

Abstract
The first rhodium catalyzed arylation of imines proceeding via C–H bond functionalization is reported. Use of a non-coordinating halide abstractor is important to obtain reactivity. Aryl branched N-Boc-amines are formed and a wide range of functionality is compatible with the reaction.
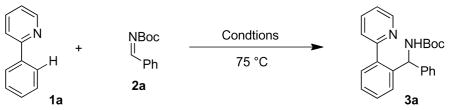
In the area of C–H bond functionalization, arylation of alkenes and alkynes have been well explored.1 In contrast, analogous additions across C–O2 or C–N3–5 multiple bonds are rare. Due to the prevalence of α-branched amines in drugs and natural products,6 we sought a method for the arylation of imines via C–H bond functionalization. The most closely related transformations are Pd-catalyzed additions to N-tosyl imines that rely on the functionalization of acidic C–H bonds.5 Herein, we report the high-yielding 2-pyridyl-directed arylation of a wide range of aromatic N-Boc-imines with a Rh(III) catalyst.
Our investigations focused on additions to N-Boc-imines due to the convenience and ease of removal of the exceedingly popular Boc protecting group. As the test substrate for arylation, we chose 2-phenylpyridine (1a) because of the pyridyl directing group’s high chemical stability and rich history in C–H functionalization with a variety of transition metals.1d,e, 7
We began our reaction exploration with Rh(III) catalysts. This type of catalyst has been proposed to activate C–H bonds via an electrophilic deprotonation mechanism to generate an aryl-Rh(III) species.8 However, upon heating 10 mol % [Cp*RhCl2]2 (Cp*: pentamethyl cyclopentadienyl) with 2-phenylpyridine and N-Boc-benzaldimine in CH2Cl2, no product was observed (entry 1, Table 1). Theorizing that the lack of reactivity may be due to the chloride ligands on the metal, which could prevent coordination of the N-Boc-imine, AgSbF6 was added as a halide abstractor and provided the desired product (3a) in 55% yield (entry 2).9 Utilizing coordinating solvents such as THF (entry 3) and DMF (entry 4) resulted in lower activity while solvents such as benzene failed due to insolubility of the catalyst system (entry 5). Analogous iridium (entry 6) and ruthenium (entry 7) based complexes were found to be inactive for this transformation. Substrate stoichiometries were also explored (entries 8 and 9) with the highest yield being achieved by employing two equivalents of the least expensive component, 2-phenylpyridine (entry 9). Under these conditions, the excess 2-phenylpyridine is recovered. Unreacted N-Boc imine is not observed and only small amounts of the hydrolyzed aldehyde product are detected. Other halide abstractors were also explored (entries 10–14) but AgSbF6 proved to be optimal (entry 9).
Table 1.
Screening of Reaction conditionsa
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | % Yieldb |
| 1 | [Cp*RhCl2]2 | none | CH2Cl2 | 0 |
| 2 | [Cp*RhCl2]2 | AgSbF6 | CH2Cl2 | 55 |
| 3 | [Cp*RhCl2]2 | AgSbF6 | THF | 30 |
| 4 | [Cp*RhCl2]2 | AgSbF6 | DMF | 0 |
| 5 | [Cp*RhCl2]2 | AgSbF6 | C6H6 | 0 |
| 6 | [Cp*IrCl2]2 | AgSbF6 | CH2Cl2 | 0 |
| 7 | [Cp*RuCl2]n | AgSbF6 | CH2Cl2 | 0 |
| 8c | [Cp*RhCl2]2 | AgSbF6 | CH2Cl2 | 60 |
| 9d | [Cp*RhCl2]2 | AgSbF6 | CH2Cl2 | 87 |
| 10d | [Cp*RhCl2]2 | AgOAc | CH2Cl2 | trace |
| 11d | [Cp*RhCl2]2 | AgOTs | CH2Cl2 | 30 |
| 12d | [Cp*RhCl2]2 | AgBF4 | CH2Cl2 | 77 |
| 13d | [Cp*RhCl2]2 | AgClO4 | CH2Cl2 | 69 |
| 14d | [Cp*RhCl2]2 | NaBPh4 | CH2Cl2 | 0 |
All reactions were performed by employing 0.05 mmol of 2-phenylpyridine, 0.05 mmol of imine in 0.5 mL of solvent with 10 mol % catalyst and 40 mol % additive.
Yields are based on NMR using 2,6-dimethoxytoluene as an internal standard.
0.1 mmol of imine.
0.1 mmol of 2-phenylpyridine.
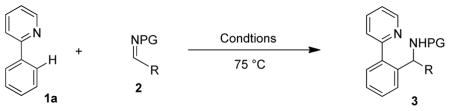
Evaluation of substituted 2-arylpyridines established that both electronically rich and deficient derivatives are effective arylation substrates and that the reaction is compatible with chloro, keto and acetanilide functional groups (Scheme 1). For 2-(3-methylphenyl)pyridine, functionalization occurred solely para to the methyl group to provide 3f. Multicyclic pyridine derivatives also serve as effective directing groups with the bicylic quinoline and tricyclic benzo[h]quinoline both providing the expected products 3g and 3h, respectively, in good yields. Notably, for all of the 2-arylpyridine substrates, products resulting from bis-arylation were not detected.
Scheme 1.

Arylpyridine substrate scopea
aAll reactions were performed by heating 2-phenylpyridine (2 equiv), imine (1 equiv), [Cp*RhCl2]2 (10 mol %), AgSbF6 (40 mol %), and CH2Cl2 (0.1 M) in a sealed tube for 20 h at 75 °C. Isolated yields are reported.
The substrate scope was further extended to a broad variety of substituted aromatic N-Boc-imines (Table 2, entries 1–12). Both electron-poor (entries 3, 5, 8 and 11) and electron-rich (entry 7) aromatic imines provided branched amine products in high yields. Substitution at the ortho- (entries 9 and 10), meta- (entry 11), and para-(entries 2–8) positions were all well tolerated. Moreover, the reaction showed excellent functional group compatibility with N-Boc-imines substituted with chloro (entries 2 and 9), nitro (entry 3), methoxy (entry 7), ester (entry 8) and even the electrophilic carboxaldehyde (entry 11) functionality all providing branched amine products in high yields. In addition, the 2-thienyl substituted branched amine product 3s was obtained by addition to a heteroaromatic imine substrate (entry 12). Of the functionalities evaluated, only the nitrile group (entry 4) gave a low yield, likely due to competitive coordination of the CN group to the Rh(III) center. In support of this explanation, a dramatic reduction in yield was observed upon addition of benzonitrile to a previously successful substrate combination (see entry 13 versus 6). While a 1:4 ratio of [Cp*RhCl2]2 to AgSbF6 gave consistently good results, we found that ratio could be decreased to 1:2 for most substrates with no effect on rates or yields (see entries 7, 9, 10 and 12). However, the reaction failed to work when the ratio was below 1:2. For more reactive imines, good yields may be obtained with 5 mol % [Cp*RhCl2]2 (entry 5). This lower catalyst loading provided a moderate reduction in yields when applied to less reactive substrates as a result of competitive imine decomposition pathways (entries 1–2, 7). Further decreasing the catalyst loading to 2.5 mol % [Cp*RhCl2]2 resulted in low yields.
Table 2.
Substrate scope of substituted iminesa
 | ||||
|---|---|---|---|---|
| Entry | R | PG | Product | % Yieldb |
| 1 | C6H5 | Boc | 3a | 82 (70)c |
| 2 | 4-ClC6H4 | Boc | 3i | 77 (64)c |
| 3 | 4-NO2C6H4 | Boc | 3j | 77 |
| 4 | 4-CNC6H4 | Boc | 3k | 50 |
| 5 | 4-CF3C6H4 | Boc | 3l | 95 (81)c |
| 6 | 4-MeC6H4 | Boc | 3m | 70 |
| 7d | 4-MeOC6H4 | Boc | 3n | 70 (51)c |
| 8 | 4-CO2MeC6H4 | Boc | 3o | 70 |
| 9d | 2-ClC6H4 | Boc | 3p | 76 |
| 10d | 2-MeC6H4 | Boc | 3q | 92 |
| 11 | 3-CHOC6H4 | Boc | 3r | 81 |
| 12d | 2-thiophene | Boc | 3s | 71 |
| 13e | 4-MeC6H4 | Boc | 3m | 27 |
| 14f | nBu | P(O)Ph2 | 3t | 0 |
| 15f | nBu | Ts | 3u | 72 |
| 16 | C6H5 | Ts | 3v | 40 |
| 17 | C6H5 | Ns | 3w | 51 |
All reactions were performed by heating 2-phenylpyridine (2 equiv), imine (1 equiv), [Cp*RhCl2]2 (10 mol %), AgSbF6 (40 mol %), and CH2Cl2 (0.1 M) in a sealed tube for 20 h at 75 °C.
Isolated yield.
Values in parentheses correspond to isolated yields after 40 h at 75 °C using [Cp*RhCl2]2 (5 mol %) AgSbF6 (20 mol %)
AgSbF6 (20 mol %).
PhCN (1 equiv) added.
2-phenylpyridine (1 equiv).
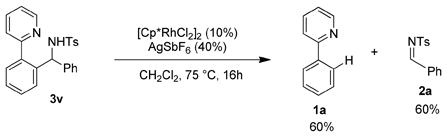
Alkyl N-Boc-imines were not effective substrates for this transformation as a result of self-condensation via imine to enamine tautomerization. We therefore explored other protecting groups for alkyl imine substrates. While N-diphenylphosphinoyl- pentaldimine proved to be unreactive (entry 14), addition to the corresponding N-tosyl-imine proceeded in good yield (entry 15). This result prompted us to also evaluate the reactivity of an aromatic N-tosyl-imine substrate, but N-tosyl-benzaldimine reacted with poor conversion to give the branched amine 3v in only 40% yield (entry 16). We considered that the moderate yield observed might possibly be attributed to the reversibility of arylation of 3v. This reversibility was demonstrated by subjecting purified 3v to the reaction conditions, which resulted in an equilibrium mixture of 3v, 2-phenylpyridine, and N-tosyl-benzaldimine (eq 1), consistent with the distribution observed in the arylation reaction (Table 2, entry 16). Significantly, while β-aryl elimination from aryl carbinols has been reported,10 to our knowledge, the corresponding transformation for branched amines has not previously been described. The mechanisms of β-aryl elimination for both the previously reported carbinols and 3v are likely to be similar, but further studies are needed. The reversibility of the reaction could be slightly shifted towards product by replacing the N-tosyl with the more electronegative N-nosyl11 group (entry 17). Notably, reversibility was not observed for the arylation products of aromatic N-Boc-imines or aliphatic N-tosyl-pentanaldimine.
 |
(1) |
A possible mechanism for the arylation reaction could involve initial electrophilic deprotonation of the ortho-phenyl C–H bond of 2-phenylpyridine to form an Ar-Rh(III) intermediate (Scheme 2).8 Coordination of the N-Boc-imine would then activate the imine for addition followed by protonolysis to regenerate the catalyst. Alternatively, rhodium could serve as a Lewis acid to activate the imine for electrophilic aromatic substitution (EAS).12 However, this latter pathway is unlikely given that only the electronically deactivated ortho position is functionalized on the electron deficient 2-phenylpyridine. Furthermore, the observation that the reaction is more efficient for electron poor 2-arylpyridines (see Scheme 1, 3b vs 3c) is inconsistent with an EAS pathway but supports an electrophilic deprotonation mechanism.
Scheme 2.

Proposed catalytic cycle
In summary, [Cp*RhCl2]2/AgSbF6 catalyzes the addition of 2-arylpyridines to N-Boc and N-sulfonyl imines via C–H bond functionalization to give branched amine products. Many commonly encountered functional groups such as ketones, aldehydes, esters, halides, trifluoromethyl, amides and nitro groups are compatible with the method. Mechanistic studies and the investigation of alternative directing groups will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant GM069559 (to J.A.E.) and by the Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, US Department of Energy under contract DE-AC02-05CH11231 (to R.G.B.). A.S.T. is grateful for an Eli Lilly Fellowship, and M.E.T. thanks the Deutsche Forschungs-gemeinschaft (DFG) for a research fellowship (Ta 733/1-1).
Footnotes
Supporting Information Available: Complete experimental details and spectral data for all compounds described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews in C–H functionalizations with alkenes and alkynes see: Satoh T, Miura M. Chem Eur J. 2010;16:11212–11222. doi: 10.1002/chem.201001363.Karimi B, Behzadnia H, Elhamifar D, Akhavan PF, Esfahani FK, Zamani A. Synthesis. 2010;9:1399–1427.Wang X, Zhou L, Lu, Wenjun Curr Org Chem. 2010;14:289–307.Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624–655. doi: 10.1021/cr900005n.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273.
- 2.Leading references on the arylation of C–O multiple bonds: Kuninobu Y, Nishina Y, Takeuchi T, Takai K. Angew Chem Int Ed. 2007;46:6518–6520. doi: 10.1002/anie.200702256.Fukumoto Y, Sawada K, Hagihara M, Chatani N, Murai S. Angew Chem Int Ed. 2002;41:2779–2781. doi: 10.1002/1521-3773(20020802)41:15<2779::AID-ANIE2779>3.0.CO;2-J.
- 3.Zhou C, Larock RC. J Am Chem Soc. 2004;126:2302–2303. doi: 10.1021/ja038687l. [DOI] [PubMed] [Google Scholar]
- 4.Leading references on the intramolecular olefination of C–N double bonds: Kuninobu Y, Kawata A, Takai K. J Am Chem Soc. 2005;127:13498–13499. doi: 10.1021/ja0528174.Fukutani T, Umeda N, Hirano K, Satoh T, Miura M. Chem Comm. 2009:5141–5143. doi: 10.1039/b910198e.
- 5.For examples of intermolecular alkylation of C–N double bonds, see: Qian B, Guo S, Shao J, Zhu Q, Yang L, Xia C, Huang H. J Am Chem Soc. 2010;132:3650–3651. doi: 10.1021/ja910104n.Aydin J, Szabó KJ. Org Lett. 2008;10:2881–2884. doi: 10.1021/ol801070n.Aydin J, Conrad CS, Szabó KJ. Org Lett. 2008;10:5175–7178. doi: 10.1021/ol8021512.
- 6.The importance of this compound class has lead to intensive development of transition metal catalyzed additions, including enantioselective catalytic additions, of aryl organometallic reagents to imines. (a) For a review on transition-metal catalyzed additions of organoboron reagents see: Miyaura N. Bull Chem Soc Jpn. 2008;81:1535–1553.For a review on Rh-catalyzed additions see: Fagnou K, Lautens M. Chem Rev. 2003;103:169–196. doi: 10.1021/cr020007u.
- 7.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Li L, Jiao Y, Brennessel WW, Jones WD. Organometallics. 2010;29:4593–4605. [Google Scholar]; (b) Li L, Brennessel WW, Jones WD. J Am Chem Soc. 2008;130:12414–12419. doi: 10.1021/ja802415h. [DOI] [PubMed] [Google Scholar]; (c) Davies DL, Al-Duaij O, Fawcett J, Giardiello M, Hilton ST, Russell DR. Dalton Trans. 2003:4132. [Google Scholar]
- 9.There have been a few recent reports of using a halide abstractor in combination with [Cp*RhCl2]2: Patureau FW, Glorius F. J Am Chem Soc. 2010;132:9982–9983. doi: 10.1021/ja103834b.Schipper DJ, Hutchinson M, Fagnou J. J Am Chem Soc. 2010;132:6910–6911. doi: 10.1021/ja103080d.Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. J Am Chem Soc. 2008;130:16474–16475. doi: 10.1021/ja806955s.
- 10.For leading references see: Nishimura T, Katoh T, Takatsu K, Shintani R, Hayashi T. J Am Chem Soc. 2007;129:14158–14159. doi: 10.1021/ja076346s.Nishimura T, Katoh T, Hayashi T. Angew Chem Int Ed. 2007;46:4937–4939. doi: 10.1002/anie.200700902.Zhao P, Incarvito CD, Hartwig JF. J Am Chem Soc. 2006;128:3124–3125. doi: 10.1021/ja058550q.Terao Y, Wakui H, Nomoto M, Satoh T, Miura M, Nomura M. J Org Chem. 2003;68:5236–5243. doi: 10.1021/jo0344034.Nishimura T, Araki H, Maeda Y, Uemura S. J Org Lett. 2003;5:2997–2999. doi: 10.1021/ol0348405.Kondo T, Kodoi K, Nishinaga E, Okada T, Morisaki Y, Watanabe Y, Mitsudo T. J Am Chem Soc. 1998;120:5587–5588.
- 11.Kan T, Fukuyama T. Chem Commun. 2004:353–359. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- 12.For recent reports and reviews of Friedel-Crafts alkylations with imines see: Poulsen TB, Jorgensen KA. Chem Rev. 2008;108:2903–2915. doi: 10.1021/cr078372e.Shirakawa S, Berger R, Leighton JL. J Am Chem Soc. 2005;127:2858–2859. doi: 10.1021/ja042522a.Uraguchi D, Sorimachi K, Terada M. J Am Chem Soc. 2004;126:11804–11805. doi: 10.1021/ja046185h.Gong Y, Kato K, Kimoto H. Bull Chem Soc Jpn. 2002;75:2637–2645.Saaby S, Fang X, Gathergood N, Jorgensen KA. Angew Chem Int Ed. 2000;39:4114–4116. doi: 10.1002/1521-3773(20001117)39:22<4114::aid-anie4114>3.0.co;2-v.Johannsen M. Chem Commun. 1999:2233.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.