

Abstract
Glycogen synthase kinase-3 (GSK-3) is a drug target under intense investigation in pharmaceutical companies and constitutes an attractive piggyback target for eukaryotic pathogens. Two different GSKs are found in trypanosomatids, one about 150 residues shorter than the other. GSK-3 short (GeneDB: Tb927.10.13780) has previously been validated genetically as a drug target in Trypanosoma brucei by RNAi induced growth retardation; and chemically by correlation between enzyme and in vitro growth inhibition. Here, we report investigation of the equivalent GSK-3 short enzymes of L. major (LmjF18.0270) and L. infantum (LinJ18_V3.0270, identical in amino acid sequences to LdonGSK-3 short) and a crystal structure of LmajGSK-3 short at 2 Å resolution. The inhibitor structure-activity relationships (SARs) of L. major and L. infantum are virtually identical, suggesting that inhibitors could be useful for both cutaneous and visceral leishmaniasis. Leishmania spp. GSK-3 short has different inhibitor SARs than TbruGSK-3 short, which can be explained mostly by two variant residues in the ATP-binding pocket. Indeed, mutating these residues in the ATP-binding site of LmajGSK-3 short to the TbruGSK-3 short equivalents results in a mutant LmajGSK-3 short enzyme with SAR more similar to that of TbruGSK-3 short. The differences between human GSK-3β (HsGSK-3β) and LmajGSK-3 short SAR suggest that compounds which selectively inhibit LmajGSK-3 short may be found.
Keywords: Glycogen synthase kinase-3, Leishmania spp., Trypanosoma brucei, African sleeping sickness, drug development, protein kinase
1. Introduction
Pathogenic protozoa kill or permanently injure millions of people living in underdeveloped regions of the world [1,2]. Parasitic diseases such as leishmaniasis not only afflict humans, but impair local economies and sicken livestock and pets [3]. Leishmania spp. are responsible for a disabling skin disease and visceral involvement that is often fatal if untreated. Due to the immune evasion mechanisms of Leishmania spp., vaccine development remains difficult and challenging [4]. Available therapies are toxic, difficult to administer, and are becoming increasingly ineffective [3,5]. Development of new anti-leishmanial therapeutics that are oral, inexpensive, and non-toxic remains an urgent priority if the debilitating effect of leishmaniasis is to be reduced. Recent advances have increased the prospect of discovering druggable enzyme targets based on biochemical and physiological differences between pathogens and host [1].
We have recently demonstrated that Trypanosoma brucei glycogen synthase kinase-3 (TbruGSK-3) short (GeneDB: Tb927.10.13780) is a promising drug target for T. brucei based on: 1) RNA interference (RNAi) data suggesting essentiality of TbruGSK-3 short for T. brucei blood-stream-form (BSF), and 2) excellent correlations between BSF cell-activity and enzyme inhibition by compounds from a focused small molecule inhibitor library [6]. As previously reported [7], there are two GSK-3 orthologs in the Leishmania spp. genomes: a short form and a long form. These are equivalent to the two orthologs found in T. brucei [6]. Humans also have two orthologs, an alpha (α) and a beta (β) form. The trypanosomatid short GSK-3 orthologs are the closest in sequence to HsGSK-3β (see Supplementary Fig. 1 for sequence alignment).
GSK-3 is a serine-threonine protein kinase under investigation for a variety of therapeutic uses in humans [8]. Glycogen synthase kinase 3 was initially defined as a regulator of glycogen metabolism [9]. It phosphorylates residues on glycogen synthase, the rate-limiting enzyme involved in glycogen synthesis [10]. GSK-3 generally acts as a secondary protein kinase, in that it phosphorylates a serine or threonine of a protein substrate that was previously phosphorylated several residues upstream. However, “glycogen synthase kinase” is somewhat of a misnomer because GSK-3 has numerous other functions. GSK-3 phosphorylates many key regulatory proteins and is itself involved in processes such as transcription and cell cycle regulation. This family of protein kinases is highly conserved and homologs have been found in mammals, yeast, mold, plants and protozoans, some of which may or may not possess glycogen or glycogen synthase but nonetheless require GSK-3 homologs activities in signaling pathways for many cellular and physiological events [7–9,11]. GSK-3 inhibition has been shown to be useful in animal models of several diseases, including diabetes mellitus [12,13] and Alzheimer's dementia [14]. The GSK-3 enzyme has also been found to be amenable to selective and safe targeting with small molecule drugs [8]. Lithium has long been used to treat mania [14] and is now known to be an inhibitor of GSK-3 activity. Long-term lithium administration at therapeutic levels for mania has not been associated with toxicity and lithium use has been associated with lower rates of cancer [15]. Several classes of GSK-3 specific small molecule inhibitors do not lead to any evident toxicity in animal models [13,16–19]. Thus, there are obvious reasons to believe that GSK-3 inhibitors could be administered to humans for treatment of parasitic diseases with minimal toxicity [16–19].
Validated drug targets for T. brucei can sometimes be extended to Leishmania species since these parasites share a conserved core proteome in large syntenic polycistronic gene clusters [20] and also share distinctive core biochemical processes. TbruGSK-3 short and L. major GSK-3 short (LmajGSK-3) short have 65 % amino acid sequence identity (Table 1, Supplementary Fig. 1). Biological evidence that GSK-3 short enzyme could be a potential target for anti-leishmanial drug development was recently described for L. donovani [7]. However, optimization of lead candidates for development of potent therapeutics remains a challenge. Detailed understanding of inhibitor SAR within the context of X-ray crystal structures and computational modeling can effectively guide optimization of known inhibitors by predicting functional groups needed to improve potency, selectivity, and alterations allowed to improve pharmacokinetic properties.
Table 1.
Comparison of amino acid identities (%) of human GSK-3β vs. T brucei GSK-3 short and LmajGSK-3 short.
| HsGSK-3β | LmajGSK-3 short | TbruGSK-3 short | |
|---|---|---|---|
| LinfGSK-3 short | 40.9 | 98.0 | 63.4 |
| HsGSK-3β | 40.9 | 43.5 | |
| LmajGSK-3 short | 63.9 |
In this report, we describe biochemical, structural, and inhibitor structure-activity relationships of Leishmania spp.GSK-3 enzymes. We expressed and purified recombinant GSK-3 short from L. major (a cause of “Old World” cutaneous leishmaniasis) and L. infantum (a cause of Mediterranean visceral leishmaniasis identical in amino acid sequence to L. donovani GSK-3 short [LdonGSK-3 short] [7]). We then solved the crystal structure of LmajGSK-3 short to a resolution of 2.0 Å. It was found that a variety of protein kinase inhibitors inhibit LmajGSK-3 short and their SAR was observed to be recapitulated with LinfGSK-3 short. Despite the high similarity in TbruGSK-3 short and LmajGSK-3 short, these enzymes have distinct structure-activity relationships (SAR) for some GSK-3 inhibitors. A structural model of TbruGSK-3 short was created based on the structure of LmajGSK-3 short which explains the disparate SAR. Specifically, the structural comparison shows that there are two key amino acids in the active site that are different from those of LmajGSK-3 short. These residues are predicted to reduce compound-protein interactions within the ATP-binding cavity in LmajGSK-3 short compared to TbruGSK-3 short. Mutation of these two amino acids in LmajGSK-3 short to the equivalent residues found in TbruGSK-3 short abolished the difference in SAR. Thus, our studies define the structural constraints for the further development of potent GSK-3 inhibitors that target T. brucei as well as for all Leishmania species.
2. Materials and Methods
2.1. Bioinformatics
The HsGSK-3β (UniProt P49841) orthologs LmjF18.0270 and LinJ18_V3.0270, from the genomes of L. major and L. infantum respectively, were identified by BLASTP [21]. A comparison of amino acid identities (%) of human GSK-3β vs. L. major, L. infantum and T. brucei GSK-3 short is shown in Table 1 and alignments of their predicted amino acid sequences are shown in Supplementary Fig. 1.
2.2. Compound Library
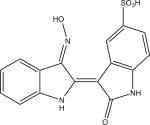
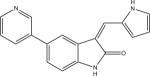
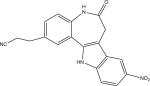
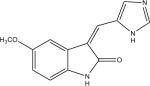
Protein kinase inhibitors purchased from Calbiochem (San Diego, CA) included GSK-3β Inhibitor VIII (Catalog no: 361557), TrkA Inhibitor (Catalog no: 648450), Angiogenesis inhibitor (Catalog no: 175580), RO-31-7549 (Catalog no: 557508), Indirubin-3′-monoxime-5-sulphonic Acid (Catalog no: 402088), JAK3 Inhibitor VI (Catalog no: 420126), Alster-paullone, 2-cyanoethyl (Catalog no: 126871), Cdk1/2 Inhibitor III (Catalog no: 217714), Hymenialdisine, Stylissa damicornis (Catalog no: 400085). The remaining inhibitors, GW8510 (Catalog no: G7791) and SU9516 (Catalog no: S1693) were obtained from Sigma Chemical Co. (St. Louis, MO). They were dissolved and stored at −20° C in 100% DMSO at a final concentration of 20 mM. Final compound concentration per well in screening assays was 10 μM, while further serial dilutions to determine IC50 values when IC50 values were <10 μM were done after the initial screening assays.
2.3. Molecular Cloning, Expression and Characterization of Parasite GSK-3 short enzymes
The complete coding region of GSK-3 short genes of L. infantum strain MHOM/BR/82/BA-2 and L. major Friedlin strain genomic DNA [21] were PCR amplified using the primers LmajGSKFwd (5'- GGGTCCTGGTTCGATGTCGCTCAACGCTGCCGCT -3'), LmajGSKRev (5'- CTTGTTCGTGCTGTTTATTACTTGCGAACTAGCTTGGCCTTC -3'). A previously described set of primers, TbruGSKLICFwd and TbruGSKLICRev, were used to amplify the ORF of TbruGSK-3 short from the TbruGSK-3(MBP)-AVA0421- expression vector [6]. These were cloned into the ligation independent cloning (LIC) site of expression vector AVA0421 [23,24]. This vector was derived from pET14b and encodes an N-terminal His(6)-Tag followed by an optimized 3C protease cleavage recognition site. The inserts were all sequenced and confirmed to correspond to GenBank entries for TbruGSK-3 short (XM_822768), LmajGSK-3 short (XM_00168238) and LinfGSK-3 short (XM_001464807). Site-directed mutagenesis of residues within the ATP-binding cavity of LmajGSK-3 short was performed on the expression plasmid using the QuikChange protocol (Stratagene, Santa Clara, CA).
Recombinant GSK-3 short proteins were expressed in E. coli BL21(DE3)* (Invitrogen, Carlsbad, CA) using Studier auto-induction protocols at 20°C [25]. Soluble TbruGSK-3, LmajGSK-3 and LinfGSK-3 short enzymes were purified by immobilized metal-affinity chromatography (IMAC) followed by size-exclusion chromatography (SEC). Soluble recombinant GSK-3 proteins were applied to a Ni2+–NTA (Qiagen, Valencia, CA) column in binding buffer composed of 20 mM HEPES pH 7.25, 500 mM NaCl, 5% glycerol, 30 mM imidazole, 0.5% CHAPS, and 1 mM TCEP. The Ni2+ column was washed with binding buffer without CHAPS, and the proteins were eluted with the same buffer supplemented with 250 mM imidazole. The eluted GSK-3 proteins were concentrated by centrifugal ultrafiltration and loaded into a 26/60 Superdex 75 SEC column. Fractions corresponding to GSK-3 were pooled and concentrated (e.g. Fig. 1A). The GSK-3 homologs were used for enzyme assays after IMAC and SEC purification.
Fig. 1.

A. SDS-PAGE coomassie blue stained gel of recombinant LmajGSK-3 short protein fractions, eluted during size exclusion chromatography (fractions 4–13 shown), demonstrating the presence of two distinct but closely spaced major bands with MW that closely match the predicted MW of LmajGSK-3 short. The M lane is molecular weight standards with the 40 and 50 kDa standards marked to the left. B. UV 260nM absorbance tracing showing peak 1 and 2 eluted from the cationic exchange column. The steady upsloping line shows increasing buffer B during elution of the cation exchange. C. SDS-PAGE coomassie blue stained protein fractions (peak 1 and peak 2) eluted from the cationic exchange. The M lane is molecular weight standards with the 40 kDa standard marked to the left. D. LC-MS tracings demonstrates the band in peak 1 correlated with mono-phosphorylated LmajGSK-3 short (41092 Da) while the major peak 2 band correlated with non-phosphorylated LmajGSK-3 short (41010 Da).
Analysis of the Ni2+ SEC purified LmajGSK-3 short by SDS-PAGE showed the presence of two distinct but closely spaced major bands with molecular weights (MW) that closely matched that predicted for LmajGSK-3 short (Fig 1A). The SEC fractions with LmajGSK-3 short were pooled and dialyzed against 20 mM HEPES pH 7.5, 200 mM NaCl, 5% glycerol and 1 mM TCEP in the presence of 3C protease at a ratio of 1:50 overnight at 4°C. The protein sample was recovered from dialysis and the salt concentration of the buffer was adjusted to 500 mM NaCl and 30 mM imidazole. The 3C-treated sample was further purified by passage through a second gravity flow 3 mL Ni2+ chromatography column to remove the His-Tag 3C protease and any uncleaved LmajGSK-3 short. The recombinant protein was further resolved by cation exchange chromatography to separate the phosphorylated and unphosphorylated versions of the protein. The Ni2+ SEC/3C cleaved protein sample was desalted into 50 mM MES pH 7.0 with 5% glycerol and 1 mM TCEP, loaded into a 6 mL Resource S column and eluted using a linear gradient to 500 mM NaCl (Buffer B) in 40 column volumes. The cation exchange column step yielded 2 major peaks (Fig. 1B). Pure samples of Ni2+ SEC purified LmajGSK-3 short after the cation exchange step were analyzed by MALDI/TOF as previously described [26]. The first peak was composed of phosphorylated-GSK and the second peak was composed of non-phosphorylated GSK (Fig. 1C–D). LC/MS/MS was used to further characterize the phosphorylation by loading protein sample into an SDS-PAGE, excising the band corresponding to LmajGSK-3 short from the gel, and digesting it with trypsin. The resulting trypsin-cleaved peptides were analyzed by LC/MS/MS.
2.4. Activity assays
Protein kinase activity of recombinant GSK-3 homologs was assayed by Scintillation Proximity Assay (SPA) [6] using streptavidin-coated beads, [γ-33P] ATP (Amersham Biosciences, Piscataway, NJ), and biotinylated peptide substrate (BioGSP-2. Peptide sequence: Biotin-C6-Tyr-Arg-Arg-Ala-Ala-Val-Pro-Pro-Ser-Pro-Ser-Leu-Ser-Arg-His-Ser-Ser-Pro-His-Gln-Ser(PO3H2)-Glu-Asp-Glu-Glu-Glu) (American Peptide Company, Inc. Sunnyvale, CA) [6,27]. Further measurement of activity and inhibition of protein kinase phosphorylation properties by small molecule inhibitors in wild type and mutant enzymes was done by a non-radioactive Kinase-Glo luciferase assay reagent (Promega, Madison, WI). This reagent evaluates protein kinase activity by measuring changes in initial ATP concentration after GSK-3 phosphorylation reaction via luminescence [28]. A filtration assay, measuring direct incorporation of [γ-33P] onto BioGSP-2 peptide substrate and its subsequent binding to a P-81 filter (Whatman, Florhan Park, NJ), was used to confirm inhibition of protein kinase phosphorylation properties, Km of ATP and Km of BioGSP-2 peptide substrate of GSK-3 enzymes. Enzyme activity assays in the presence of 3.2 μM BioGSP-2; 8.2 nM LmajGSK-3, LmajEGSK-3, LmajFGSK-3 or LmajFEGSK-3; or 2.8 nM LinfGSK-3; 2.7 nM TbruGSK-3 with or without inhibitor(s) were performed in a buffered solution containing 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2.6H2O, 5 mM DTT (dithiothreitol), 0.1 mg/mL BSA plus 2 U/mL heparin. The reaction was initiated with addition of 2 μM ATP and incubated for 90 minutes at 30°C. The Km for ATP and peptide substrate (BioGSP-2) was measured in a filtration assay based on the incorporation of [γ-33P] into the peptide after 90 minutes at 30°C and its subsequent binding to a P-81 filter (Whatman, Florhan Park, NJ) [6]. Human GSK-3β, enzyme assay buffer, and control wells with no peptide substrate were included in each assay plate as internal positive and negative controls. Kinase-Glo luminescence and SPA reaction results were read as counts per second and counts per minute, respectively, on a Chameleon 425-104 multi-label plate scintillation counter (Hidex, Oy, Turku Finland).
2.5. Protein Crystallization
Purified LmajGSK-3 short protein was screened at the high-throughput crystallography facility at Hauptman-Woodward Institute [29] as previously described [30]. After 4 weeks, there was a crystal. The initial conditions were subsequently optimized for crystal growth using vapor diffusion from sitting drops. Crystals for diffraction measurement were grown by adding 1 μL protein at 15 mg/mL to 1 μL reservoir solution consisting of 70% v/v Tacsimate™ (Hampton Research, Aliso Viejo, CA), 0.1 M bis-Tris, pH 7.0. Tacsimate™ contains 1.8305 M malonic acid, 0.25 M ammonium citrate tribasic, 0.12 M succinic acid, 0.3 M DL-malic acid, 0.4 M sodium acetate trihydrate, 0.5 M sodium formate, and 0.16 M ammonium tartrate dibasic.
2.6. X-ray diffraction and structure determination
Crystals of LmajGSK-3 short were frozen directly in liquid nitrogen. Data were collected to 2.0 Å resolution from a single crystal on a MarMosaic 300 mM CCD detector on beamline 23 ID-D at the Advanced Photon Source, Argonne IL, at a wavelength of 0.9184 Å. Data were processed and scaled using HKL2000 [31]. Data collection statistics are given in Table 2. The structure was solved by molecular replacement using the HsGSK-3β structure (PDB 1GNG) [32] after removal of loops and ligands. Molrep [32,34] placed two copies of the model into the asymmetric unit yielding a correlation coefficient 0.48. Iterative manual model building using Coot [35] and refinement in REFMAC continued until R and Rfree reached 0.226 and 0.224, respectively. Model quality was validated using Coot and MolProbity [35,36]. The structure has been deposited in the Protein Data Bank with access code 3E3P.
Table 2.
Data-collection and refinement statistics for L. major GSK-3 short
| Wavelength (Å) | 0.9184 |
| Resolution (Å) | 50 – 2.0 (2.00 – 2.07) |
| Space group | C2221 |
| Unit cell (Å) | a = 70.2, b = 155.7, c = 158.8. |
| Total number of observations | 358269 |
| Unique reflections | 54336 (3879) |
| Rmerge | 0.087 (0.691) |
| Completeness (%) | 92.3 (66.6) |
| Mean I/σ(I) | 20.1 (1.7) |
| Redundancy | 6.6 (5.0) |
| Wilson B factor (Å2) | 32.1 |
| Refinement | |
| Resolution (Å) | 40 –2.0 |
| R/Rfree | 0.226/0.271 |
| Rmsd bonds (Å) | 0.022 |
| Rmsd angles (°) | 1.968 |
| Protein atoms | 5415 |
| Water molecules | 196 |
| Mean B protein (Å2) | 46.1 |
| Mean B other atoms (Å2) | 39.8 |
| Residues in most favored regions (%) | 96.1 |
| Residues in allowed regions (%) | 100.0 |
| Residues in disallowed regions (%) | 0 |
2.7. Modeling of glycine-rich loop
In order to be able to perform docking studies with inhibitors, Modeller (v8.2) [37] was used to generate coordinates for the glycine-rich loop and the hinge regions of LmajGSK-3 short, which are disordered in the crystal structure. The template for modeling was HsGSK-3β (PDB_ID: 1PYX) [38].
2.8. Modeling of inhibitors
The inhibitors used in the assays were docked into the LmajGSK-GH model using the GOLD software (Genetic Optimization of Ligand Docking, GOLD 4.0.1, CCDC, UK) [39]. The ligand-binding site in LmajGSK-3 was defined as all residues within a 10 Å radius of the main chain carbonyl oxygen atom of Glu 101. The number of poses for each inhibitor was limited to 10, and they were clustered so that conformations within 1.5 Å RMSD (root mean square difference) of each other were considered identical. The individual binding poses of each ligand were ranked according to the GOLD score. The top ranked conformation of each ligand was selected and visualized using Hermes 1.0.1 (Hermes 1.0.1, CCDC, UK) [39] to examine the mode of protein-inhibitor binding, then examined individually.
Prior to docking inhibitors into the LmajGSK-3 short structure, we verified that the GOLD software was able to reproduce the correct pose of the substrate analog phosphoaminophosphonic acid-adenylate ester ANP. The binding mode of ANP is known when bound to human GSK (PDB 1PYX) [38]. The RMSD between the docked pose of ANP in the LmajGSK-3 short structure and its bound conformation in the crystal structure 1PYX [38] was 0.56 Å when the protein structures were superimposed. Thus, it seems likely that the docking results with LmajGSK-3 short are reasonable.
3. Results
3.1. Leishmania spp. GSK-3 short: Expression, Protein kinase Activity, and Structure-Activity Relationships of Inhibitors
The short GSK-3 orthologs of T. brucei, L. major and L. infantum were analyzed in a protein kinase assay using a phosphopeptide commonly used to assay GSK-3 enzymes [6] and found to be active in transferring phosphate group from ATP to the peptide substrate. The Km values of the two substrates, ATP and BioGSP2 (Table 3), were determined by varying the concentrations of both substrates at constant enzyme concentration for each GSK-3 homolog.
Table 3.
Recombinant Glycogen Synthase Kinase-3 short kinetic characteristics
| TbruGSK-3 | LmajGSK-3 | HsGSK-3b | LinfGSK-3 | |
|---|---|---|---|---|
| Km(ATP) (μM) | 9.42 ± 2 | 8.66 ± 3 | 9.25 ± 0.84 | 6.86 ± 1.7 |
| Km (BioGSP-2) (μM) | 6.56 ± 3.6 | 6.61 ± 2.42 | 6.83 ± 0.86 | 5.41 ± 0.65 |
Eleven commercially available protein kinase inhibitors were assayed for activity against LmajGSK-3 short and LinfGSK-3 short and compared for activity against TbruGSK-3 short (Table 4) using an ATP consumption assay in which changes in initial ATP concentration were monitored by luciferase and light production (Kinase-Glo). The inhibitor results demonstrate that the SARs for the Leishmania spp. GSK-3 short are nearly identical. Surprisingly, the SAR of TbruGSK-3 short was distinct from the Leishmania spp. GSK-3 short for some of these inhibitors. The most obvious example is the GW8510 inhibitor, a potential candidate for further chemical and structural optimization. GW8510 was earlier identified as a versatile scaffold for the development of an effective neuroprotective compound that could have therapeutic value in the treatment of human neurodegenerative diseases [40]. GW8510's IC50 value for TbruGSK-3 short was 21 nM compared to 840 nM for LmajGSK-3 short and 628 nM for LinfGSK-3 short (Table 4, Fig. 2). The two most potent inhibitors of LmajGSK-3, GW8510 and CDK 1/2 inhibitor III showed competition for the ATP binding site. This was demonstrated by a reduction in velocity when increasing concentrations of the inhibitors were added to the reaction (Fig. 3).
Table 4.
Comparative inhibition of GSK-3 short from various species, mutants of LmajGSK-3 short, and HsGSK-3β. Displayed are the mean IC50s (μM) and standard deviation of the mean in parentheses.
| Inhibitors: Mean IC50 in μM (SD) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Enzyme | GW8510 | GSK-3β Inhibitor VIII | TrkA Inhibitor | Angio-genesis inhibitor | Ro-31-7549 | Indirubin-3'-monoxime-5-sulphonic Acid | JAK3 Inhibitor VI | Alster-paullone, 2-cyanoethyl | SU9516 | Cdk1/2 Inhibitor III | Hymeni-aldisine, Stylissa dami-cornis |
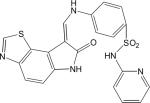
|
|
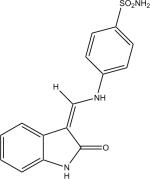
|
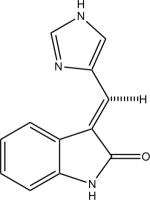
|
|
|
|
|
|
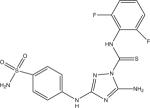
|
|
|
| LinfGSK-3 | 0.63 (0.04) | no activity | no activity | 13.59 (0.89) | 1.52 (0.20) | 2.22 (0.07) | 4.04 (2.49) | 1.04 (0.40) | 1.87 (0.09) | 0.26 (0.09) | 18.25 (3.83) |
| LmajGSK-3 | 0.84 (0.15) | no activity | no activity | 18.30 (0.68) | 1.63 (0.66) | 2.84 (1.01) | 6.79 (0.07) | 1.00 (0.22) | 3.04 (0.24) | 0.32 (0.09) | 13.13 (4.51) |
| LmajEGSK-3 | 0.33 (0.04) | no activity | no activity | 18.02 (2.77) | 1.55 (0.46) | 8.63 (0.88) | 5.82 (0.92) | 4.76 (1.97) | 3.15 (0.29) | 0.46 (0.15) | 10.42 (0.07) |
| LmajFGSK-3 | 0.09 (0.01) | no activity | 16.43 (5.84) | 8.47 (0.38) | 1.08 (0.07) | 0.93 (0.15) | 2.43 (0.05) | 0.31 (0.04) | 1.40 (0.35) | 0.15 (0.05) | 16.07 (2.77) |
| LmajEFGSK-3 | 0.03 (0.01) | 13.31 (1.08) | 6.15 (2.48) | 6.76 (0.74) | 0.89 (0.17) | 2.29 (0.37) | 6.22 (2.62) | 0.87 (0.29) | 1.29 (0.27) | 0.18 (0.06) | 9.25 (0.55) |
| TbruGSK-3 | 0.02 (0.01) | 4.30 (0.90) | 2.81 (0.03) | 2.44 (0.47) | 1.34 (0.48) | 0.48 (0.02) | 1.70 (0.48) | 1.05 (0.29) | 0.59 (0.09) | 0.12 (0.04) | 3.95 (0.47) |
| HsGSK-3b | 0.02 (0.01) | 0.80 (0.16) | 13.93 (2.57) | 1.62 (0.20) | 0.004 (0.001) | 0.06 (0.01) | 1.16 (0.37) | 0.02 (0.01) | 0.41 (0.14) | 0.21 (0.05) | 0.10 (0.02) |
Fig. 2.

Titration of GW8510 and the varying forms of LmajGSK-3 short (and mutant forms), LinfGSK-3 short, and TbruGSK-3 short. LmajE and LmajF represent mutants of LmajGSK-3 short with substitutions in the Y102F (LmajF) and D105E (LmajE) sites (Table 4) to represent residues similar to that of TbruGSK-3 short. LmajFE1 represents preparations of double mutants with both Y102F and D105E substitutions.
Fig. 3.

Untransformed plots for the effects of different concentrations of ATP competitive inhibitors GW8510 and Cdk1/2 inh. III on the velocity of LinfGSK-3 short (A) and LmajGSK-3 short (B).
3.2. Phosphorylation and Mass spectrometry analysis of LmajGSK-3 short
Analysis of LmajGSK-3 short, prepared in E. coli, showed the presence of two protein forms with MW suggesting a phosphorylated form and an unphosphorylated form (Fig. 1A). The recombinant protein was further resolved by cation exchange chromatography, yielding 2 major peaks (Fig. 1B). Analysis by SDS-PAGE showed that both peaks were composed of a 41-kDa protein band, which matched the predicted MW of 3C-cleaved LmajGSK-3 short (Fig. 1C). MALDI/TOF analysis of the resulting peaks showed that the first peak was composed of phosphorylated-GSK and the second peak was composed of non-phosphorylated GSK (Fig. 1D). LC/MS/MS analysis showed the presence of 4 peptides with phosphorylated amino acids corresponding to Ser2 (MSPLNAAAAADER), Ser232 (GDNSPAGQLHEIVR), Tyr264 (LNPSHTDVDLYPNSK) and Tyr305 (MKPYPEALCHPYDELHDPATK). Both phosphorylated and non-phosphorylated LmajGSK-3 short were highly active in the kinase activity assay. There was no detectable doubly phosphorylated LmajGSK-3 short and none of the detected phosphorylation sites were within the activation loop. This does not however rule out the possibility of physiological phosphorylation within the activation loop since activation phosphorylation may occur in Leishmania spp. by other kinases.
3.3. Overall structure description
The structure of the non-phosphorylated form of LmajGSK-3 short was solved to 2.0 Å resolution by molecular replacement (Table 2). Residues 7–23 and 34–353 are well defined by electron density for monomer A and 7–23 and 33–353 for monomer B. The protein (Fig. 4) has a typical two-domain kinase fold [41] in which each domain is connected via a linker or hinge region (residues 102–106). The β-strand domain at the N-terminal end consists of a β sheet of five antiparallel strands in the order of 1–2–3–5–4 (residues 20–22, 35–39, 45–52, 94–101, and 79–86, respectively). The β strands 1–3 lie between two helices α1 (10–18) and α2 (59–72). The α-helical domain at the C-terminal end consists of 17 helices (Fig. 5). The ATP-binding site and the activation loop (residues 169–187) lie between the two domains near the hinge. The missing residues of 24–33 make up the glycine-rich loop, which forms the top of the active site (Fig. 4 and 5).
Fig. 4.

Crystal structure of LmajGSK-3 short modeled with the CDK1/2 Inhibitor III. The α-helical and β-strand domains are colored cyan and blue, respectively. The hinge and activation loop of the active site are highlighted in yellow. The glycine-rich loop that is disordered in the crystal structure is depicted as magenta dots. The CDK1/2 Inhibitor III is absent from the crystal structure, but drawn here to depict the location of the inhibitor-binding site.
Fig. 5.

Overlay of the LmajGSK-3 short crystal structure (blue) and model (grey). The modeled glycine-rich loop is not visible in the crystal structure and is colored in red. The right panel is rotated 30 degrees compared to the left.
3.4. Active site comparison
The ATP-binding site that can be occupied by inhibitors consists of five areas (Table 5): 1) the adenine site next to the hinge region of the enzyme, 2) a hydrophobic pocket close to the N6 of the adenine ring whose size is controlled by the so-called gatekeeper residue, 3) a pocket next to N3 of the adenine ring, 4) a fairly open region where the ribose binds, and 5) the binding site for triphosphate plus Mg (II) [42]. Most inhibitors only occupy some of these areas [42].
Table 5.
ATP binding site analysis
| L. major | L. infantum | T. brucei | Human | |
|---|---|---|---|---|
| Adenine pocket | A26 | A26 | A26 | I62 |
| V34 | V34 | V34 | V70 | |
| A47 | A47 | A47 | A83 | |
| E101(bb) | E101(bb) | E102 (bb) | D133 (bb*) | |
| Y102 | Y102 | F103 | Y134 | |
| V103(bb) | V103(bb) | V104(bb) | V135(bb) | |
| L158 | L158 | L159 | L188 | |
| Gatekeeper pocket | V77 | V77 | V77 | V110 |
| M100 | M100 | M101 | L132 | |
| “Near adenine N3” pocket | P104 | P104 | P105 | P136 |
| D105 | D105 | E106 | E137 | |
| T106 | T106 | T107 | T138 | |
| R109 | R109 | R110 | R141 | |
| Ribose region | G27 | G27 | G27 | G63 |
| H155(bb) | H155(bb) | H156(bb) | Q185(bb) | |
| PPP+Mg(II) pocket | Q28(bb) | Q28(bb) | Q28(bb) | N64(bb) |
| G29 | G29 | G29 | G65 | |
| K49 | K49 | K49 | K85 | |
| K153 | K153 | K154 | K183 | |
| N156 | N156 | N157 | N186 | |
| D170 | D170 | D171 | D200 |
bb = the residue contributes to the binding site only through the backbone atoms.
The 21 residues lining each of these five areas in LmajGSK-3 short and their counterparts in the related TbruGSK-3 and HsGSK-3β are listed in Table 5. Only 3 residues differ between LmajGSK-3 short and HsGSK-3β. They line the adenine, gatekeeper, and the N3 pockets. Ala 26 of Lmaj and Tbru, which originates from the so-called glycine-rich loop, sits above the adenine plane and is replaced by an isoleucine in the human enzyme. In principle, the presence of a smaller residue in the parasite enzymes provides more space for larger, bulkier inhibitors. However, most protein kinase inhibitors cannot exploit this difference because they mimic the adenine by a planar ring system that forms hydrogen bonds to the hinge region analogous to those formed by the N1 and N6 of adenine, thereby presenting their planar ring perpendicular to Ala 26, a direction which is unreachable by substitutents attached to such scaffolds.
Another residue in the adenosine pocket, Tyr 102 in LmajGSK-3 short, which originates from the hinge region of the bilobal enzyme, might offer opportunities for selectivity in T. brucei, which has a phenylalanine in that position. Unfortunately the homologous residue in HsGSK-3β is also Tyr 134 (Table 5), thus selectivity cannot be obtained between human and LmajGSK-3 short. In the gatekeeper position of LmajGSK-3 and TbruGSK-3 there is a methionine (Met 100 or 101, respectively), whereas HsGSK-3β gatekeeper is a Leucine (Leu 132). Both Met and Leu residues have approximately the same van der Waals volume, but maybe the difference between an unbranched and a branched hydrophobic residue can be exploited. Finally, in a pocket near N3 of the adenine ring, LmajGSK-3 but not TbruGSK-3, possesses Asp 105 instead of the Glu 137 as seen in the human enzyme. Comparison of the crystal structures reveals a difference at this position. Glu 137 of the human enzyme makes a salt bridge with Arg 141, but the shorter Asp 105 of LmajGSK-3 is apparently unable to partner with Arg 109 and instead forms a hydrogen bond with the backbone carbonyl of Pro 104, thereby partially filling the pocket. Thus, LmajGSK-3 short is expected to be more restrictive to inhibitor size in this region compared to the human and the T. brucei enzymes.
3.5. The SAR of inhibition of LmajGSK-3 short by compounds can be explained by molecular modeling
Despite repeated attempts, we were unable to obtain LmajGSK-3 short structures co-crystallized with inhibitors. Since the glycine-rich loop was missing in the structure, yet is known to interact with inhibitors, we created a model of the GSK-3 short (Fig. 5). The inhibitors listed in Table 4 were docked with GOLD into the LmajGSK-3 short model. Their predicted poses agree with those observed experimentally in various crystal structures of these inhibitors in complex with HsGSK-3β. For example, in the case of CDK1/2 inhibitor III the diamino-substituted, triazole is predicted to bind to the adenine pocket and form hydrogen bonds with the backbone carbonyl of Glu 101 and the backbone nitrogen and carbonyl of Val 103 (Fig. 4 and 6A). The attached phenylsulfonamide is predicted to sit in the pocket near adenine N3. The difluorophenyl is predicted to occupy the PPP pocket and form hydrogen bonds to the amidino group of Arg 109 and the backbone of Ala 26. This CDK1/2 inhibitor III has been observed in several protein kinase structures in the same binding pose: with casein kinase 1 gamma 3 [PDB: 2CHL], ste20-like kinase [PDB: 2J51; 2JFL] [43], and CAM4K [PDB: 2W4O; unpublished SGC structure].
Fig. 6.

A) Model of LmajGSK-3 short with CDK 1/2 inhibitor III showing sidechain interactions to Arg 109 and backbone interactions to Glu 101, Val 103, and Ala 26. B) Model of LmajGSK-3 short with GW8510 showing similar interactions as in CDK 1/2 inhibitor III, but with additional backbone interactions to Met 25.
In the case of GW8510, the thiazolo-indolinone ring of the inhibitor is predicted to sit in the adenine pocket and form hydrogen bonds with the backbone carbonyl of Glu 101 and the backbone nitrogen of Val 103 (Fig. 6B). The attached p-aminophenyl is predicted to make a hydrogen bond to the backbone carbonyl of Val 103. The terminal pyridine group is predicted to sit against Ala 26 and to form additional hydrogen bonds to Met 25 compared to CDK1/2 inhibitor III. These additional interactions may account for GW8510's enhanced potency as an inhibitor, compared with CDK 1/2 inhibitor III. The GW8510 inhibitor was observed in the same pose as in human CDK2 [PDB: 1FVV] [44].
3.6. Differences in the SAR between Leishmania spp. GSK-3 and T. brucei can be explained by 2 residues that line the ATP-binding pocket
As noted above, there were two amino acid variations that caused differences in the ATP-binding pocket of Leishmania spp. GSK-3 and T. brucei GSK-3 short, namely Tyr102Phe [Y102F] and Asp105Glu [D105E] (1st AA is from LmajGSK-3 short, # based on LmajGSK-3 short, 2nd AA is the homologous AA in TbruGSK-3 short). We hypothesized that these residues in the LmajGSK-3 short might inhibit the binding of inhibitors, such as GW8510 and CDK1/2 Inhibitor III, thus explaining the more potent inhibition of GW8510 and CDK1/2 Inhibitor III against TbruGSK-3 short vs. LmajGSK-3 short. To test this hypothesis, we made recombinant LmajGSK-3 short mutants with single substitutions Y102F or D105E and with both substitutions. All of these mutants were found to be enzymatically active. The mutation Y102F of LmajGSK-3 short had a partial effect, conferring more than 10-fold sensitivity to GW8510 inhibition, while mutation D105E had nearly 2 fold effect (Table 4, Fig. 2). However, the double mutant (Y102F and D105E) was remarkably sensitive to GW8510 and CDK1/2 Inhibitor III, with a value roughly equal to that of the IC50 of TbruGSK-3 short (Table 4, Fig. 2). The double LmajGSK-3 short mutant had a decreased IC50, compared to that of wild type LmajGSK-3 short, for GW8510 and CDK1/2 Inhibitor III (Table 4, Fig. 1). GW8510 and CDK1/2 inhibitor III were predicted to have limited contact with Tyr 102 (Y102) and Asp105 (D105) amino acids in LmajGSK-3 short wild type enzyme and clearly these interactions increase with mutant Phe 102 (F102) and Glu 105 (E105) residues. The greatest single mutation effect was observed when the mutants were tested against GW8510. The LmajGSK-3 short IC50 value of 840 nM for GW8510 was reduced to 93 nM for the LmajGSK-3 short Y102F mutant. These observations support our inhibitor-binding mode model that suggests a better interaction in the adenine binding pocket between the aromatic rings of GW8510 with the hydrophobic phenylalanine residue relative to the less hydrophobic and more acidic tyrosine residue occupying the same position in WT LmajGSK-3. However, it is notable that HsGSK-3β has the same tyrosine residue as LmajGSK-3 short at the homologous residue, but it is rotated 180 degrees away from the inhibitor-binding site. Thus other dynamic processes must explain the increased affinity for GW8510 for binding HsGSK-3β vs. wild type LmajGSK-3 short. The observations also suggest that compounds that fill more space and hydrogen bonds where they contact Asp 105 will be more successful inhibitors of LmajGSK-3 short. Therefore, potency of selected compounds may be improved by making analogues with a bulkier functional group having tighter contact with Asp 105 of LmajGSK-3 short.
4. Discussion and Conclusion
Increasing interest in GSK-3 enzymes [6,7,11] for anti-parasitic drug development warrants detailed understanding of their structure and SAR. This study presents the first solved structure of a parasitic protozoan GSK-3 enzyme, i.e. LmajGSK-3 short. There are 2 orthologs of GSK-3 enzyme in Leishmania spp. and T. brucei, GSK-3 short and GSK-3 long. We focused on the short orthologs of Leishmania spp. (LmajGSK-3 short (GeneDB LmjF18.0270) and LinfGSK-3 short (GeneDB_LinJ18.0270)) since the orthologous LdonGSK-3 short (98% sequence identity with LmajGSK-3 short and 100% identity with LinfGSK-3 short) and the TbruGSK-3 short were recently shown to be potential drug targets [6,7]. Similarly, there was no obvious evidence of overlap in functional characteristics that can suggest compensation of roles when TbruGSK-3 short or TbruGSK-3 long gene products were separately ablated by RNAi [6]. Furthermore, similarities in the ATP binding domains of GSK-3 short and GSK-3 long may be sufficient enough for an inhibitor to hit both targets at the same time. By extrapolating from the TbruGSK-3 genes, it would be anticipated that inhibitors of Leishmania spp GSK-3 short would be sufficient to stop the growth and proliferation of these parasites. Moreover, Xingi et al [7] recently identified LdonGSK-3 short as the predominant intracellular target of 6-Br-5methylindirubin-3'oxime (5-Me-6-Bio) in L. donovani and provided genetic evidence for a potential role in cell-cycle progression and apoptosis-like death (note that our results from LinfGSK-3 short inhibitors will be the same for that of LdonGSK-3 short as the amino acid sequences of these enzymes are identical). Thus, there is reasonable evidence that GSK-3 short is a promising drug target for Leishmania spp. and their GSK-3 long activities may not provide sufficient compensation to reverse potent inhibitors' effects
All 11 protein kinase inhibitors are predicted to bind within the enzyme active site at the ATP-binding pocket of LmajGSK-3 short. Direct evidence of inhibitor competition for ATP binding site was demonstrated by a decrease in velocity when different concentrations of the most active compounds CDK1/2 Inhibitor III and GW8510 were added to the reaction across increasing concentrations of ATP (Fig. 3). LmajGSK-3 short has a hydrophobic cavity in its active site, and it is expected that these inhibitors are capable of interacting with the hydrophobic pocket and more sub-sites near this pocket; thus, further optimization could lead to more potent inhibitors and possibly leishmania-specific inhibitors. Such an expectation is consistent with our bioassay results, as exemplified by species inhibitory differences among all inhibitors (Table 4).
The compounds CDK1/2 Inhibitor III and GW8510 have lowest IC50 values against LmajGSK-3 short. All attempts to co-crystallize these and other inhibitors with LmajGSK-3 short failed. However the structure and molecular modeling suggests that CDK1/2 Inhibitor III and GW8510 occupy almost the whole active pocket of GSK-3 and interact with conserved residues in the hinge region via hydrogen bonding, thus explaining their potency (Fig. 6A and B).
It is important to note that many of the compounds in this study are known to inhibit multiple human protein kinases; hence, off–target effects are a potential drawback of these compounds. However, with our knowledge and understanding of compound interaction with residues within the ATP-binding cavity described above, there is a greater possibility of designing inhibitors that will be relatively specific for Leishmania spp GSK-3. A screen for new scaffolds inhibiting LmajGSK-3 short, using high-throughput screening of focused protein kinase inhibitor libraries, might yield new leads that have selective activity against LmajGSK-3 short.
Alternatively, non-selective compounds that effectively target both Leishmania GSK-3 and HsGSK-3β may be suitable for anti-leishmanial chemotherapy, as GSK-3 inhibitors have not been found to be toxic when administered to rodents [13,16–19]. Indeed, GSK-3 inhibitors are being actively studied by pharmaceutical companies for the therapy of Alzheimer's, diabetes, and mania [13–19].
Recent studies have shown that HsGSK3β inhibition is associated with the inhibition of inflammation by attenuating activation of the pro-inflammatory transcription factor NFκB. HsGSK3β inhibition has also been shown to induce secretion of the anti-inflammatory cytokine IL-10 [45–47]. Thus, anti-inflammatory effects of HsGSK3β inhibitors may reduce the inflammatory pathology of leishmaniasis. Both anti-parasitic and anti-inflammatory properties of GSK3 inhibitors would aid in the successful treatment of leishmaniasis.
In conclusion, the GSK-3 short enzymes of L. major and L. infantum have been recombinantly expressed and the crystal structure of LmajGSK-3 short was solved. Our structural and docking studies combined with mutational studies demonstrate a solid understanding of the binding mode of various inhibitors and delineate ways to predict inhibitors that might be useful for the therapy of leishmaniasis. The inhibitor SAR of Lmaj GSK-3 short and LinfGSK-3 short (and by inference, L. donovani) are virtually identical, suggesting that compounds inhibiting GSK-3 short could be useful for the therapy of both cutaneous and visceral leishmaniasis.
Summary.
This study presents the first solved structure of a parasitic protozoan GSK-3 enzyme and concluded that LmajGSK-3 short structure reported here will aid in the design and development of potent GSK-3 inhibitors for all Leishmania species.
Pictogram.

Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (awards GM64655, AI067921, AI080625) and UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR). K.K.I. was supported by a US National Institutes of Health grant from the Fogarty International Center 2D43 TW000924. We thank Molly C. Reid, Isolde LeTrong and Eric Larson for assistance with data collection. GM/CA CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357. K.K. Inampudi thanks the Cambridge Crystallographic Data Centre for providing the GOLD software.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Hammarton TC, Mottram JC, Doerig C. The cell cycle of parasitic protozoa: potential for chemotherapeutic exploitation. Prog Cell Cycle Res. 2003;5:91–101. [PubMed] [Google Scholar]
- [2].Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5:873–82. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- [3].Sibley CH, Hunt SY. Drug resistance in parasites: can we stay ahead of the evolutionary curve? Trends Parasitol. 2003;19:532–7. doi: 10.1016/j.pt.2003.09.009. [DOI] [PubMed] [Google Scholar]
- [4].Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev. 2005;18:293–305. doi: 10.1128/CMR.18.2.293-305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Allocco JJ, Donald R, Zhong T, Lee A, et al. Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro. Int.J.Parasitol. 2006;36:1249–59. doi: 10.1016/j.ijpara.2006.06.013. [DOI] [PubMed] [Google Scholar]
- [6].Ojo KK, Gillespie JR, Riechers AJ, et al. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob Agents Chemother. 2008;52:3710–7. doi: 10.1128/AAC.00364-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xingi E, Smirlis D, Myrianthopoulos V, et al. 6-Br-5methylindirubin-3 ' oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: Exploitation of GSK-3 for treating leishmaniasis. International Journal for Parasitology. 2009;39:1289–303. doi: 10.1016/j.ijpara.2009.04.005. [DOI] [PubMed] [Google Scholar]
- [8].Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- [9].Plyte SE, Hughes K, Nikolakaki E, et al. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114:147–62. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- [10].Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur J Biochem. 1983;130:227–34. doi: 10.1111/j.1432-1033.1983.tb07140.x. [DOI] [PubMed] [Google Scholar]
- [11].Droucheau E, Primot A, Thomas V, et al. Plasmodium falciparum glycogen synthase kinase-3: molecular model, expression, intracellular localisation and selective inhibitors. Biochim Biophys Acta. 2004;1697:181–96. doi: 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- [12].Coghlan MP, Culbert AA, Cross DA, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- [13].Ring DB, Johnson KW, Henriksen EJ, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. 2003;52:588–95. doi: 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- [14].Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–7. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- [15].Cohen Y, Chetrit A, Cohen Y, et al. Cancer morbidity in psychiatric patients: influence of lithium carbonate treatment. Med Oncol. 1998;15:32–6. doi: 10.1007/BF02787342. [DOI] [PubMed] [Google Scholar]
- [16].Cuzzocrea S, Mazzon E, Esposito E, et al. Glycogen synthase kinase-3beta inhibition attenuates the development of ischaemia/reperfusion injury of the gut. Intensive Care Med. 2007;33:880–93. doi: 10.1007/s00134-007-0595-1. [DOI] [PubMed] [Google Scholar]
- [17].Cuzzocrea S, Di Paola R, Mazzon E, et al. Glycogen synthase kinase 3 beta inhibition reduces the development of nonseptic shock induced by zymosan in mice. Shock. 2007;27:97–107. doi: 10.1097/01.shk.0000235084.56100.71. [DOI] [PubMed] [Google Scholar]
- [18].Dugo L, Abdelrahman M, Murch O, et al. d Glycogen synthase kinase-3 beta inhibitors protect against the organ injury and dysfunction caused by hemorrhage and resuscitation. Shock. 2006;25:485–91. doi: 10.1097/01.shk.0000209545.29671.31. [DOI] [PubMed] [Google Scholar]
- [19].Cuzzocrea S, Crisafulli C, Mazzon E, et al. Inhibition of glycogen synthase kinase-3beta attenuates the development of carrageenan-induced lung injury in mice. Br J Pharmacol. 2006;149:687–702. doi: 10.1038/sj.bjp.0706902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].El Sayed NM, Myler PJ, Blandin G, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–9. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- [21].Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- [22].Ivens AC, Peacock CS, Worthey EA, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–42. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mehlin C, Boni E, Buckner FS, et al. Heterologous expression of proteins from Plasmodium falciparum: results from 1000 genes. Mol Biochem Parasitol. 2006;148:144–60. doi: 10.1016/j.molbiopara.2006.03.011. [DOI] [PubMed] [Google Scholar]
- [24].Alexandrov A, Vignali M, LaCount DJ, et al. A facile method for high-throughput co-expression of protein pairs. Mol Cell Proteomics. 2004;3:934–8. doi: 10.1074/mcp.T400008-MCP200. [DOI] [PubMed] [Google Scholar]
- [25].Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- [26].Napuli AJ, Alzhanova DV, Doneanu CE, et al. The 64-kilodalton capsid protein homolog of Beet yellows virus is required for assembly of virion tails. J Virol. 2003;77:2377–84. doi: 10.1128/JVI.77.4.2377-2384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–9. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ojo KK, Larson ET, Keyloun KR, et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010 doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luft JR, Collins RJ, Fehrman NA, et al. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–9. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- [30].Arakaki TL, Buckner FS, Gillespie JR, et al. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol Microbiol. 2008;68:37–50. doi: 10.1111/j.1365-2958.2008.06131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallogr. 1997;276(Pt A):307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- [32].Bax B, Carter PS, Lewis C, et al. The structure of phosphorylated GSK-3 beta complexed with a peptide, FRATtide, that inhibits beta-catenin phosphorylation. Structure. 2001;9:1143–52. doi: 10.1016/s0969-2126(01)00679-7. [DOI] [PubMed] [Google Scholar]
- [33].Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- [34].Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography. 1997;30:1022–5. [Google Scholar]
- [35].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [36].Lovell SC, Davis IW, Arendall WB, III, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- [37].Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- [38].Bertrand JA, Thieffine S, Vulpetti A, et al. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J Mol Biol. 2003;333:393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- [39].Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- [40].Johnson K, Liu L, Majdzadeh N, et al. Inhibition of neuronal apoptosis by the cyclin-dependent kinase inhibitor GW8510: identification of 3' substituted indolones as a scaffold for the development of neuroprotective drugs. J Neurochem. 2005;93:538–48. doi: 10.1111/j.1471-4159.2004.03004.x. [DOI] [PubMed] [Google Scholar]
- [41].Scapin G. Structural biology in drug design: selective protein kinase inhibitors. Drug Discov Today. 2002;7:601–11. doi: 10.1016/s1359-6446(02)02290-0. [DOI] [PubMed] [Google Scholar]
- [42].Liao JJ. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem. 2007;50:409–24. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- [43].Pike ACW, Rellos P, Niesen FH, et al. Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. Embo Journal. 2008;27:704–14. doi: 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Davis ST, Benson BG, Bramson HN, et al. Prevention of chemotherapy-induced alopecia in rats by CDK inhibitors. Science. 2001;291:134–7. doi: 10.1126/science.291.5501.134. [DOI] [PubMed] [Google Scholar]
- [45].Spinnler K, Mezger M, Steffens M, et al. Role of glycogen synthase kinase 3 (GSK-3) in innate immune response of human immature dendritic cells to Aspergillus fumigatus. Med Mycol. 2010;48:589–97. doi: 10.3109/13693780903420625. [DOI] [PubMed] [Google Scholar]
- [46].Klamer G, Song E, Ko KH, et al. Using small molecule GSK3beta inhibitors to treat inflammation. Curr Med Chem. 2010;17:2873–81. doi: 10.2174/092986710792065090. [DOI] [PubMed] [Google Scholar]
- [47].Meijer L, Thunnissen AM, White AW, et al. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.