

Abstract
AIMS
The objective of the present study was to assess the pharmacokinetics of riluzole in patients with spinal muscular atrophy (SMA).
METHODS
Fourteen patients were enrolled in an open-label, nonrandomized and repeat-dose pharmacokinetic study. All participants were assigned to receive 50 mg riluzole orally for 5 days. Riluzole plasma concentrations were determined from samples obtained at day 5.
RESULTS
The pharmacokinetic analysis demonstrated that a dose of 50 mg once a day was sufficient to obtain a daily total exposure [AUC(0,24 h) = 2257 ng ml−1 h] which was comparable with results obtained in adult healthy volunteers or ALS patients in whom a dose of 50 mg twice a day is recommended. The pharmacokinetic simulation demonstrated that the administration of 50 mg twice a day could result in higher concentrations, hence reduced safety margin.
CONCLUSION
The dose of 50 mg once a day was chosen for the clinical trial evaluating the efficacy of riluzole in SMA patients.
Keywords: pharmacokinetics, riluzole, spinal muscular atrophy
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Spinal muscular atrophy (SMA) is a neuromuscular disorder of childhood.
Riluzole is an anti-excitatory agent recommended for the treatment of amyotrophic lateral sclerosis (ALS).
Riluzole pharmacokinetics are well documented in patients with ALS.
WHAT THIS STUDY ADDS
Riluzole pharmacokinetics have never documented in patients with SMA.
This study showed that the administration of 50 mg riluzole once a day to patients with SMA leads to total riluzole daily exposure comparable with that obtained after the administration of 50 mg twice a day in healthy volunteers or ALS patients.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive disease, clinically manifested by slowly progressive muscle weakness caused by a degeneration of anterior horn cells. This common neuromuscular disorder of childhood occurs in 1 in 6000 to 10 000 births [1].
Current classification of SMA forms, that has been mapped to chromosome 5q13, is based on clinical criteria [2]: the earlier the beginning of symptoms, the more severe is the disease. Spinal muscular atrophy type I is characterized by onset before 6 months of age, failure to achieve sitting without support and severe respiratory insufficiency. Spinal muscular atrophy type II (chronic SMA) is usually symptomatic between 6 and 18 months, but may start earlier. Those patients ultimately attain independent sitting when placed and may live into adulthood. Spinal muscular atrophy III (juvenile SMA or Kugelberg-Welander disease) becomes symptomatic after the age of 18 months, and all patients walk independently. Patients with SMA III have a normal life span.
Hypotheses regarding the pathogenesis of SMA include: defective inhibition of apoptosis, glutamate cytotoxicity and lack of neurotrophic factors in nerve or muscle [3].
Clinical trials were or are currently being conducted to evaluate the effect of pharmacological therapy in SMA. Few studies have evaluated the effect of drugs that mainly act by increasing SMN2-derived RNA and proteins levels, such as valproic acid and phenylbutyrate.
Other clinical trials have evaluated the safety and efficacy of other drugs acting as neuroprotectors. Cell bodies from bulbar and spinal motor neurons receive afferents from glutamate neurons. These neurons are therefore particularly exposed to glutamate, an excitatory amino acid neurotransmitter with high neurotoxic potential. A glutamate inhibitor is currently recommended for treatment of amyotrophic lateral sclerosis (ALS) to slow the progression of the disease [4, 5].
Riluzole is a neuroprotective drug that is thought to act by blocking excitatory amino acid mediated neurotransmission and thus attenuating excitotoxicity [6]. Riluzole is the first drug to have been shown to be of benefit in the treatment of ALS. It is given as a 50 mg oral tablet twice daily. The drug is well and rapidly absorbed from the gastrointestinal tract. The relationship between administered dose and plasma concentrations is linear and the absolute bioavailability is ∼60%, although there is considerable inter-individual variability, presumably due to differences in the extent of the first-pass effect [7].
Steady-state plasma concentrations are achieved after ∼5 days, and peak plasma concentrations are ∼500 ng ml−1. Riluzole is metabolized by phase I oxidative enzymes, principally CYP1A2, followed by glucuronide conjugation and it is eliminated in the urine [8].
Haddad et al. showed that riluzole can attenuate the disease course of SMA after the onset of neuromuscular defects and may warrant further investigation in a therapeutic trial in SMA [9].
Currently, only one phase 1 controlled trial evaluating the efficacy and tolerance of riluzole in 10 young patients (seven treated, three placebo) with SMA type I has been reported [10]. Riluzole was safe in young children and may have a mitigating effect on the natural course of the disease. However, this was a limited study with insufficient power to show a difference between the two groups. Also, no pharmacokinetic data concerning the oral administration of riluzole in children has yet been published.
The aim of this study was the determination of steady state main pharmacokinetic parameters of riluzole after its oral administration at 50 mg day−1 in young patients (age between 9–17 years) with SMA.
Methods
Subjects
Fourteen patients participated in this open label, nonrandomized, repeat-dose, preliminary pharmacokinetic study. The study was conducted in one clinical centre (APHP, Raymond Poincaré Hospital, Garches, France). This study was nested in an efficacy clinical study organized as a prospective, double-blind, placebo-controlled multi-centre trial in 141 SMA patients at present ongoing.
Patients were at least 6 years old and younger than 20 years at the time of enrolment and fulfilled international classification criteria for SMA II or III with a mobility score ≤ 12 on the MFM scale [11].
Eligibility was determined on the basis of medical history, a physical examination, an electrocardiogram and standard laboratory tests including haematology and blood chemistry.
Patients were excluded if they met any of the following criteria: treatment with riluzole, treatment with any hepatotoxic drugs, renal, cardiac and hepatic insufficiency.
All subjects received a daily dose of 50 mg of riluzole administered as a 50 mg capsule for 5 days. At day 5, they stayed for 24 h at the hospital and blood samples (approximately 2 ml) were drawn into lithium heparin tubes before and at 1 h, 5 h, 9 h, 14 h and 24 h after the administration of the riluzole daily dose. These samples were then centrifuged and plasma was decanted and frozen at −20°C until analysis.
Informed consent from parents was obtained for all the patients and recruitment began after the formal approval of the protocol by the ethics committee (St Germain en Laye, France). APHP was the promoter of this study.
Assay method
Riluzole was quantified in plasma samples, after protein precipitation, using a validated liquid chromatography-mass spectrometry (LC/MS/MS) method. At the moment of assay, 100 µl of plasma was exactly measured. Protein precipitation was carried out using 150 µl of acetonitrile containing diazepam-D5 as the internal standard (I.S.). The samples were vortex mixed, then centrifuged. The supernatant was transferred to micro vials and 5 µl were analyzed by the chromatographic system. The analysis was achieved by reversed phase high performance liquid chromatography using a C-8 ACQUITY column maintained at 40°C. The mobile phase was nebulized using an electrospray source. The mass spectrometry (Quatro premier) was programmed to transmit the protonated molecules [M + H]+ at m/z 235.2 for riluzole and m/z 290.2 for I.S. via the first quadripole filter (Q1), with collision fragmentation in Q2 while monitoring, Q3, the product ions at m/z 216.2 (riluzole) and m/z 198.2 (I.S.). Micromass software (Waters, USA) was used to acquire chromatographic data, integrate the peaks and calculate plasma concentrations.
Calibration curves were obtained by fitting the peak area ratio (riluzole : I.S.) to standard concentrations using the weighted (1/concentration) linear least squares regression. The calibration curve was linear over the concentration range 2–500 ng ml−1 with determination coefficient <0.99. When a plasma sample concentration was above 500 ng ml−1, the sample was diluted with human blank plasma and re-assayed.
Based on quality control samples, the overall relative standard deviation (an index of precision) was less than 11.6%. The overall relative error (an index of accuracy) was within ± 13.9%. The lower limit of quantification (LLOQ) was validated at 2 ng ml−1.
Pharmacokinetic analysis
The noncompartmental model independent analysis was performed using WinNonLin® Pro v.4.1 (Pharsight Corporation, USA), to estimate riluzole main pharmacokinetic parameters at the steady state. Data were used to estimate individual maximal concentration (Cmax) and time necessary to reach maximal concentration (tmax). In addition, for each treatment the elimination rate constant (λz), and the area under the concentration–time curve from time 0 to 24 h of blood sampling [AUC(0,24 h)] were estimated. λz was estimated as the slope of the log-linear terminal portion of the plasma concentration vs. time curve, determined using unweighted linear least-squares regression analysis. The best number of concentrations was chosen as that giving the highest coefficient of determination, as recommended. Area under concentration–time curve was computed from 0 to 24 h using the log-linear trapezoidal method. Additionally, from these estimated parameters, several pharmacokinetic parameters were derived. Terminal elimination half-life was calculated as t1/2 = ln2/λz.
Compartmental modelling approach
Studied pharmacokinetic models
In order to describe the absorption after oral administration of riluzole, two absorption kinetic models were compared. First, the one-compartment model with first-order absorption and elimination rates was tested. The second model was a one-compartment model with zero-order absorption rate and first order elimination rate.
Estimation method
WinNonLin® software was used to fit the data of each subject using two combinations of models: a one-compartment open model with either zero order or first order absorption. The initial estimates of the pharmacokinetic parameters were computed by WinNonLin® using curve stripping.
The pharmacokinetic parameters were Vd, λz and an absorption parameter: Ka in the case of first order absorption. In the case of zero order absorption, t was fixed and determined as time necessary to reach the maximal concentration for each subject individually.
From these parameters, several derived pharmacokinetic parameters were computed: area under the curve [AUC(0,24 h)], time needed to reach maximal concentration (tmax) and maximal concentration (Cmax).
For each concentration, an additive error was assumed arising from a zero mean Gaussian distribution, with a heteroscedastic variance. Errors on two different concentrations were assumed to be uncorrelated. The error included error of the analytical method and error due to the pharmacokinetic model misspecification.
The Gauss-Newton method with Levenberg modification was used to provide the ‘least square’ estimates. Data were weighted using a constant coefficient of variation error model based on model predicted plasma concentration 1/ŷ2. As using observed data as weights is problematic since they are measured with error, predicted values are used instead. In this manner, any measurement error or random variability in the data is controlled.
Comparison of models
The Akaike Information Criterion (AIC) was used to identify the best combination of models, since the first order and the zero order absorption models are not nested. This criterion can be viewed as the sum of a measure of the goodness of fit, and of a penalty function proportional to the number of estimated parameters in the model. For each combination of models, the criterion for all subjects was computed. The combination of models with the smallest AIC is the most adequate according to a parsimony principle.
Pharmacokinetic simulation
The individual parameter estimates from the final chosen model were used to compare two dosing regimens, 50 mg once daily and 50 mg twice daily with dosing interval τ = 12 h.
Statistical analysis
The influence of sex, age, weight, body surface area and type of SMA on riluzole pharmacokinetic parameters [AUC(0,24 h), Cmax, half-life of elimination] were determined using multivariate linear regression analysis.
Statistical calculations were performed with R 5.2.0 software (R development core team 2007).
Results
Fourteen patients (five male, nine female) were enrolled in the study. Table 1 summarizes the demographic data. Only one subject was excluded from the pharmacokinetic analysis due to blood sampling difficulties. None of patients experienced adverse effects.
Table 1.
Subject demographics
| SMA II subjects | SMA III subjects | |
|---|---|---|
| Number | 8 | 6 |
| Gender | ||
| Male | 3 | 2 |
| Female | 5 | 4 |
| Age (years) | ||
| Mean ± SD | 12.8 ± 2.85 | 13.5 ± 2.34 |
| Range | 9–17 | 11–17 |
| Weight (kg) | ||
| Mean ± SD | 28 ± 13 | 30 ± 7.3 |
| Range | 17–58 | 19–39 |
| Body surface area (m2) | ||
| Mean ± SD | 0.99 ± 0.27 | 1.04 ± 0.17 |
| Range | 0.74–1.6 | 0.78–1.26 |
Figure 1 shows the mean plasma concentration vs. time profile of riluzole at steady state. The individual and mean pharmacokinetic parameters in young patients are summarized in Table 2. The individual pharmacokinetic data showed inter-individual variability in pharmacokinetic parameters with CV% varying from 45.5% to 71%;
Figure 1.
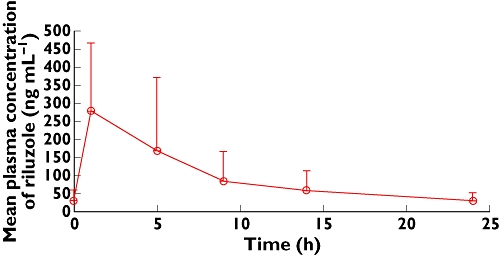
Mean pharmacokinetic profile of riluzole at steady state
Table 2.
Estimated and derived pharmacokinetic parameters of riluzole obtained with the noncompartmental approach
| Subjects | AUC(0,24 h) (ng ml−1 h) | Cmax (ng ml−1) | tmax(h) | Half-life (h) |
|---|---|---|---|---|
| SMA II | ||||
| 2001 | 1764 | 145 | 5 | 13.4 |
| 2002 | 1441 | 145 | 5 | 7.04 |
| 2003 | 2568 | 404 | 1 | 22.6 |
| 2004 | 1643 | 444 | 1 | 9.2 |
| 2005 | 7312 | 831 | 5 | 7.9 |
| 2006 | 1624 | 206 | 1 | 9.7 |
| 2007 | 2256 | 173 | 5 | 10.5 |
| 2008 | 2085 | 542 | 1 | 3.5 |
| Mean ± SD | 2587 ± 1946 | 361 ± 243 | 3* (1–5) | 10.5 ± 5.7 |
| CV% | 75.2 | 67.4 | 54.1 | |
| CI | (1238, 3825) | (192, 554) | (6.55, 17.0) | |
| SMA III | ||||
| 3001 | 2078 | 396 | 1 | 11.9 |
| 3002 | 1764 | 539 | 1 | 8.6 |
| 3003 | 2420 | 618 | 1 | 8.6 |
| 3004 | 547 | 62 | 1 | 6.0 |
| 3005 | 1842 | 314 | 1 | 8.6 |
| Mean ± SD | 1730 ± 709 | 385 ± 216 | 1 | 8.8 ± 2.1 |
| CV% | 41 | 56 | 24 | |
| CI | (1109, 2839) | (196, 582) | (6.9, 15.6) | |
| All subjects | ||||
| Mean ± SD | 2257 ± 1667 | 359 ± 234 | 1 (1–5) | 7.8 ± 4.8 |
| CV% | 74 | 65 | 61 |
AUC, area under the curve; tmax, time to reach maximal concentration; Cmax, maximal concentration.
Median value.
The mean plasma concentrations of riluzole increased until 1 h, and thereafter, plasma concentrations of riluzole declined. The mean apparent terminal half-life (t1/2) was 9.75 h.
A delay in the time to reach the maximal concentration was observed in the group of patients with SMA II as compared with patients with SMA III (median tmax 3 h vs. 1 h), with no significant effect on the total exposure and the maximal concentration.
One-compartment open models were studied in order to describe the pharmacokinetics of riluzole.
For each combination of models, individual fittings of data were obtained. Figure 2 shows the pharmacokinetic curve fitted by the model for a typical subject.
Figure 2.
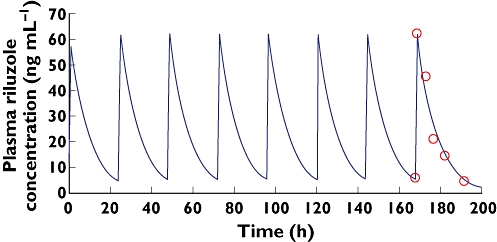
Plot of time course of riluzole plasma concentrations in a typical subject after oral administration of 50 mg once daily. Solid lines represent the pharmacokinetic curve predicted by the one compartment model with zero order absorption. Observed ( ); Predicted (
); Predicted ( )
)
After oral administration, riluzole concentrations were better predicted by a zero order absorption model than by a first order absorption model.
These graphical results were confirmed by the AIC for all subjects (4.29 vs. −18.2) for first order and zero order absorption, respectively.
For the chosen models, no trend was noticed in the graphics of standardized residuals vs. predicted concentrations or vs. time. The pharmacokinetic parameters estimated with the iterative reweighted least-squares method are summarized in Table 3.
Table 3.
Estimated and pharmacokinetic parameters of riluzole obtained with the compartmental modelling analysis
| Subjects | Vd (ml) | Estimation CV% | λz (h−1) | Estimation CV% |
|---|---|---|---|---|
| SMA II | ||||
| 2001 | 396 298 | 20.1 | 0.067 | 14.7 |
| 2002 | 396 159 | 42.7 | 0.075 | 29.7 |
| 2003 | 186 254 | 33.8 | 0.089 | 21.3 |
| 2004 | 194 255 | 45.0 | 0.122 | 22.5 |
| 2005 | 70 882 | 40.9 | 0.091 | 25.4 |
| 2006 | 326 259 | 16.1 | 0.089 | 10.1 |
| 2007 | 349 578 | 22.2 | 0.062 | 16.9 |
| 2008 | 409 043 | 12.0 | 0.089 | 7.5 |
| SMA III | ||||
| 3001 | 191 888 | 32.0 | 0.110 | 17.4 |
| 3002 | 165 102 | 45.8 | 0.140 | 20.6 |
| 3003 | 134 425 | 38.9 | 0.133 | 18.3 |
| 3004 | 826 723 | 10.7 | 0.109 | 5.9 |
| 3005 | 237 130 | 26.0 | 0.103 | 14.8 |
Vd, volume of distribution of central compartment; λz, elimination constant
The standard errors of individual estimates were also obtained, except for the time of absorption t, which was fixed and determined as the tmax estimated using the noncompartmental approach.
Discussion
This study aimed to characterize the pharmacokinetic profile of riluzole after oral administration in SMA patients.
The pharmacokinetics of riluzole were determined after steady-state concentration had been reached. Two types of pharmacokinetic analysis were carried out to determine the dose to be administered in this population, with regards to the total systemic exposure, absorption and elimination of riluzole.
Noncompartmental analysis
This analysis showed that riluzole reaches rapidly its maximal concentration after oral administration of 50 mg capsules (median tmax = 1 h). Four SMA II patients had a delay in riluzole absorption (experimental tmax = 5 h). Three of them had a Cmax two-fold lower than the mean Cmax, while the fourth had a Cmax two-fold higher than the mean Cmax. However, riluzole total exposure in this subject was three-fold higher than the mean total exposure, which could explain the higher Cmax, despite the delayed absorption. One subject, with SMA III, had a four-fold lower total exposure than mean total exposure, accompanied by a four-fold decrease in the Cmax as compared with group mean.
This variability could be due, in part, to fluctuations in CYP1A2 activity, the predominant isoenzyme in phase I riluzole metabolism [8, 12].
Although, the tmax values in the SMA II and SMA III group, respectively, were not significantly different, which was probably due to subject numbers in the SMA III group, the absorption of riluzole seemed to be delayed in the SMA II group. This delay has no consequent significant effect on the other pharmacokinetic parameters [AUC(0,24 h), Cmax and half-life of elimination], which were not significantly different between the SMA II and SMA III groups.
The mean Cmax of the whole group studied, as compared with other clinical studies conducted in healthy adults [13–15], appeared to be higher, with a high variability, even if a delay was observed to obtain the Cmax in SMA patients. However, this delay could be probably due to the sampling schedule followed in this study.
AUC(0,24 h) appears to be two-fold higher than AUC(0,24 h) obtained in other clinical studies conducted at steady state in healthy volunteers and patients with ALS, in which the mean total exposure ranged between 654 and 1470 ng ml−1 h [7, 13–15] (Table 4).
Table 4.
Main riluzole pharmacokinetic parameters obtained in clinical studies conducted in healthy volunteers and patients with ALS
| PK results | ||||||
|---|---|---|---|---|---|---|
| Reference | Population | Administration | Dose | tmax (h) | Cmax (ng ml−1) | AUC (ng ml−1 h) |
| Leliboux et al. [13] | Healthy young | Oral/R | 50 mg twice daily | 0.75 | 271 | 1029 |
| Healthy elderly | Oral/R | 50 mg twice daily | 0.75 | 244 | 869 | |
| Groenveld et al. [14] | Patients | Oral/R | 50 mg twice daily | – | 231 | 48.7* |
| Leliboux et al. [7] | Healthy | Oral/S | 25 mg | 1.1 | 52 | 207 |
| Healthy | Oral/S | 50 mg | 0.9 | 180 | 537 | |
| Healthy | Oral/S | 100 mg | 1.6 | 282 | 1195 | |
| Healthy | Oral/R | 25 mg | 0.8 | 77 | 295 | |
| Healthy | Oral/R | 50 mg | 0.8 | 137 | 654 | |
| Healthy | Oral/R | 100 mg | 1.3 | 357 | 1483 | |
| Groenveld et al. [15] | Patients | Oral/R | 50 mg twice daily | – | 183 | 1473 |
AUC, area under the curve; tmax time to reach maximal concentration; Cmax maximal concentration; λz elimination constant; t1/2 elimination half-life; S, Single dose study; R: Repeated dose study.
AUC normalized to weight.
These results suggest that riluzole total daily exposure after the administration of 50 mg once daily in children, is comparable with that obtained after the administration of 50 mg twice daily in adult healthy volunteers and ALS patients.
Compartmental analysis
The best model fitting the individual data was evaluated as follows: sum of AIC for all subjects, coefficient of variation of parameters estimation.
The zero order absorption model had the smaller sum of AIC as compared with the first order absorption. In addition, the estimation CV%s were acceptable ‘between 5.87%–45.8%’.
The model underestimated Cmax as compared with Cmax estimated by NCA which could be due to the choice of the structural model. Riluzole exhibits bi-exponential decline as has been already reported [7, 12].
Bruno et al. chose a one compartment model with first order absorption and elimination, as the best population model for riluzole, although the two compartment model substantially improved the fit, as both models provided very similar clearance estimates [12].
In our study, although the examination of data on a semi-log scale could suggest a bi-exponential decline, the number of points per subject prevented the fitting using a two compartment model.
However, the chosen model provided good estimates of pharmacokinetic parameters.
The final chosen model parameters were used to simulate individual plasma concentration–time profiles following the oral administration of 50 mg riluzole twice daily. Figure 3, shows two simulated pharmacokinetic profiles following the oral administration of 50 mg riluzole once daily and twice daily in a typical subject. This simulation demonstrates that the administration of 50 mg riluzole twice daily would result in a higher accumulation of the drug, with higher maximal and trough concentrations, and hence a reduced safety margin. However, the benefit/risk balance appears to be in favoor of a 50 mg once daily regimen, taking into account the pharmacokinetic comparison with results obtained from clinical trials conducted in adults [14].
Figure 3.
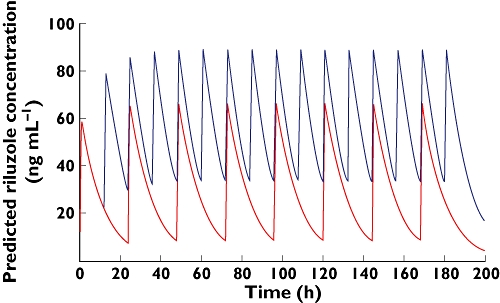
Simulated pharmacokinetic profiles following the oral administration of 50 mg riluzole once daily and twice daily in a typical subject. once daily ( ); twice daily (
); twice daily ( )
)
Different hypotheses could be proposed to explain these findings. To investigate the effect of demographic variables, a multilevel linear regression was applied to test the correlation between the main pharmacokinetic parameters [AUC(0,24 h), Cmax, half-life of elimination] and the demographic variables (age, body weight, body surface area). The results obtained from this analysis showed that there was no correlation between the studied demographic co-variables and the main pharmacokinetic parameters (Fisher test, P > 0.05). As a consequence, riluzole AUC(0,24 h), Cmax and half-life were not affected by the patient's age, weight or body surface area.
A clinical trial evaluating riluzole efficacy in children with obsessive compulsive disorder has been published recently [16]. In this study, riluzole was administered to children from 8 to 17 years old, at doses increased by 10 mg every few days, and the maximal daily dose was set at 120 mg. All the children included in this study (n = 6) reached the dose of 50 mg of riluzole twice daily. The authors reported that riluzole was well tolerated although some adverse events, such as drowsiness and transient elevations of liver enzymes, were noticed. Although no pharmacokinetic evaluation was realized during this study, the observations of the tolerability of riluzole and dosing results added to our pharmacokinetic findings suggest that modifications in the pharmacokinetics of riluzole in patients with SMA are due to the physiopathology more than other demographic variables such as age, weight and gender.
Concerning the metabolism, reduced riluzole elimination in children could explain the increased total exposure and peak concentration. This increase is usually accompanied by a prolongation of the terminal half-life. In this study, the elimination half-life was comparable with that obtained in clinical trials conducted in healthy volunteers and ALS patients [7]. In addition, during our study, all treatments influencing riluzole metabolism by CYP1A2 were avoided, which minimized the probability of drug–drug interactions.
Concerning the protein binding effect, plasma protein leads to higher drug plasma concentrations at steady state with a reduced volume of distribution. For all low extraction ratio drugs, regardless of route of administration, and for all drugs administered orally and eliminated primarily by the liver, total exposure is independent of protein binding and no dosing adjustment will need to be made. Only high extraction ratio drugs given intravenously and oral drugs eliminated by nonhepatic high extraction ratio routes will exhibit changes in unbound drug exposure when protein binding changes [17].
Riluzole is highly bound to albumin (98%), and it is eliminated principally in the urine. Change in albumin concentrations could lead to a change in riluzole plasma concentrations. Nevertheless, it has never been reported that a change in albumin concentration in SMA patients was observed. In addition, in this study, albumin was not determined on the day of the pharmacokinetic study.
Drug absorption might be affected by any disease that causes changes in intestinal blood flow, gastro-intestinal motility, stomach emptying time, gastric pH that can affect drug solubility, intestinal pH that affects the extent of ionization, the permeability of the gut wall, digestive enzyme secretion or alteration of normal gutI flora. One or more, of these physiological changes could be observed in patients with SMA, which could lead to modification in riluzole absorption profile in addition to high variability.
Although the compartmental analysis showed that the zero order absorption appears to be a best model to fit our data, in both SMA II and SMA III groups, this type of absorption could not be confirmed physiologically by data obtained from this study due to the small number of points in the absorption phase.
However, zero order absorption usually occurs when the drug is absorbed by a saturable process or a zero-order controlled release delivery system is used. This could suggest the implication of an active transport phenomenon. Several studies have reported an interaction between riluzole drug efflux and the P-gp and BCRP transporters [18, 19]. The findings of studies realized in mice suggest that riluzole diffusion is regulated by P-gp at the blood brain barrier level. Meanwhile, interactions between intestinal P-gp and riluzole are still not well documented [19]. The hypothesis that riluzole absorption is regulated by intestinal P-gp needs to be confirmed by further investigations.
The findings of the compartmental analysis, added to those obtained from the noncompartmental approach, suggest that the probable reason leading to enhanced exposure and absorption of riluzole in children with SMA, is related to the absorption step more than other stages of its kinetics, and probably the severity of the pathology could be related to modification in absorption profiles in these patients. Nevertheless, these observations need to be confirmed by a further clinical and biological investigation.
In conclusion, this study showed that the administration of 50 mg riluzole once a day to patients with SMA led to total riluzole daily exposure comparable with that obtained after the administration of 50 mg twice a day in healthy volunteers or ALS patients. This dose was chosen for the clinical trail evaluating the efficacy of riluzole in SMA patients.
Competing interests
There are no competing interests to declare.
We would like to thank the Association Française de lutte contre les Myopathies (AFM), and the Délégation à la Recherche Clinique et au Développement for their technical and logistical help. Riluzole had been graciously offered by Sanofi-Aventis France.
REFERENCES
- 1.Mostacciuolo ML, Danieli GA, Trevisan C, Muller E, Angelini C. Epidemiology of spinal muscular atrophies in a sample of the Italian population. Neuroepidemiology. 1992;11:34–8. doi: 10.1159/000110905. [DOI] [PubMed] [Google Scholar]
- 2.Munsat TL, Skerry L, Korf B, Pober B, Schapira Y, Gascon GG, al-Rajeh SM, Dubowitz V, Davies K, Brzustowicz LM. Phenotypic heterogeneity of spinal muscular atrophy mapping to chromosome 5q11.2-13.3 (SMA 5q) Neurology. 1990;40:1831–6. doi: 10.1212/wnl.40.12.1831. [DOI] [PubMed] [Google Scholar]
- 3.Greensmith L, Vrbova G. Possible strategies for treatment of SMA patients: a neurobiologist's view. Neuromuscul Disord. 1995;5:359–69. doi: 10.1016/0960-8966(94)00090-v. [DOI] [PubMed] [Google Scholar]
- 4.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–91. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 5.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347:1425–31. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 6.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47(Suppl 4):S233–241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 7.Le Liboux A, Lefebvre P, Le Roux Y, Truffinet P, Aubeneau M, Kirkesseli S, Montay G. Single- and multiple-dose pharmacokinetics of riluzole in white subjects. J Clin Pharmacol. 1997;37:820–7. doi: 10.1002/j.1552-4604.1997.tb05630.x. [DOI] [PubMed] [Google Scholar]
- 8.Sanderink GJ, Bournique B, Stevens J, Petry M, Martinet M. Involvement of human CYP1A isoenzymes in the metabolism and drug interactions of riluzole in vitro. J Pharmacol Exp Ther. 1997;282:1465–72. [PubMed] [Google Scholar]
- 9.Haddad H, Cifuentes-Diaz C, Miroglio A, Roblot N, Joshi V, Melki J. Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve. 2003;28:432–7. doi: 10.1002/mus.10455. [DOI] [PubMed] [Google Scholar]
- 10.Russman BS, Iannaccone ST, Samaha FJ. A phase 1 trial of riluzole in spinal muscular atrophy. Arch Neurol. 2003;60:1601–3. doi: 10.1001/archneur.60.11.1601. [DOI] [PubMed] [Google Scholar]
- 11.Berard C, Payan C, Fermanian J, Girardot F. [A motor function measurement scale for neuromuscular diseases – description and validation study] Rev Neurol (Paris) 2006;162:485–93. doi: 10.1016/s0035-3787(06)75039-1. [DOI] [PubMed] [Google Scholar]
- 12.Bruno R, Vivier N, Montay G, Le Liboux A, Powe LK, Delumeau JC, Rhodes GR. Population pharmacokinetics of riluzole in patients with amyotrophic lateral sclerosis. Clin Pharmacol Ther. 1997;62:518–26. doi: 10.1016/S0009-9236(97)90047-3. [DOI] [PubMed] [Google Scholar]
- 13.Le Liboux A, Cachia JP, Kirkesseli S, Gautier JY, Guimart C, Montay G, Peeters PA, Groen E, Jonkman JH, Wemer J. A comparison of the pharmacokinetics and tolerability of riluzole after repeat dose administration in healthy elderly and young volunteers. J Clin Pharmacol. 1999;39:480–6. [PubMed] [Google Scholar]
- 14.Groeneveld GJ, Van Kan HJ, Kalmijn S, Veldink JH, Guchelaar HJ, Wokke JH, Van den Berg LH. Riluzole serum concentrations in patients with ALS: associations with side effects and symptoms. Neurology. 2003;61:1141–3. doi: 10.1212/01.wnl.0000090459.76784.49. [DOI] [PubMed] [Google Scholar]
- 15.Groeneveld GJ, van Kan HJ, Lie AHL, Guchelaar HJ, van den Berg LH. An association study of riluzole serum concentration and survival and disease progression in patients with ALS. Clin Pharmacol Ther. 2008;83:718–22. doi: 10.1038/sj.clpt.6100382. [DOI] [PubMed] [Google Scholar]
- 16.Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17:761–7. doi: 10.1089/cap.2007.0021. [DOI] [PubMed] [Google Scholar]
- 17.Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71:115–21. doi: 10.1067/mcp.2002.121829. [DOI] [PubMed] [Google Scholar]
- 18.Milane A, Fernandez C, Vautier S, Bensimon G, Meininger V, Farinotti R. Minocycline and riluzole brain disposition: interactions with p-glycoprotein at the blood-brain barrier. J Neurochem. 2007;103:164–73. doi: 10.1111/j.1471-4159.2007.04772.x. [DOI] [PubMed] [Google Scholar]
- 19.Milane A, Tortolano L, Fernandez C, Bensimon G, Meininger V, Farinotti R. Brain and plasma riluzole pharmacokinetics: effect of minocycline combination. J Pharm Pharm Sci. 2009;12:209–17. doi: 10.18433/j36c78. [DOI] [PubMed] [Google Scholar]