

Abstract
Prolactin (Prl) and progesterone (P) cooperate synergistically during mammary gland development and tumorigenesis. We hypothesized that one mechanism for these effects may be through mutual induction of receptors (R). EpH4 mouse mammary epithelial cells stably transfected with PR-A express elevated levels of PrlR mRNA and protein compared to control EpH4 cells that lack the PR. Likewise, T47D human breast cancer cells treated with P overexpress the PrlR and activate PrlR promoter III. PrlR promoter III does not contain a classical P response element but contains several binding sites for transcription proteins, including C/EBP, Sp1 and AP1, which may also interact with the PR. Using promoter deletion and site directed mutagenesis analyses as well as gel shift assays, cooperative activation of the C/EBP and adjacent Sp1A, but not the Sp1B or AP1, sites by P is shown to confer P responsiveness leading to increased PrlR transcription.
Keywords: prolactin receptor promoter, prolactin, progesterone, mammary
1. Introduction
Development of the mammary gland occurs through the action of a cascade of hormones and growth factors. Paramount among these signaling molecules are the hormones prolactin (Prl) estrogen (E), and progesterone (P) (Hovey et al., 2002). Each works through its concomitant receptor, PrlR, ER, and PR, respectively. In the mouse, mammary ductal elongation occurs through the action of E while lateral branching occurs through the interaction of E plus P. Tertiary ductal branching and alveolar development result from signaling pathways downstream of the PR and PrlR. We found that transfection of PR into EpH4 mouse mammary epithelial cells increased branching morphogenesis in collagen gels through induction of the homeobox gene Msx2 (Satoh et al., 2007). Additionally, the cooperative effects of these three hormones may occur in part due to mutual induction of each other’s receptors. While Prl upregulates PR mRNA in the mammary gland (Ormandy et al., 1997) and activates ERα in T47D cells (Gonzalez et al., 2009), PrlR mRNA is induced by P in T47D cells (Tseng and Zhu, 1998) and by E in MCF7 cells (Dong et al., 2006).
Previously, we showed that PR and PrlR are co-expressed temporally and spatially in the developing mouse mammary gland (Hovey et al., 2001). As such, they work synergistically to induce DNA synthesis leading to ductal branching and lobuloalveolar formation. This synergy was also reported by Morabito et al. (Morabito et al., 2008) who showed that the distal enhancer of the MMTV-LTR is activated when PrlR and PR signaling pathways converge at adjacent Stat5 and Mammary Gland Specific Complex (MGSC) response elements, respectively. Activated GATA proteins are part of the MGSC. In an effort to further explore the mechanism by which P and Prl act synergistically in mammary gland development, we examined the mechanism by which P influences expression of the PrlR.
Mouse and human PrlR each have one long and several short isoforms (Davis and Linzer, 1989;Hu et al., 2001;Trott et al., 2003). In each species, the major cell-associated receptors contain the same extracellular and transmembrane domains (Clevenger et al., 2003), with splice variants from the remaining exons resulting in different intracellular/signaling domains. The PrlR gene in both mice and humans has several (5 for mouse, 6 for human) exon 1s (Dong et al., 2006;Ormandy et al., 1998). Each exon 1 has its own promoter, several of which have been identified. The promoter/exon 1 combination that is utilized may depend on which transcription factors are present in a particular tissue at a particular time (Ormandy et al., 1998). The most generic of these promoters and the one that is functional in both the mouse and human mammary gland is PrlR promoter region III (Dong et al., 2006;Ormandy et al., 1998). The human promoter in this region (−439/−179) is 81% similar in sequence to the mouse promoter, as is the rat PIII promoter (Hu et al., 1996). However, the region of the PIII promoter of interest to us is the proximal part containing Sp1 and C/EBP response elements; these particular sequences are 100% homologous between the human and mouse.
Herein we examine the induction of PrlR expression by P/PR in both mouse and human mammary cells. We find that P/PR induces expression of the PrlR through tethering to the activated transcription factors C/EBP (CCATT/Enhancer Binding Protein ) and Sp1 (Specificity Protein 1). Our understanding of the relationship of Prl/PrlR and P/PR is important in view of the fact that these hormone/receptor interplays may be involved in tumorigenesis as well as in normal growth.
2. Materials and Methods
2.1 Cell culture
The human breast epithelial cancer cell line T47D and derivatives, T47DY, T47DYA and T47DYB, were routinely maintained in RPMI 1640 media supplemented with 5% fetal bovine serum (FBS; Lonza, Walkersville, MD), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Invitrogen, Gaithersburg, MD) and insulin (10 μg/ml) (Sigma, St. Louis, MO). Cells were plated at a density of 250, 000 cells/ml in growth media and sequentially stepped down to media containing charcoal stripped serum (CSS, 1% to 0.1%; Gemini Bio-Products, W. Sacramento, CA) over two days in order to deplete the cells of endogenous steroid hormones. Cells were incubated in the presence of media containing 0.1% CSS for an additional 48 hr prior to exposure to either 10−8 or 10−7 M P (Sigma, St. Louis, MO). For RNA or protein isolation, cells were treated with P for 24 or 48 hr; for nuclear extracts, cells were exposed to P for 0.5, 1, 2, or 4 hr.
EpH4 cells, a mouse mammary epithelial cell line, were transfected and clones selected as previously described (Satoh et al., 2007). Seven EpH4-PR clones were isolated from the EpH4 transfected lines; all clones expressed similar levels of PrlR message. One clone was selected for subsequent experimental work. EpH4-EV or EpH4-PR cells were routinely maintained in DMEM Growth Media (DMEM/GM) containing 10% FBS, 2mM L-glutamine (Invitrogen), 20 mM HEPES (Sigma), penicillin (100 U/ml) and streptomycin (100 μg/ml) and neomycin (G418; 400 μg/ml). For RNA and protein isolation, cells were plated at a density of 100, 000 cells/ml in DMEM/GM in the absence of antibiotics and grown for 48 hr to approximately 70–90% confluence. Cells were then rinsed with PBS and media changed to DMEM/CSS with and without P (10−8 or 10−6 M) for the indicated times.
P was serially diluted in media starting from a 1 mM stock solution prepared in 100% ethanol. Control cells were treated with serially diluted ethanol as vehicle control.
2.2 Semi-quantitative RT-PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen). Following DNase treatment, 1 μg RNA was used for each 25 μl reverse transcriptase reaction as described (Satoh et al., 2007). For all PCR analyses, the number of cycles used was selected at the linear expression level for each gene tested. PCR products were resolved by gel electrophoresis, quantified using NIH Image J and expressed relative to the corresponding housekeeping gene. PCR primers used and conditions for amplification of the mPrlR gene were as previously validated and described (Hovey et al., 2001). Isoform specific plasmids were used as positive controls in all cases.
2.3 Western blot analysis
EpH4-EV, EpH4-PR or T47D cells were collected, homogenized (50 mM Tris-HCl, pH 7.5, 50 mM sodium chloride, 1 mM DTT, 1mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin and 0.1 mM sodium orthovanadate), sonicated, and protein concentration determined (Bradford, 1976). Proteins were subjected to 10% SDS-PAGE, transferred to Hybond nitrocellulose ECL membrane and probed with mPrlR antibody (M2-5 for EpH4 cells; 1:250 dilution (Vonderhaar et al., 1985); or hPRLR antibody (Invitrogen; 2 μg/ml). Tubulin was used as loading control (Sigma). Immunoreactivity was determined using enhanced chemiluminescence (ECL Plus; GE Healthcare, Pittsburgh, PA).
2.4 Mouse PrlR promoter constructs
The mouse PrlR promoter III (824-PIII; GeneBank accession #AC074172; a kind gift from Dr. Robert Callahan, NCI) including the 5’ flanking region/exon 1 (Dong et al., 2006) and contained within a pGL3 basic vector (Promega, Madison, WI) was PCR cloned with 5 Prime (Gaithersburg, MD) using XhoI/HindIII primers to make the −439/−179-PIII (-439-PIII) construct. Primers (Table 1A) were used at a concentration of 6 μM each with 50 ng of 824-PIII. Deletion mutants (-386-PIII, -398-PIII) were made using the -439-PIII template and primers (Table 1A) at a concentration of 10 μM each. PCR products were resolved on a 2% agarose gel, excised, and extracted with a MinElute Gel Extraction kit (Qiagen, Valencia, CA). Purified products were digested with XhoI/HindIII and ligated with T4 Rapid Ligation kit (Roche Applied Science, Indianapolis, IN), purified, cloned, and sequence verified.
Table 1.
| A.PIII deletion primers | Primer sequence (5’ – 3’) |
|---|---|
| PIII-XhoI forward | CGGCTCGAGACTGTTTTGCCTCCAAGCAAGGA |
| PIII-HindIII reverse | ACCAAGCTTTTTTCAGTGACAGGTAAAGCTGTTTCC |
| -386-PIII forward | CGGCTCGAGTAGTGAAGAAAGAGTGAACAAG |
| -398-PIII forward | CGGCTCGAGGACTCCTCCTCTAGTGAAGAA |
| B. PIII site-directed mutagenesis primers | Primer sequence (5’ – 3’)* |
| AP1 forward reverse | CGCCTTCCTGCTCTGTCTAACTAACTCCTCTCCTGCGTTCTGG CCAGAACGCAGGAGAGGAGTTAGTTAGACAGAGCAGGAAGGCG |
| Sp1A forward reverse | GCATGTTGCAACACTGACTACTCATCTAGTGAAGAAAGAG CTCTTTCTTCACTAGATGAGTAGTCAGTGTTGCAACATGC |
| Sp1B forward reverse | GAGCTGGGCTTTCCACGCATTCCTGCTCTGTCTCACTC GTGAGACAGAGCAGGAATGCGTGGAAAGCCCAGCTC |
| C/EBP forward reverse | CCAGCAAGGAACATGCATGCTACAACACTGACTCCTCCTCTAG CTAGAGGAGGAGTCAGTGTTGTAGCATGCATGTTCCTTGCTGG |
| Double forward reverse | CAGCAAGGAACATGCATGTTACAATACTGACTACTCATCTAG CTAGATGAGTAGTCAGTATTGTAACATGCATGTTCCTTGCTC |
| Triple forward reverse | CAGCAAGGAACATGCATGTTACAATACTGACTACTCATCTAG CTAGATGAGTAGTCAGTATTGTAACATGCATGTTCCTTGCTC |
| * Template for all site-directed mutagenesis constructs is -439-PIII deletion mutant; template for triple had Sp1B site already mutated. | |
| C. Gel shift oligos | Primer sequence (5’ – 3’) |
| Sp1A forward reverse | ATGTTGCAACACTGACTCCTCCTCTAGTGAAGA TCTTCACTAGAGGAGGAGTCAGTGTTGCAACAT |
| Sp1A mutated forward reverse | ATGTTGCAACACTGACTACTCATCTAGTGAAGA TCTTCACTAGATGAGTAGTCAGTGTTGCAACA |
| Sp1B forward reverse | GAGCTGGGCTTTCCCCGCCTTCCTGCTCTGTCT AGACAGAGCAGGAAGGCGGGGAAAGCCCAGCTC |
| Sp1B mutated forward reverse | GAGCTGGGCTTTCCACGCATTCCTGCTCTGTCT AGACAGAGCAGGAATGCGTGGAAAGCCCAGCTC |
| C/EBP forward reverse | AGGAACATGCATGTTGCAACACTGACTCCT AGGAGTCAGTGTTGCAACATGCATGTTCCTTGC |
| C/EBP mutated forward reverse | GCAAGGAACATGCATGTTACAATACTGACTCCT AGGAGTCAGTATTGTAACATGCATGTTCCTTGC |
Primer pairs (Table 1B) for single site-directed mutagenesis were constructed to contain specific transcription factor binding site mutations using the QuikChange II XL Site-Directed Mutagenesis kit (Stratagene, LaJolla, CA). Reactions were carried out as directed and DNA was sequence verified. Subsequent mutations were performed as above on the -439-PIII construct to create either a double (C/EBP and Sp1A) or triple mutant (C/EBP with both Sp1A and Sp1B); all sequences were verified.
2.5 Luciferase and β-galactosidase reporter assays
T47D cells were plated in 35mm dishes at a density of 125, 000 cells/ml in RPMI1640 with 5% FBS. After overnight attachment, each well was transiently transfected using FuGENE6 with 1 μg β-galactosidase reporter construct and pGL3 basic vector containing either 824-PIII or PIII-mutated constructs. The following day, media were replaced with RPMI1640 with 1% CSS in the presence or absence of 10−7 M P. After an additional 24 hr, cells from replicate wells were then collected in 1x Reporter Lysis Buffer (Luciferase Assay System, Promega) and assayed for luciferase activity on a Lumat LB9507 luminometer. β-galactosidase activity was assayed using an aliquot from the above lysed cells using Promega’s β-galactosidase Enzyme Assay System as instructed by the manufacturer. Activity was measured at 405nm. Luciferase activity was compared to β-galactosidase units in order to normalize results among transfections.
2.6 Gel shift assay
Oligonucleotide probes were made corresponding to specific sites identified on the mPrlR promoter PIII (Table 1C). Single stranded oligos were resuspended in STE (10mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA) buffer and annealed at equimolar amounts. Probes were end-labeled with γ32P-ATP (3000Ci/mmol; PerkinElmer, Waltham, MA) and T4 Polynucleotide Kinase by the phosphorylation reaction described in Gel Shift Assay Systems (Promega). Unincorporated nucleotide was removed using G-25 Sephadex columns (GE Healthcare).
2.7 Preparation of nuclear extracts for DNA binding reactions
T47D cells were plated as outlined above for RNA and protein isolation. Cells were collected, resuspended, and lysed as previously described (Goldhar et al., 2005). Nuclear extracts were frozen at −80° C until ready for use. Radiolabeled consensus oligonucleotide was incubated with nuclear extract using the Gel Shift Assay system (Promega) and run out on 6% DNA Retardation polyacylamide gels (Invitrogen), Gels were dried on Whatman filter paper and exposed to BioMax film.
2.8 Cross-linking to Protein A beads
Protein A-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) were washed and incubated with either PR (Santa Cruz; sc-538) or Sp1 (Santa Cruz, sc-59) antibody for 1 hr at room temperature. Beads were washed twice with 0.2M sodium borate, pH 9, then mixed, end-over-end, with 5.2 mg/ml dimethyl pimelimidate (Sigma) in 0.2M sodium borate, pH 9, for 1 hr at room temperature. Following three washes with 0.2M ethanolamine (Sigma), beads were resuspended in PBS.
Nuclear extracts (450 μg) were added to the cross-linked beads and incubated for 1.5 hr at 4° C while mixing. Beads were washed in buffer containing protease inhibitors, collected, resuspended in Tris-Glycine SDS Sample Buffer (Invitrogen) and boiled. Resulting supernatant was loaded onto a gel for western blot analysis and probed with Sp1 antibody.
2.9 Statistics
Data are expressed as the mean ± SEM. The statistical significance of the difference between groups was determined using Student’s t-test or ANOVA as appropriate. P ≤ 0.05 was considered significant.
3. Results
The spatial/temporal co-localization of the PR and PrlR in the developing mouse mammary gland (Hovey et al., 2001) led us to examine the expression of the PrlR isoforms in EpH4 cells stably transfected with PR. EpH4 cells were originally derived from the mid-pregnant murine mammary gland (Pinkas and Leder, 2002;Reichmann et al., 1992) and lack PR (Satoh et al., 2007). Transfection of the EpH4 cells with cDNA for the mouse PR resulted in expression of PR-A (Satoh et al., 2007), which is known to act in an unliganded manner (Jacobsen et al., 2002). As shown in Figs. 1A and 1B, there is at least a 10-fold increase (range 10–80-fold) in expression of the mRNA for the PrlR in EpH4-PR cells compared to control EpH4-EV or untransfected EpH4 parental cells; the most abundant PrlR, the long form (Prl-LF), shows a 10-fold increase, while short forms, PrlR-SF2 and PrlR-SF3, demonstrates a 28- and 79-fold increase, respectively. No additional effect was observed with 10−6M P. Neither EpH4-EV nor EpH4-PR cells express PrlR-SF1.
Fig. 1.

Increased expression of PrlR mRNA and protein in EpH4-PR cells and T47D cells treated with progesterone.
A. Parental EpH4, EpH4-EV and EpH4-PR cells were plated and maintained in DMEM growth media containing 10% FBS for 48 hr. Media was then changed to 5% CSS and, 24 hr. later, cells were collected. RNA was extracted, DNase-treated, and subjected to RT-PCR using PCR primers specific for mouse PrlR extracellular domain and G3PDH. PCR products were resolved by gel electrophoresis and quantified using NIH Image J. Densitometric histograms from three separate determinations are expressed relative to G3PDH. * = p 0.001 relative to EpH4-EV.
B. EpH4-EV and EpH4-PR cells were plated and maintained in growth media containing 10% FBS for 48 hr. Media were then changed to 5% CSS ± 10−6M P and 24 hr. later cells were collected. RNA was extracted, DNase-treated, and subjected to RT-PCR using PCR primers specific for mouse PrlR LF, SF1, SF2, SF3, and G3PDH. PCR products were resolved by gel electrophoresis. No message was observed for SF1. Data shown are representative of 3 separate experiments.
C. EpH4-EV and EpH4-PR cells were plated, grown, and treated as in (A) above. Cells were then collected in homogenization buffer and whole cell lysates were subjected to western blot analysis with M2.5 antibody that detects the extracellular domain of the mouse PrlR. Nitrocellulose blots were stripped and probed with anti-tubulin as a loading control.
D. T47D, T47DY, T47DYA and T47DYB cells were plated in RPMI growth media containing 5% FBS. The next day, cells were switched to media containing 1% CSS for 24hr and then to media containing 0.1% CSS ± 10−7 M P for up to 48 hr. Whole cell lysates were subject to western blot analysis with antibody detecting the extracellular domain of the PrlR. Data shown are after 24 hr treatment with P. Blots were stripped and probed with anti-tubulin as a loading control.
Using an antibody specific for the common extracellular domain of the mouse PrlRs, we examined PrlR protein expression in EpH4-EV and EpH4-PR cells. Figure 1C shows that whereas there is a weak signal for PrlR-LF in the EpH4-EV cells, there is a significant amount in the EpH4-PR cells. Addition of P to the medium had no effect on the expression of the PrlR-LF (not shown) suggesting that the increase in PrlR expression in the EpH4-PR cells was due to unliganded PR-A. PrlR-LF is the only isoform detected; this antibody is unable to detect any SFs of the mouse PrlR.
Western blot analysis for human PrlR was performed on extracts of T47D cells. T47D cells contain both the A and B forms of the PR. Derivatives of this cell line are available that lack PR (T47DY) or contain only the A (T47DYA) or B (T47DYB) form. T47D and T47DYA cells have the highest baseline expression of PrlR-LF, possibly due to the unliganded action of PR-A; exogenous P does not increase the level of expression (Fig. 1D). On the other hand, T47DYB cells have a lower baseline but show a 1.7-fold induction of PrlR-LF with 10−7 M P treatment for 24 hr. Similar results were seen as early as 6 hr and continued to 48 hr (not shown). T47DY cells lacking PR demonstrate a baseline PrlR expression, but are unresponsive to added P. PrlR expression was linear at protein concentrations examined (25–100 μg, not shown).
Examination of the sequence of the PIII region of the mPrlR promoter showed that it does not contain a canonical P responsive element (PRE) but it does contain several transcription factor binding sites including an AP1, a C/EBP and two SP1 sites (Hu et al., 1998) through which the PR may act (Fig. 2). These sites are all in the proximal promoter -439-PIII. Response elements for C/EBP and Sp1A are proximally spaced in this region suggesting that these transcription factors may both tether to PR while simultaneously binding to the promoter. The sequences of the mouse and human PrlR PIII promoter are 81% homologous (Hu et al., 1998) allowing us to use the mouse PrlR PIII promoter for studies in human cells. Since the EpH4-PR cells express only the PR-A, we utilized the human T47D breast cancer cell line that expresses both PRA and PRB, to further examine the effects of exogenous P on activation of the promoter. The T47D cells treated with 10−7 M P show a 2-fold increase in the activity of the full-length 824-PIII-luciferase construct (Fig. 3A), while the T47DYB cells give a 4-fold induction of the 824-PIII promoter with P treatment. Neither theT47DY nor the T47DYA show an increase in activation of the 824-PIII promoter activity with P treatment.
Fig. 2.
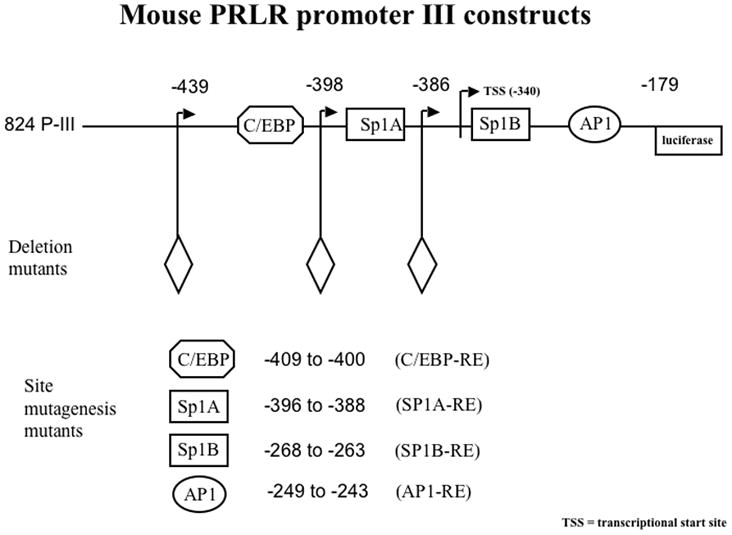
The functional PIII promoter for the PrlR gene. Full-length promoter region III of the mouse PrlR is 824 bp (henceforth referred to as 824-PIII). 824-PIII contains the 5’ flanking region of the promoter as part of the non-coding first exon of the PrlR gene. 824-PIII lacks a PRE; other putative DNA responsive elements that may tether to PR include C/EBP, Sp1A and Sp1B, and AP1 among others. Diamond denotes the 5’ end of the deletion.
Fig. 3.

Screening for progesterone responsive regions in the 824-PIII promoter by deletion and site directed mutagenesis analyses.
A. T47D, T47DY, T47DYA and T47DYB cells were plated separately in RPMI growth media with 5% FBS and left to attach overnight. Cells were transfected with the 824-PIII-luciferase construct for 24 hours. Cells were then washed twice with 1X PBS prior to culture for 24 hr in media containing 1% CSS without (closed bars) or with (open bars) 10−8 M P. Data are mean ± SEM for luciferase activity and are corrected for transfection efficiency as described in Materials and Methods. * = p 0.001.
B. T47D cells were plated as in (A) and transfected with the indicated full length 824-PIII or deletion mutant -439-PIII promoter-luciferase constructs . Cells were treated as in (A) in media containing 1% CSS without (closed bars) or with (open bars) 10−8 M P. Data are mean ± SEM for the luciferase activity and are corrected for transfection efficiency as described in Materials and Methods. ** = p 0.05.
C. T47D cells were transfected with the indicated promoter-luciferase deletion constructs. Treatment was as in (A) in media containing 1% CSS without (closed bars) or with (open bars) 10−7 M P. Data are mean ± SEM for the luciferase activity and are corrected for transfection efficiency as described in Materials and Methods. * = p 0.001.
D. T47D cells were transfected with the indicated promoter-luciferase site-directed mutagenesis constructs and treated as in (A) prior to treatment in media containing 1% CSS without (closed bars) or with (open bars) 10−7 M P. Two binding sites (C/EBP and Sp1A) are disrupted in the “Double” and three (C/EBP, Sp1A and Sp1B) in the “Triple” constructs. Data are mean ± SEM for the luciferase activity and are corrected for transfection efficiency as described in Materials and Methods. * = p 0.001; ** = p 0.05.
The region of the mPrlR promoter responsive to P was further investigated by deletion and site directed mutagenesis analyses. T47D cells were transfected with various deletion or site directed mutants of the mPRL PIII-luciferase promoter construct prior to treatment with 10−7 M P. A construct of PIII-luciferase (-439-PIII; Fig. 2) that lacks the 5’ flanking region but retains the C/EBP, Sp1A, Sp1B and AP1 response elements has the same baseline and inducible luciferase activity as does the intact 824-PIII-luci construct (Fig. 3B). Of the other deletion mutants examined, the loss of C/EBP alone (-398-PIII; Fig. 2) or in combination with Sp1A (-386-PIII; Fig. 2) lowers the baseline of the luciferase activity but retains a partial P response; the retention of the Sp1A site in the absence of the C/EBP site gives an 1.8-fold increase in luciferase activity with P treatment compared to a 3-fold increase when both transcription sites are present (Fig. 3C). Deletion of both the C/EBP and Sp1A sites results in the loss of P induction entirely (Fig 3C). These data suggest that the C/EBP site contributes to the P activation of the promoter, possibly through interaction with the Sp1A site.
A series of site-directed mutagenesis constructs were used to confirm that P acts through C/EBP and Sp1A. In all cases, comparable results were obtained using either deletion or site-directed mutagenesis mutants of the promoter. Fig. 3D shows reduced basal and inducible activity for C/EBP or Sp1A site directed mutants. The C/EBP-Sp1A “double” mutant that retained the remainder of the PIII promoter with intact Sp1B and AP1 sites has a reduced baseline but also shows complete loss of a P response. These data are comparable to the effects seen with its deletion mutant counterpart, -386-PIII (Fig. 3C). A similar comparison with the C/EBP site-directed mutagenesis mutant and its deletion counterpart -398-PIII is observed. In constructs with mutated Sp1B or AP1 sites, the P induction of luciferase activity is comparable to that of the non-mutated promoter construct (Fig. 3D). Thus, neither Sp1B nor AP1 are essential to the effect of P on induction of expression of the PrlR. Similarly, the triple mutant, with mutated Sp1B site as well as mutated C/EBP and Sp1A sites, also lacked inducibility by P.
The effects of P on the binding of C/EBP and Sp1A on the PIII promoter were examined in nuclear extracts of T47D cells treated with P using EMSA. As shown in Figure 4A, when nuclear extracts of P treated (1, 2, 4 hr) and non-treated cells are incubated with an Sp1A oligonucleotide probe, hormone treatment (lanes 2, 3, 4) increases binding when compared to the untreated extracts (Fig. 4A). The specificity of binding is shown by competition with a 250-fold excess of unlabeled Sp1A oligonucleotide (lane 5) and the lack of competition from the unrelated NFκB oligonucleotide. Similar results are obtained with C/EBP (Fig. 4B). C/EBP binding to the promoter is increased at 1 and 2 hr (lanes 3 and 4) of 10−8M P treatment of T47D cells. The specificity of binding is seen by competition with unlabeled C/EBP oligonucleotide (lane 5). Similar to the results of Hu et al. (Hu et al., 1998), complexes are not consistently formed when the Sp1B consensus sequence is used as a probe (Fig. 4C).
Fig. 4.

Effect of P on recruitment of nuclear proteins from T47D cells to transcription factor response sites on the PrlR promoter.
A. T47D cells were plated in growth media containing 5% FBS. Over a 4 day period, serum concentration was stepped down, first to 1% and then to 0.1% CSS. On day 5, cells were treated with 10−8 M P or vehicle for 0, 1, 2 or 4 hr. Nuclear proteins were incubated with a 32P-labeled oligonucleotide probe with sequence identical to the Sp1A response site prior to electrophoresis. A faint protein complex bound to probe in the absence of P (0 hr; lane 1) showed induction with P treatment with time (lanes 2–4). Specificity of binding is demonstrated by competition with excess unlabeled Sp1A oligo (lane 5) and lack of competition with the unlabeled unrelated NFκB oligo (lane 6).
B. T47D cells were plated and cultured as in (A). On day 5, cells in media containing 0.1% CSS were treated with 10−8 M P or vehicle for 0, 0.5, 1, or 2 hr. Nuclear proteins were incubated with a 32P-labeled oligonucleotide probe with sequence identical to the C/EBP response site prior to electrophoresis. A protein complex bound to probe in the absence of P (0 hr; lane 1) showed induction with P treatment with time (lanes 2–4). Specificity of binding is demonstrated by competition with excess unlabeled C/EBP oligo (lane 5).
C. Nuclear extracts obtained as in (B) were incubated with 32P-labeled oligonucleotide probe with sequence identical to the Sp1B response site. Specific complexes for Sp1B do not appear to be upregulated by P treatment. Specificity of binding is demonstrated by competition with an excess of unlabeled Sp1B oligo (lane 5) and lack of competition with the unlabeled mutated Sp1B oligo (lane 6).
Similar increased binding of Sp1A and C/EBP to the nucleus is shown in EpH4-PR cells that contain activated PR-A as compared to EpH4-EV cells that lack PR. Fig. 5A demonstrates that nuclear extracts of EpH4-EV cells (lane 1) have non-perceptible Sp1 proteins binding to the Sp1A oligonucleotide. Binding is substantial, however, in nuclear extracts from EpH4-PR cells (lane 5). Specificity of the binding is observed with EpH4-PR cell extracts by competition by a 25-fold excess unlabeled Sp1A oligonucleotide (lane 6), but no competition using a 250-fold molar excess of unlabeled mutated Sp1A oligonucleotide (lane 7) and partial blocking of binding with Sp1 antibody (lane 8). Binding to the C/EBP oligonucleotide probe by extracts from EpH4-EV and EpH4-PR cells appears similar (Fig. 5B, lanes 1 and 4, respectively). However, while excess unlabeled C/EBP oligonucleotide abolished the binding of the extracts, competition with unlabeled mutant C/EBP oligonucleotide, which should not interfere with transcription factor binding (lanes 3 and 6), shows much more signal in the EpH4-PR lane, indicating the presence of more activated C/EBP in EpH4-PR cells. This result agrees with previous findings that PRB alone can activate C/EBP (Jacobsen et al., 2002). As was the case for T47D cells, Sp1B oligonucleotide (Fig. 5C) did not give consistent results when incubated with extracts from EpH4-EV and EpH4-PR cells.
Fig. 5.

Analysis of DNA binding proteins present in EpH4-EV and EpH4-PR cells.
A. Nuclear extracts were prepared from EpH4-EV (lanes 1–4) and PR (lanes 5-8) cells that were first grown in media containing 10% FBS and then switched to media containing 5% CSS for 24 hr. Extracts were incubated in all cases with 32P-Sp1A oligonucleotide probe either alone (lanes 1 and 5) or with an excess of unlabeled Sp1A oligo (lanes 2 and 6), an excess of unlabeled mutant Sp1A oligo (lanes 3 and 7) or with Sp1 antibody (lanes 4 and 8) to determine specificity.
B. The nuclear extracts from (A) were incubated in all cases with 32P-C/EBP oligonucleotide probe either alone (lanes 1 and 4) or with an excess of unlabeled C/EBP oligo (lanes 2 and 5) or an excess of unlabeled mutant C/EBP oligo (lanes 3 and 6) to determine specificity.
C. The nuclear extracts from (A) were incubated in all cases with 32P-Sp1B oligonucleotide probe either alone (lanes 1 and 5) or with an excess of unlabeled Sp1B oligo (lanes 2 and 6), an excess of unlabeled mutant Sp1B oligo (lanes 3 and 7) or with Sp1 antibody (lanes 4 and 8) to determine specificity.
To determine whether the effect of P occurs directly on the level of activated Sp1 in P treated T47D cells and whether the activated Sp1 was associated directly with the PR, immunoprecipitation and western blot analyses were performed. Whole cell lysates of T47D cells with and without P treatment for 24 hr show equivalent amounts of total Sp1 protein (Fig 6A). Immunoprecipitates of nuclear extracts of T47D cells that had been incubated with Sp1 antibody-conjugated agarose beads, subjected to gel electrophoresis and probed with Sp1 antibody, show a minimal increase in activated Sp1 after P treatment (Fig 6B). However, when the nuclear extracts were incubated with PR antibody-conjugated beads, electrophoresed and probed with Sp1 antibody (Fig. 6C) more Sp1 is bound to PR when cells were treated with 10−8 or 10−7 M P. While the formation of non-specific complexes cannot be ruled out, the data suggest that PR and Sp1 may be associated in an active complex that may then associate with the PrlR promoter. The complex interplay of PR with various transcription factors including Sp1 to regulate the expression of the PrlR is an important area for further studies on P and Prl action in normal mammary gland development and breast cancer.
Fig. 6.

Interaction of SP1 protein with progesterone receptor.
T47D cells were plated and grown for 24 hr in media containing 5% FBS. Media was changed to 1% CSS, then to 0.1% CSS, and followed by treatment with either ethanol vehicle or 10−8 or 10−7 M P for 1 hr.
A. Nuclear extracts were subjected to gel electrophoresis and western blot analysis performed using antibody against Sp1.
B. T47D cells were grown as in (A) with final incubation with either ethanol vehicle or 10−8 M P for 1 hr. Nuclear extracts from both treatments were incubated with Sp1 antibody conjugated to Protein A agarose beads. Bound proteins were released by boiling and subjected to western blot analysis probed with the Sp1 antibody.
C. Nuclear extracts from cells treated with either ethanol vehicle or 10−8 or 10−7 M P for 1 hr were incubated with PR antibody conjugated to Protein A agarose beads. Bound proteins were released by boiling and subjected to western blot analysis probed with the Sp1 antibody.
4. Discussion
The synergistic action of P and Prl in mammary gland development (Hovey et al., 2001) and breast cancer cells (Morabito et al., 2008) presents a unique opportunity to investigate mechanisms by which these two hormones influence each other’s action. Thus we hypothesized that mutual induction of receptors may account, at least in part, for this synergy. Prl induction of PR, in the mouse mammary gland but not in tumors, was previously reported based on binding activity (Koseki et al., 1987) whereas in human breast cancer cells, Prl increased expression of the PR mRNA (Ormandy et al., 1997). Similarly, P has been shown to increase Prl binding in cultured human breast cancer cell lines (Leroy-Martin and Peyrat, 1989;Murphy et al., 1986) and in primary cultures of mouse mammary cells (Sakai et al., 1979).
The mouse PrlR gene has five exon 1s while the human gene has six (Dong et al., 2006;Hu et al., 1999;Ormandy et al., 1998). Several promoters have been identified that are associated with each exon suggesting that the promoter/exon 1 combination utilized in a given target tissue may depend on which transcription factors are active (Ormandy et al., 1998). PrlR promoter region III is the most generic and the one that is functional in the mammary gland in both the mouse and in humans (Hu et al., 1998;Ormandy et al., 1998). In this study, we examined the proximal part of the PIII promoter containing Sp1 and C/EBP response elements; these particular sequences are 100% homologous between the human and mouse (Hu et al., 1999). A study of the rat PrlR promoter region III showed that it has a C/EBPβ and a nearly adjacent Sp1 site in its proximal region (Hu et al., 1998). The proximal human promoter retains these response elements and is, in fact, 81 % similar in sequence to the rat promoter (Dong et al., 2006), as is the mouse PIII promoter (Ormandy et al., 1998). The rat generic PrlR PIII promoter is constitutively activated by C/EBP and Sp1 in liver and gonadal cells (Hu et al., 1996;Moldrup et al., 1996); the human PrlR promoter in MCF7 breast cancer cells is activated by E acting through its receptor which tethers to these same transcription factors (Dong et al., 2006). Moreover, these transcription factors have been shown to act cooperatively on other promoters such as that of CDllc in myeloid cells (Lopez-Rodriguez et al., 1997) and the placenta-specific trophoblast gene PLAC-1 (Koslowski et al., 2009).
C/EBPβ is essential to mouse mammary gland development (Seagroves et al., 1998;Seagroves et al., 2000) as well as other processes such as mouse adipocyte differentiation (Tang and Lane, 1999). C/EBPβ −/− mice display overexpression and disruption of cellular PR expression that coincides with a 10-fold decrease in mammary cell proliferation following steroid stimulation (Seagroves et al., 2000). The mammary glands of these mice also display similar changes in the level and pattern of PrlR in addition to aberrant ductal morphogenesis and decreased lobular alveolar development (Grimm et al., 2002). Herein, we have shown that both PR-A and PR-B are capable of inducing expression of PrlR, the former in an unliganded manner and the latter upon binding P. In the mouse, PR-A is associated with ductal elongation; PR-B is associated with lobuloalveolar development (Shyamala et al., 2000). They are thus temporally and spatially separated (Aupperlee et al., 2005). In the human breast the ratio of PR-A to PR-B controls the cell’s response to P. In the normal breast, the ratio of PRA to PRB is the same, with prediction of 50% heterodimers (Jacobsen et al., 2002;Lange et al., 2008). As a lesion progresses from normal to invasive, the odds of predominance of one isoform or the other increases thus suggesting that loss of coordinate expression of PR isoforms is an early event in breast carcinogenesis (Mote et al., 2002). In many breast cancers the PR-A:PR-B ratio is higher, where PR-A is associated with a less differentiated and more aggressive tumor (Bamberger et al., 2000). Furthermore, because PRB is more transcriptionally active than PRA, tumors with a high PRA:PRB are more hormone refractive. PRB is often a stronger activator of target genes such as cyclin D1 that is overexpressed in many breast cancers (Graham et al., 1995). The role of these isoforms is indeed complex (Kariagina et al., 2008).
The steric role that PR plays in hormone-induced PrlR upregulation can be surmised. Most steroid responsive promoters such as that for β-casein (Wyszomierski and Rosen, 2001) or Prl (Duan et al., 2008) contain steroid specific responsive elements along with other transcription factor binding sites. However, there is no PRE or ½ PRE on the PIII region of the PrlR promoter. Instead the PR utilizes C/EBP and an adjacent Sp1A sites to activate the PrlR promoter. PR acts as a docking protein in the cytoplasm (Hagan et al., 2009). A phosphorylation cascade occurs through c-src and MAP kinase (Faivre et al., 2008;Faivre and Lange, 2007) where PR and Sp1 are phosphorylated. Phosphorylated PR translocates to the nucleus, where it also acts as a docking protein (Faivre et al., 2008;Faivre and Lange, 2007) and can tether to phosphorylated Sp1; Sp1 can bind to other transcription factors such as CBPp3000 (Owen et al., 1998) or, in the case of the PrlR promoter, C/EBP-β. We have shown in our stably transfected EpH4 cells that only express PR-A that the levels of PrlR are elevated even in the absence of hormone treatment. This is in agreement with findings of others (Bamberger et al., 1996) that PR-A location is primarily nuclear and constitutively active. In T47D cells that contain both PR-A and PR-B, elevated baseline PrlR levels may result from the action of the unliganded PR-A. PR-A relies on phosphorylated Sp1 already present in the nucleus to induce PrlR (Owen et al., 1998). Nevertheless some PR-A remains in the cytoplasm and can still bind ligand (Jacobsen et al., 2002;Owen et al., 1998;Scarpin et al., 2009).
PR-B signaling is both nuclear and cytoplasmic. In cytoplasmic signaling, liganded PR-B first activates c-src and MAP kinase as well as activating Wnt 1 and EGFR, before it is itself phosphorylated and transported to the nucleus (Faivre and Lange, 2007). Once in the nucleus, phosphorylated PR-B interacts with transcription factors such as Sp1 (Faivre et al., 2008;McGowan et al., 2007) to turn on target genes. We have also shown that T47DYB cells expressing only PR-B respond to exogenous P with a 1.7-fold increase in PrlR protein expression. This effect can be compared to the 1.7-fold increase in PrlR mRNA when PRA was added to T47DYA cells (Jacobsen et al., 2002). Upregulation of PrlR protein in T47DYB cells occurred as early as 6 hr of P exposure and correlates with an increase of p21 and cyclin D1 protein expression when T47DYB cells were treated with the progestigen R5020 for 8 hr (Faivre et al., 2008).
Activation of both full-length PIII and the deletion mutant, -439-PIII that lacks the 5’ flanking region, showed similar baseline activation and P responsiveness in T47D cells. The latter construct contains all of the P responsive regions including both the C/EBP and Sp1 sites. C/EBPs interact with steroid receptors such as the glucocorticoid receptor and with transcription factor Stat 5 (Wyszomierski and Rosen, 2001) or ER and Sp1 (Dong et al., 2006) to turn on target genes. Sp1 is a ubiquitous transcription factor often involved in the expression of developmentally controlled genes (Koslowski et al., 2009). Sp1 proteins are overexpressed in breast tumor compared with non-tumor cells and may be used equally in induction or repression of genes (Wu et al., 2009). We found base pair mutations in the C/EBP and Sp1A sites reduced activity in T47D cells by 30–50%. Mutating or deleting both C/EBP and Sp1A further reduced promoter activity and eliminated induction by P. A virtually identical reduction in basal activation response was found when these binding sites on the rat PrlR promoter were disrupted in gonadal cells (Hu et al., 1996). Lopez-Rodriguez et al (Lopez-Rodriguez et al., 1997) similarly describe that C/EBP and Sp1 act on the CD11 promoter as well as the minimal Prl promoter in hematopoietic and epithelial cells. In our study, both Sp1A and C/EBP were upregulated in T47D cells treated with P and in EpH4-PR cells compared to EpH4-EV cells.
5. Conclusion
These results show that PR can act in a non-classical manner to induce expression of the PrlR through the cooperative action of C/EBP and an adjacent Sp1. Since both PR and PrlR have been implicated in breast cancer, these two factors could be targets for cancer therapy.
Acknowledgments
This research was supported by the Center for Cancer Research, an Intramural Research Program of the National Cancer Institute.
Abbreviations
- Prl
prolactin
- P
progesterone
- PrlR
prolactin receptor
- PR
progesterone receptor
- E
estrogen
- EV
empty vector
- LF
long form
- SF
short form
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- C/EBP
CCATT/Enhancer Binding Protein
- Sp1
Specificity Protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone receptor isoforms A and B: temporal and spatial differences in expression during murine mammary gland development. Endocrinology. 2005;146:3577–3588. doi: 10.1210/en.2005-0346. [DOI] [PubMed] [Google Scholar]
- 2.Bamberger AM, Bamberger CM, Gellersen B, Schulte HM. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Natl Acad Sci U S A. 1996;93:6169–74. doi: 10.1073/pnas.93.12.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bamberger AM, Milde-Langosch K, Schulte HM, Loning T. Progesterone receptor isoforms, PR-B and PR-A, in breast cancer: correlations with clinicopathologic tumor parameters and expression of AP-1 factors. Horm Res. 2000;54:32–7. doi: 10.1159/000063434. [DOI] [PubMed] [Google Scholar]
- 4.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 5.Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis JA, Linzer DIH. Expression of multiple forms of the prolactin receptor in mouse liver. Mol Endocrinol. 1989;3:674–680. doi: 10.1210/mend-3-4-674. [DOI] [PubMed] [Google Scholar]
- 7.Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006;281:18825–36. doi: 10.1074/jbc.M512826200. [DOI] [PubMed] [Google Scholar]
- 8.Duan R, Ginsburg E, Vonderhaar BK. Estrogen stimulates transcription from the human prolactin distal promoter through AP1 and estrogen responsive elements in T47D human breast cancer cells. Mol Cell Endocrinol. 2008;281:9–18. doi: 10.1016/j.mce.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Faivre EJ, Daniel AR, Hillard CJ, Lange CA. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol Endocrinol. 2008;22:823–37. doi: 10.1210/me.2007-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27:466–80. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldhar AS, Vonderhaar BK, Trott JF, Hovey RC. Prolactin-induced expression of vascular endothelial growth factor via Egr-1. Mol Cell Endocrinol. 2005;232:9–19. doi: 10.1016/j.mce.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez L, Zambrano A, Lazaro-Trueba I, Lopez E, Gonzalez JJ, Martin-Perez J, Aranda A. Activation of the unliganded estrogen receptor by prolactin in breast cancer cells. Oncogene. 2009;28:1298–308. doi: 10.1038/onc.2008.473. [DOI] [PubMed] [Google Scholar]
- 13.Graham JD, Yeates C, Balleine RL, Harvey SS, Milliken JS, Bilous AM, Clarke CL. Characterization of progesterone receptor A and B expression in human breast cancer. Cancer Res. 1995;55:5063–8. [PubMed] [Google Scholar]
- 14.Grimm SL, Seagroves TN, Kabotyanski EB, Hovey RC, Vonderhaar BK, Lydon JP, Miyoshi K, Hennighausen L, Ormandy CJ, Lee AV, Stull MA, Wood TL, Rosen JM. Disruption of steroid and prolactin receptor patterning in the mammary gland correlates with a block in lobuloalveolar development. Mol Endocrinol. 2002;16:2675–91. doi: 10.1210/me.2002-0239. [DOI] [PubMed] [Google Scholar]
- 15.Hagan CR, Faivre EJ, Lange CA. Scaffolding actions of membrane-associated progesterone receptors. Steroids. 2009;74:568–72. doi: 10.1016/j.steroids.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, Sundararajan K, Vonderhaar BK. Transcriptional and spatiotemporal regulation of prolactin receptor mRNA and cooperativity with progesterone receptor function during ductal branch growth in the mammary gland. Dev Dyn. 2001;222:192–205. doi: 10.1002/dvdy.1179. [DOI] [PubMed] [Google Scholar]
- 17.Hovey RC, Trott JF, Vonderhaar BK. Establishing a framework for the functional mammary gland: from endocrinology to morphology. J Mammary Gland Biol Neoplasia. 2002;7:17–38. doi: 10.1023/a:1015766322258. [DOI] [PubMed] [Google Scholar]
- 18.Hu Z, Zhuang L, Dufau ML. Multiple and tissue-specific promoter control of gonadal and non-gonadal prolactin receptor gene expression. J Biol Chem. 1996;271:10242–6. doi: 10.1074/jbc.271.17.10242. [DOI] [PubMed] [Google Scholar]
- 19.Hu ZZ, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. J Biol Chem. 2001;276:41086–94. doi: 10.1074/jbc.M102109200. [DOI] [PubMed] [Google Scholar]
- 20.Hu ZZ, Zhuang L, Dufau ML. Prolactin receptor gene diversity: structure and regulation. Trends Endocrinol Metab. 1998;9:94–102. doi: 10.1016/s1043-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 21.Hu ZZ, Zhuang L, Meng J, Dufau ML. Transcriptional regulation of the generic promoter III of the rat prolactin receptor gene by C/EBPbeta and Sp1. J Biol Chem. 1998;273:26225–35. doi: 10.1074/jbc.273.40.26225. [DOI] [PubMed] [Google Scholar]
- 22.Hu ZZ, Zhuang L, Meng J, Leondires M, Dufau ML. The human prolactin receptor gene structure and alternative promoter utilization: the generic promoter hPIII and a novel human promoter hP(N) J Clin Endocrinol Metab. 1999;84:1153–6. doi: 10.1210/jcem.84.3.5659. [DOI] [PubMed] [Google Scholar]
- 23.Jacobsen BM, Richer JK, Schittone SA, Horwitz KB. New human breast cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem. 2002;277:27793–800. doi: 10.1074/jbc.M202584200. [DOI] [PubMed] [Google Scholar]
- 24.Kariagina A, Aupperlee MD, Haslam SZ. Progesterone receptor isoform functions in normal breast development and breast cancer. Crit Rev Eukaryot Gene Expr. 2008;18:11–33. doi: 10.1615/critreveukargeneexpr.v18.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koseki Y, Cole D, Matsuzawa A, Costlow ME. Prolactin regulation of estrogen and progesterone receptors in normal and neoplastic mouse mammary tissue. Jpn J Cancer Res. 1987;78:1105–11. [PubMed] [Google Scholar]
- 26.Koslowski M, Tureci O, Biesterfeld S, Seitz G, Huber C, Sahin U. Selective activation of trophoblast-specific PLAC1 in breast cancer by CCAAT/enhancer-binding protein beta (C/EBPbeta) isoform 2. J Biol Chem. 2009;284:28607–15. doi: 10.1074/jbc.M109.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange CA, Sartorius CA, Abdel-Hafiz H, Spillman MA, Horwitz KB, Jacobsen BM. Progesterone receptor action: translating studies in breast cancer models to clinical insights. Adv Exp Med Biol. 2008;630:94–111. [PubMed] [Google Scholar]
- 28.Leroy-Martin B, Peyrat JP. Modulation of prolactin receptors (PRL-R) by lactogenic and steroid hormones in human breast cancer cells in long-term tissue culture (T-47D) Anticancer Res. 1989;9:631–6. [PubMed] [Google Scholar]
- 29.Lopez-Rodriguez C, Botella L, Corbi AL. CCAAT-enhancer-binding proteins (C/EBP) regulate the tissue specific activity of the CD11c integrin gene promoter through functional interactions with Sp1 proteins. J Biol Chem. 1997;272:29120–6. doi: 10.1074/jbc.272.46.29120. [DOI] [PubMed] [Google Scholar]
- 30.McGowan EM, Russell AJ, Boonyaratanakornkit V, Saunders DN, Lehrbach GM, Sergio CM, Musgrove EA, Edwards DP, Sutherland RL. Progestins reinitiate cell cycle progression in antiestrogen-arrested breast cancer cells through the B-isoform of progesterone receptor. Cancer Res. 2007;67:8942–51. doi: 10.1158/0008-5472.CAN-07-1255. [DOI] [PubMed] [Google Scholar]
- 31.Moldrup A, Ormandy C, Nagano M, Murthy K, Banville D, Tronche F, Kelly PA. Differential promoter usage in prolactin receptor gene expression: hepatocyte nuclear factor 4 binds to and activates the promoter preferentially active in the liver. Mol Endocrinol. 1996;10:661–71. doi: 10.1210/mend.10.6.8776726. [DOI] [PubMed] [Google Scholar]
- 32.Morabito JE, Trott JF, Korz DM, Fairfield HE, Buck SH, Hovey RC. A 5' distal palindrome within the mouse mammary tumor virus-long terminal repeat recruits a mammary gland-specific complex and is required for a synergistic response to progesterone plus prolactin. J Mol Endocrinol. 2008;41:75–90. doi: 10.1677/JME-08-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72:163–72. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- 34.Murphy LJ, Murphy LC, Stead B, Sutherland RL, Lazarus L. Modulation of lactogenic receptors by progestins in cultured human breast cancer cells. J Clin Endocrinol Metab. 1986;62:280–7. doi: 10.1210/jcem-62-2-280. [DOI] [PubMed] [Google Scholar]
- 35.Ormandy CJ, Binart N, Helloco C, Kelly PA. Mouse prolactin receptor gene: Genomic organization reveals alternative promoter usage and generation of isoforms via alternative 3 '-exon splicing. DNA and Cell Biology. 1998;17:761–770. doi: 10.1089/dna.1998.17.761. [DOI] [PubMed] [Google Scholar]
- 36.Ormandy CJ, Hall RE, Manning DL, Robertson JFR, Blamey RW, Kelly PA, Nicholson RI, Sutherland RL. Coexpression and cross-regulation of the prolactin receptor and sex steroid hormone receptors in breast cancer. J Clin Endocrinol Metab. 1997;82:3692–3699. doi: 10.1210/jcem.82.11.4361. [DOI] [PubMed] [Google Scholar]
- 37.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273:10696–701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 38.Pinkas J, Leder P. MEK1 signaling mediates transformation and metastasis of EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition. Cancer Res. 2002;62:4781–90. [PubMed] [Google Scholar]
- 39.Reichmann E, Schwarz H, Deiner EM, Leitner I, Eilers M, Berger J, Busslinger M, Beug H. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–16. doi: 10.1016/s0092-8674(05)80060-1. [DOI] [PubMed] [Google Scholar]
- 40.Sakai S, Bowman PD, Yang J, McCormick K, Nandi S. Glucocorticoid regulation of prolactin receptors on mammary cells in culture. Endocrinology. 1979;104:1447–9. doi: 10.1210/endo-104-5-1447. [DOI] [PubMed] [Google Scholar]
- 41.Satoh K, Hovey RC, Malewski T, Warri A, Goldhar AS, Ginsburg E, Saito K, Lydon JP, Vonderhaar BK. Progesterone enhances branching morphogenesis in the mouse mammary gland by increased expression of Msx2. Oncogene. 2007;26:7526–34. doi: 10.1038/sj.onc.1210555. [DOI] [PubMed] [Google Scholar]
- 42.Scarpin KM, Graham JD, Mote PA, Clarke CL. Progesterone action in human tissues: regulation by progesterone receptor (PR) isoform expression, nuclear positioning and coregulator expression. Nucl Recept Signal. 2009;7:e009. doi: 10.1621/nrs.07009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seagroves TN, Kmacik S, Raught B, Gay J, Burgess-Beusse B, Darlington GJ, Rosen JM. C/EPBβ, but not C/EPBα, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev. 1998;12:1917–1928. doi: 10.1101/gad.12.12.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seagroves TN, Lydon JP, Hovey RC, Vonderhaar BK, Rosen JM. C/EBPβ (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Molecular Endocrinology. 2000;14:359–368. doi: 10.1210/mend.14.3.0434. [DOI] [PubMed] [Google Scholar]
- 45.Shyamala G, Yang X, Cardiff RD, Dale E. Impact of progesterone receptor on cell-fate decisions during mammary gland development. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3044–3049. doi: 10.1073/pnas.97.7.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang QQ, Lane MD. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev. 1999;13:2231–41. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Alternative splicing to exon 11 of human prolactin receptor gene results in multiple isoforms including a secreted prolactin-binding protein. J Mol Endocrinol. 2003;30:31–47. doi: 10.1677/jme.0.0300031. [DOI] [PubMed] [Google Scholar]
- 48.Tseng L, Zhu HH. Progestin, estrogen, and insulin-like growth factor-I stimulate the prolactin receptor mRNA in human endometrial stromal cells. J Soc Gynecol Investig. 1998;5:149–55. doi: 10.1016/s1071-5576(97)00116-0. [DOI] [PubMed] [Google Scholar]
- 49.Vonderhaar BK, Bhattacharya A, Alhadi T, Liscia DS, Andrew EM, Young JK, Ginsburg E, Bhattacharjee M, Horn TM. Isolation, characterization and regulation of the prolactin receptor. J Dairy Sci. 1985;68:466–488. doi: 10.3168/jds.S0022-0302(85)80847-X. [DOI] [PubMed] [Google Scholar]
- 50.Wu F, Ivanov I, Xu R, Safe S. Role of SP transcription factors in hormone-dependent modulation of genes in MCF-7 breast cancer cells: microarray and RNA interference studies. J Mol Endocrinol. 2009;42:19–33. doi: 10.1677/JME-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyszomierski SL, Rosen JM. Cooperative effects of STAT5 (signal transducer and activator of transcription 5) and C/EBPbeta (CCAAT/enhancer-binding protein-beta) on beta-casein gene transcription are mediated by the glucocorticoid receptor. Mol Endocrinol. 2001;15:228–40. doi: 10.1210/mend.15.2.0597. [DOI] [PubMed] [Google Scholar]