of the MT1-MMP Hpx domain and demonstrated that this form of dimerization is crucial to express proMMP-2 activation and collagen degradation activities by full-length MT1-MMP on the cell surface. The next important step is to understand the exact molecular arrangement of full-length MT1-MMP and the dynamics of the molecule on the cell surface. Such information would provide further insights into the mechanisms of MT1-MMP action, which may contribute to identify specific means to control MT1-MMP-mediated cellular invasion and tissue damage seen in diseases such as cancer and arthritis.
Acknowledgments
We are grateful to professor R. Fässler and professor R. Huber for constant support of this study.
This work was supported, in whole or in part, by National Institutes of Health Grant AR40994. This work was also supported by Cancer Research UK Project Grant C1507/A2670, an Arthritis Research UK (formerly Arthritis Research Campaign) core grant for the Kennedy Institute of Rheumatology, Wellcome Trust Programme Grant 075473, and European Union Projects QLK3-CT-2002-02136 (MMP profiling), LSH-2002-2.2 (cancer degradome), and LSHG-2006-018830 (CAMP).
The atomic coordinates and structure factors (code 3C7X) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- MT1
- membrane type 1
- MMP
- matrix metalloproteinase
- Hpx
- hemopexin
- TM
- transmembrane
- DTY/KAF
- D385K/T412A/Y436F
- RWD/AAK
- R339A/W340A/D391K
- DTY/DTF
- Y436F
- DTY/AAF
- D385A/T412A/Y436F
- DTY/DAF
- T412A/Y436F
- MM
- molecular mass
- BN-PAGE
- blue native PAGE
- EGFP
- enhanced green fluorescent protein
- MT1F
- FLAG-tagged MT1-MMP.
REFERENCES
- 1. Sabeh F., Shimizu-Hirota R., Weiss S. J. (2009) J. Cell Biol. 185, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Itoh Y. (2006) IUBMB Life 58, 589–596 [DOI] [PubMed] [Google Scholar]
- 3. Visse R., Nagase H. (2003) Circ. Res. 92, 827–839 [DOI] [PubMed] [Google Scholar]
- 4. Tallant C., Marrero A., Gomis-Rüth F. X. (2010) Biochim. Biophys. Acta 1803, 20–28 [DOI] [PubMed] [Google Scholar]
- 5. Li Y., Aoki T., Mori Y., Ahmad M., Miyamori H., Takino T., Sato H. (2004) Cancer Res. 64, 7058–7064 [DOI] [PubMed] [Google Scholar]
- 6. Ohuchi E., Imai K., Fujii Y., Sato H., Seiki M., Okada Y. (1997) J. Biol. Chem. 272, 2446–2451 [DOI] [PubMed] [Google Scholar]
- 7. Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. (1994) Nature 370, 61–65 [DOI] [PubMed] [Google Scholar]
- 8. Knäuper V., Will H., López-Otin C., Smith B., Atkinson S. J., Stanton H., Hembry R. M., Murphy G. (1996) J. Biol. Chem. 271, 17124–17131 [DOI] [PubMed] [Google Scholar]
- 9. Kajita M., Itoh Y., Chiba T., Mori H., Okada A., Kinoh H., Seiki M. (2001) J. Cell Biol. 153, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belkin A. M., Akimov S. S., Zaritskaya L. S., Ratnikov B. I., Deryugina E. I., Strongin A. Y. (2001) J. Biol. Chem. 276, 18415–18422 [DOI] [PubMed] [Google Scholar]
- 11. Rozanov D. V., Hahn-Dantona E., Strickland D. K., Strongin A. Y. (2004) J. Biol. Chem. 279, 4260–4268 [DOI] [PubMed] [Google Scholar]
- 12. Deryugina E. I., Bourdon M. A., Jungwirth K., Smith J. W., Strongin A. Y. (2000) Int. J. Cancer 86, 15–23 [DOI] [PubMed] [Google Scholar]
- 13. Deryugina E. I., Ratnikov B. I., Postnova T. I., Rozanov D. V., Strongin A. Y. (2002) J. Biol. Chem. 277, 9749–9756 [DOI] [PubMed] [Google Scholar]
- 14. Endo K., Takino T., Miyamori H., Kinsen H., Yoshizaki T., Furukawa M., Sato H. (2003) J. Biol. Chem. 278, 40764–40770 [DOI] [PubMed] [Google Scholar]
- 15. Itoh Y., Seiki M. (2006) J. Cell. Physiol. 206, 1–8 [DOI] [PubMed] [Google Scholar]
- 16. Taniwaki K., Fukamachi H., Komori K., Ohtake Y., Nonaka T., Sakamoto T., Shiomi T., Okada Y., Itoh T., Itohara S., Seiki M., Yana I. (2007) Cancer Res. 67, 4311–4319 [DOI] [PubMed] [Google Scholar]
- 17. Okada Y., Morodomi T., Enghild J. J., Suzuki K., Yasui A., Nakanishi I., Salvesen G., Nagase H. (1990) Eur. J. Biochem. 194, 721–730 [DOI] [PubMed] [Google Scholar]
- 18. Strongin A. Y., Collier I., Bannikov G., Marmer B. L., Grant G. A., Goldberg G. I. (1995) J. Biol. Chem. 270, 5331–5338 [DOI] [PubMed] [Google Scholar]
- 19. Itoh Y., Takamura A., Ito N., Maru Y., Sato H., Suenaga N., Aoki T., Seiki M. (2001) EMBO J. 20, 4782–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lehti K., Lohi J., Juntunen M. M., Pei D., Keski-Oja J. (2002) J. Biol. Chem. 277, 8440–8448 [DOI] [PubMed] [Google Scholar]
- 21. Itoh Y., Ito N., Nagase H., Evans R. D., Bird S. A., Seiki M. (2006) Mol. Biol. Cell 17, 5390–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itoh Y., Ito N., Nagase H., Seiki M. (2008) J. Biol. Chem. 283, 13053–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miller M. C., Manning H. B., Jain A., Troeberg L., Dudhia J., Essex D., Sandison A., Seiki M., Nanchahal J., Nagase H., Itoh Y. (2009) Arthritis Rheum. 60, 686–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ingvarsen S., Madsen D. H., Hillig T., Lund L. R., Holmbeck K., Behrendt N., Engelholm L. H. (2008) Biol. Chem. 389, 943–953 [DOI] [PubMed] [Google Scholar]
- 25. Itoh Y., Kajita M., Kinoh H., Mori H., Okada A., Seiki M. (1999) J. Biol. Chem. 274, 34260–34266 [DOI] [PubMed] [Google Scholar]
- 26. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P., Round A., Fiedler S., Hermes C., Svergun D. (2007) J. Appl. Crystallogr. 40, S190–S194 [Google Scholar]
- 27. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 28. Svergun D. I. (1992) J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 29. Svergun D., Barberato C., Koch M. H. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 30. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Storoni L. C., McCoy A. J., Read R. J. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 [DOI] [PubMed] [Google Scholar]
- 32. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 33. Turk D., Podobnik M., Kuhelj R., Dolinar M., Turk V. (1996) FEBS Lett. 384, 211–214 [DOI] [PubMed] [Google Scholar]
- 34. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 35. Swamy M., Siegers G. M., Minguet S., Wollscheid B., Schamel W. W. (2006) Sci. STKE 2006, pl4. [DOI] [PubMed] [Google Scholar]
- 36. Uekita T., Itoh Y., Yana I., Ohno H., Seiki M. (2001) J. Cell Biol. 155, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anilkumar N., Uekita T., Couchman J. R., Nagase H., Seiki M., Itoh Y. (2005) FASEB J. 19, 1326–1328 [DOI] [PubMed] [Google Scholar]
- 38. Albrecht-Buehler G. (1977) Cell 11, 395–404 [DOI] [PubMed] [Google Scholar]
- 39. Gomis-Rüth F. X. (2003) Mol. Biotechnol. 24, 157–202 [DOI] [PubMed] [Google Scholar]
- 40. Gomis-Rüth F. X. (2004) in Handbook of Metalloproteins (Messerschmidt A., Bode W., Cygler M. eds) pp. 631–646, John Wiley & Sons, Ltd., Chichester, UK [Google Scholar]
- 41. Li J., Brick P., O'Hare M. C., Skarzynski T., Lloyd L. F., Curry V. A., Clark I. M., Bigg H. F., Hazleman B. L., Cawston T. E., et al. (1995) Structure 3, 541–549 [DOI] [PubMed] [Google Scholar]
- 42. Libson A. M., Gittis A. G., Collier I. E., Marmer B. L., Goldberg G. I., Lattman E. E. (1995) Nat. Struct. Biol. 2, 938–942 [DOI] [PubMed] [Google Scholar]
- 43. Gohlke U., Gomis-Rüth F. X., Crabbe T., Murphy G., Docherty A. J., Bode W. (1996) FEBS Lett. 378, 126–130 [DOI] [PubMed] [Google Scholar]
- 44. Cha H., Kopetzki E., Huber R., Lanzendörfer M., Brandstetter H. (2002) J. Mol. Biol. 320, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 45. Gomis-Rüth F. X., Gohlke U., Betz M., Knäuper V., Murphy G., López-Otín C., Bode W. (1996) J. Mol. Biol. 264, 556–566 [DOI] [PubMed] [Google Scholar]
- 46. Wittig I., Braun H. P., Schägger H. (2006) Nat. Protoc. 1, 418–428 [DOI] [PubMed] [Google Scholar]
- 47. Wu Y. I., Munshi H. G., Sen R., Snipas S. J., Salvesen G. S., Fridman R., Stack M. S. (2004) J. Biol. Chem. 279, 8278–8289 [DOI] [PubMed] [Google Scholar]
- 48. Rosenblum G., Van den Steen P. E., Cohen S. R., Grossmann J. G., Frenkel J., Sertchook R., Slack N., Strange R. W., Opdenakker G., Sagi I. (2007) Structure 15, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 49. Wang P., Nie J., Pei D. (2004) J. Biol. Chem. 279, 51148–51155 [DOI] [PubMed] [Google Scholar]
- 50. Cao J., Kozarekar P., Pavlaki M., Chiarelli C., Bahou W. F., Zucker S. (2004) J. Biol. Chem. 279, 14129–14139 [DOI] [PubMed] [Google Scholar]
- 51. Jozic D., Bourenkov G., Lim N. H., Visse R., Nagase H., Bode W., Maskos K. (2005) J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]