

Abstract
AIMS
To predict the concentration and target inhibition profiles of the survivin inhibitor antisense oligonucleotide LY2181308 in humans.
METHODS
An indirect pharmacokinetic/pharmacodynamic (PK/PD) model was built to predict the inhibition of survivin mRNA and protein in humans following LY2181308 dosing. Plasma and tissue PK data from cynomolgus monkeys were analyzed by non-linear mixed effect modelling techniques. Human PK parameters were predicted using allometric scaling. Assumptions about the pharmacodynamic parameters were made based upon the target and tumour growth inhibition data from mouse xenograft models. This enabled the prediction of the clinical PK/PD profiles.
RESULTS
Following a 750 mg dose, LY2181308 tumour concentrations ranging from 18.8 to 54 µg g−1 were predicted to lead to 50 to 90% target inhibition. In humans, LY2181308 tumour concentrations from 13.9 to 52.8 µg g−1 (n = 4, LY2181308 750 mg) were observed associated with a median survivin mRNA and protein inhibition of 20% ± 34 (SD) (n = 9) and 23% ± 63 (SD) (n = 10), respectively. The human PK parameters were adequately estimated: central Vd, 4.09 l (90% CI, 3.6, 4.95), distribution clearances, 2.54 (2.36, 2.71), 0.0608 (0.033, 0.6) and 1.67 (1.07, 2.00) l h−1, peripheral Vds, 25 900 (19 070, 37 200), 0.936 (0.745, 2.07) and 2.51 (1.01, 2.922) l, mean elimination clearance 23.1 l h−1 (5.6, 33.4) and mean terminal half-life, 32.7 days (range 22–52 days).
CONCLUSION
The model reasonably predicted LY2181308 PK in humans. Overall, the integration of preclinical PK/PD data enabled to appropriately predict dose and dosing regimen of LY2181308 in humans with pharmacologically relevant survivin inhibition achieved at 750 mg.
Keywords: antisense oligonucleotide, dose prediction, model
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The plasma pharmacokinetics (PK) of second generation antisense oligonucleotides (ASOs) are fairly well understood with well conserved pharmacokinetic properties across species, moderate clearance and extensive volume of distribution.
WHAT THIS STUDY ADDS
The tissue distribution and exposure and the pharmacodynamic (PD) or target inhibition following ASO administration are not yet well understood leading to still fairly empiric early clinical development strategies. This paper illustrates how preclinical PK and PD data were used to predict more accurately the ASO tissue exposure in humans and to support the early clinical development strategy.
Introduction
Prior to starting a first-in-human dose (FHD) study in oncology patients, there is a considerable uncertainty on what dose or dose regimen is needed to achieve relevant anti-tumour effects without exposing patients to undue safety risks. One way to reduce this uncertainty consists in building a predictive pharmacokinetic/pharmacodynamic (PK/PD) model by integrating preclinical pharmacokinetic and efficacy data [1]. Such an approach proved useful to predict the efficacious plasma exposure for cytotoxic anti-tumour compounds. Another and complementary approach is based on the integration of PK and PD data in a model which describes the quantitative relationship between drug exposure or concentration and pharmacological effect as measured by biomarkers [2]. Whether these types of PK/PD models can be applied to predict activity of novel anti-cancer therapies such as kinase inhibitors or second generation antisense oligonucleotide (SG-ASO) in humans is not known. LY2181308 is an SG-ASO with a 2′-O-methoxyethyl modified 18-mer structure, and is designed to target specifically the mRNA of survivin, a member of the inhibitor of apoptosis protein (IAP) family [3]. Survivin is expressed in a wide range of human cancers and is generally not expressed in normal adult tissue [4]. Over-expression of survivin protein in cancer tissue is associated with a poorer prognosis [5] and consequently survivin represents an attractive molecular target for therapeutic intervention.
In order to predict the PK and PD (target inhibition) response in humans, following administration of LY2181308, the first step is to understand what PK data might be critical to explain the PD response. The PK of SG-ASOs is well conserved across species and across SG-ASOs of similar length [6]. This information from previous studies indicates that both plasma and tissue PK of SG-ASOs, such as LY2181308, exhibit important differences between the plasma and tissue concentration–time curves. In plasma, the pharmacokinetic profiles of SG-ASOs are characterized by multiple (at least three) decreasing phases. In tissue, following a rise of concentration over the first 24 h after administration, the tissue concentration–time curves of SG-ASOs are usually characterized by a monophasic decreasing phase [6]. The multiphasic plasma PK profile primarily represents the distribution of the SG-ASOs from plasma into tissue whereas the monophasic PK profile in the tissues represents the elimination of SG-ASOs. This elimination is believed to be mediated through degradation by various endo- and exonucleases. The elimination rate of SG-ASOs in tissue ultimately influences tissue concentrations, which subsequently determines the pharmacodynamic effect. This effect occurs within the tissue cells through hybridization to the targeted mRNA.
For this reason, preclinical LY2181308 tissue pharmacokinetic and target inhibition data were gathered to discover the PK/PD relationship for LY2181308. Because of the unique expression pattern of survivin, the PK/PD evaluation in patients measured both LY2181308 concentration and the reduction in survivin mRNA or protein expression in human tumour tissue samples after administration of LY2181308.
We here present a PK/PD model which utilized cynomolgus monkey data (plasma and tissue PK) and mouse xenograft data (tissue PK and tumour PD) to predict the dose and dosing regimen of LY2181308 in humans. The model had three main objectives: to predict the drug concentrations at the tumour site, to predict the range of LY2181308 concentrations needed for efficacious target inhibition and to predict the target inhibition profile. In the subsequent FHD trial, we used the human PK and PD information to continually re-evaluate the PK/PD model and to confirm the original prediction.
Methods
Preclinical data
Preclinical pharmacokinetic data
LY2181308 plasma and tissue pharmacokinetic data in mice and cynomolgus monkey were used.
In cynomolgus monkeys, plasma PK data from a single dose study (20 mg kg−1 intravenously (i.v.) over 3 h infusion, n = 3 animals) and a multiple dose study (45 mg kg−1 i.v. infusion over 72 h as a loading dose (starting day 1) followed by 4 mg kg−1 i.v. infusion over 3 h twice a week for seven doses as a maintenance dose (starting day 12 until day 33), n = 6 animals) were used (Table 1). In addition, various tissues (including liver, kidney, lung, muscle, spleen, bone marrow) from these monkeys were collected for PK assessment at necropsy 24 h and 5 days post last dose in the single and multiple dose studies, respectively (Table 1).
Table 1.
Pharmacokinetic sampling scheme
| Time relative to the start of the last infusion administered | |
|---|---|
| Mice single dose*†‡ | 4, 8 , 24, 48, 72, 96 h |
| Monkey single dose¶ | Predose, 1, 3§, 3.25, 3.5, 4, 4, 6, 10, 27† h |
| Monkey multiple dose | Predose, 24, 48, 72§, 72.25, 72.5, 74, 78, 82, 96, 120 h |
| Loading dose** | |
| Monkey multiple dose** | On day 18 : Predose, 3§, 24 h |
| Maintenance dose | On day 33 : Predose, 3§, 24, 120† h |
PK and PD sampling scheme.
various tissues sampled for PK analysis.
six animals per time point.
end of infusion samples.
three animals
six animals.
In mice, a single dose (5, 20 and 50 mg kg−1 i.v. bolus) study was carried out. PK data in tissues were collected during the single dose study and were available for plasma, liver, kidney, lung and muscle (Table 1).
Preclinical pharmacodynamic data in mice
In addition, target inhibition and anti-tumour efficacy data from the U87MG glioblastoma mouse xenograft model [3] were used to supplement the proposed PD model of LY2181308. In vivo target inhibition data following a single 50 mg kg−1 i.v. dose (sampling time in Table 1) was used. Tumour growth delay data following multiple doses (50 mg kg−1 intraperitoneally (i.p.) as a loading dose followed by 25 mg kg−1 i.p. every other day for 12 doses as a maintenance dosing regimen) were also integrated into the model. Tumour volume was measured every 4 days for the 25-day treatment period and then every 5 days until 11 days after treatment discontinuation. The formula used to calculate tumour volume was as follows:
where L is mid axis length and W is mid axis width [3].
Clinical PK and PD data
The FHD study has been published elsewhere and was a multiple dose escalation trial to determine the safety, pharmacokinetic and pharmacodynamic properties of LY2181308 to support selection of the phase II dose [7–9]. Patients first received LY2181308 as a 3 h infusion on day 1, day 2 and day 3 (loading doses) followed by a weekly maintenance dose starting on day 8. The following doses were investigated: 100 mg (n = 1 patient), 200 mg (n = 1), 400 mg (n = 4), 600 mg (n = 3), 750 mg (n = 26), 900 mg (n = 3) and 1000 mg (n = 2). Plasma samples were collected on day 1 at 3 h (end of infusion), day 3 (pre-dose, 3 (end of infusion), 3.25, 3.5, 4, 6, 7 to 8 and 27 to 60 h post-dose), day 8 (pre-dose), day 15 (pre-dose, 3 (end of infusion), 9 and 24 h post-dose) and day 22 (pre-dose) leading to a total of 16 samples per patient. Tumour biopsies were collected in the 48–96 h time window (study days 5–7) following the end of the loading dose regimen on day 3. Biopsy samples from patients were prioritized for bioanalytical assays: first, the PD effect of LY2181308 (survivin protein and mRNA content), second, LY2181308 tumour concentrations and third, apoptosis pathway proteins.
Bio-analytical methods for LY2181308 measurements and survivin reduction
The human plasma LY2181308 concentrations were assayed using a validated hybridization ELISA method (precision 7–12% and accuracy 87–104% in the clinic). The lower limit of quantitation of this assay was 39.6 ng ml−1. This enabled characterization of LY2181308 plasma PK profiles over the weekly dosing interval. There were not any clinical plasma samples with LY2181308 concentrations below limit of quantitation. For human tissue biopsies, concentrations of LY2181308 were determined by a validated LC/MS/MS method with associated precision/accuracy of 11–14%/92–107%. Concentrations of LY2181308 in mouse and cynomolgus monkey plasma were determined by a validated hybridization ELISA with precision and accuracy in mouse plasma of 3–9% and 96–117%, and in monkeys of 4–15% and 99–106%. LY2181308 tissue concentrations were determined by a validated capillary gel electrophoresis method in mouse and monkey tissues with associated precision/accuracy of 2–14%/78–104% for mice and 2–16%/87–108% for monkeys. Survivin mRNA and protein expression analysis in the tumour biopsies were assessed using a branched DNA [10] and immunochemistry (IHC) [11] assay, respectively [9].
Pharmacokinetic/pharmacodynamic analysis method: (PK/PD) model
In 2004, prior to the initiation of the FHD study, the preclinical plasma and tissue PK data from cynomolgus monkeys were analyzed and the resulting PK model, model A (Figures 1 and 3 and Appendix 1) was scaled to humans using allometric scaling per bodyweight [6] (Table 2). This model enabled us to predict human plasma and tissue LY2181308 concentrations. Secondly, the mouse tissue PK and PD data (target inhibition and efficacy data) from the U87MG xenograft model were used as supplementary information to determine a) the level of target inhibition required to achieve anti-tumour growth effects,b) the concentration of LY2181308 needed to achieve this efficacious target inhibition and c) the turn-over rate or half-life of survivin mRNA and protein. Third, this information in mice was used to infer values of PD parameters in humans which enabled us to link LY2181308 tissue concentration to target inhibition within a PK/PD model, model C (Figures 1 and 2 and Appendix 1). The PK and PK/PD models were used to simulate human LY2181308 tissue concentrations and target inhibition profiles, thus enabling the prediction of an efficacious clinical dose range. Finally, clinical PK and PD data were collected during the FHD study and the predictions were continually re-assessed.
Figure 1.
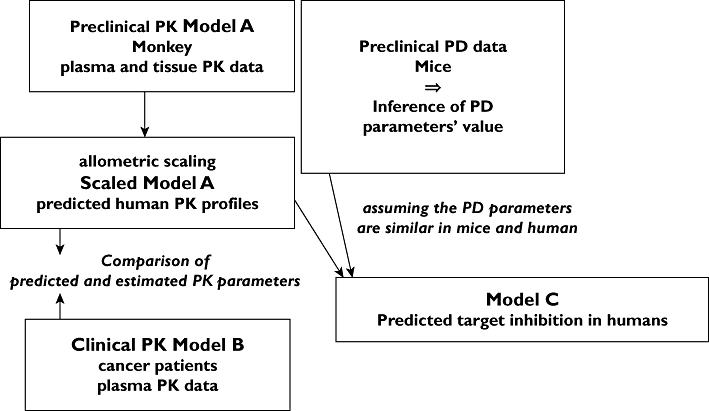
Modelling strategy scheme
Figure 3.
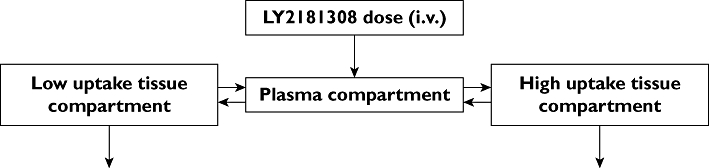
Schematic representation of the preclinical monkey pharmacokinetic model
Table 2.
Allometric scaling equations
| Equation 1* | PK parameter = a* Weightb; (b = 1) |
| Equation 2* | PK parameter in humans = PK parameter in monkey* (Weight human/Weight monkey)b; (b = 1) |
Assuming weight of 5 kg monkey and 75 kg human.
Figure 2.
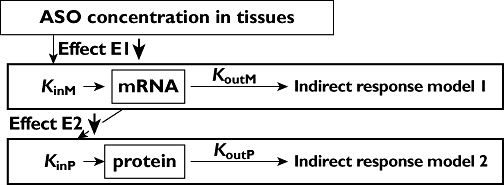
Schematic representation of the pharmacodynamic/pharmacokinetic model. Foot notes: KinM and KoutM: rate constant of synthesis and degradation of survivin mRNA assumed to be equal in the absence of LY2181308. KinP and KoutP: rate constant of synthesis and degradation of survivin protein assumed to be equal in the absence of LY2181308. LY2181308 inhibits the target through an inhibitory Emax model, E1 E2. Please refer to Appendix 1 for more information on the model equations
Pharmacokinetic model
A non-linear mixed effect modelling technique implemented in nonmem version V was used to a) analyze the monkey preclinical PK plasma and tissue data (Figures 1 and 3 and Appendix 1 model A), b) simulate LY2181308 plasma and tissue PK profiles in human (1000 Monte-Carlo simulations) (Figures 1 and 3, Table 2 and Appendix 1 scaled model A) and c) analyze human plasma LY2181308 data (Figures 1 and 6 and Appendix 1 model B, nonmem version VI was used to analyze the log transformed human PK data [12]). The discrepancy in the nonmem version used is explained by the fact that at the time of the preclinical data analysis, the more recent version VI of nonmem was not installed in a validated way at Lilly. nonmem (ICON corporation) version V level 1.1 and VI level 1.2 for Linux were installed on a server running with a Portland Group Fortran compiler. This methodology enables the estimation of mean PK parameter values as well as several levels of variability (or random effect), more precisely inter-individual (at the population level) and intra-individual (at the individual level) variability. Model parameters were estimated using the first order conditional estimation method with interaction [13–15]. The inter-individual and intra-individual (or residual) variability were coded as an exponential and proportional relationship, respectively. The aim of the modelling analysis was to describe the PK data through a multi-compartmental model. Three and/or four compartmental PK models with elimination from the peripheral compartment with a similar elimination rate constant were fitted to the PK data. This approach was justified based on the fact that SG-ASOs distribute extensively into tissues with different rates of uptake depending on the tissue type [6]. The criteria for model selection included goodness of fit plots (posterior predictive check), standard errors on the estimates and the objective function. Posterior predictive checks were performed as follows: the final models (model A, scaled model A, model B, model C) fixed and random parameters were used to simulate 1000 PK or PD profiles using Monte Carlo simulation (Appendix 1). Then statistical calculation was applied to these simulated data to derive the mean median 5th and 95th percentiles PK or PD profiles. Furthermore on the final clinical PK model a bootstrap analysis using Perl-speaks-NONMEM (PsN) software was performed on 500 re-sampled data sets to determine the 90% confidence interval for the parameters estimates.
Figure 6.
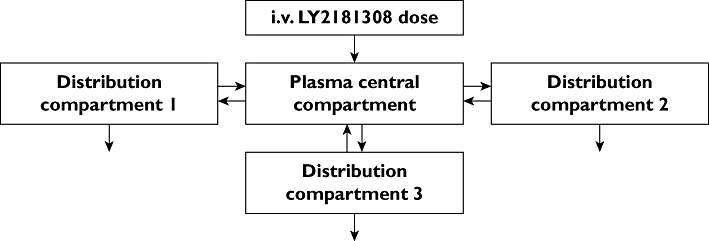
Schematic representation of the clinical LY2181308 pharmacokinetic model
In addition to the modelling analysis, the human LY2181308 plasma concentrations data were also analyzed using a standard non-compartmental method (WinNonlin Enterprise 5.2, Pharsight Corporation 1998–2009).
Pharmacodynamic model
A sequence of two indirect response models, model C, was used to describe the effect of LY2181308 on the targets (survivin mRNA and survivin protein) in human (Figures 1 and 2 and Appendix 1). To predict the outcome of this PD model, Monte-Carlo simulations were carried out (nonmem version V) using the PD parameters presented in Table 3. These PD parameters were derived from the preclinical mice data [3]. More precisely, the in vivo multiple dose efficacy experiment in the mouse xenograft model showed that a 50 mg kg−1 loading dose followed by 25 mg kg−1 every other day for 12 doses (as maintenance doses) led to efficacy with significant tumour growth delay. In addition, a 60 to 70% inhibition of survivin protein within 24 to 48 h post dose was observed after a dose of 50 mg kg−1 (single dose mouse study). These findings indicated that survivin protein inhibition of 60% (or greater) may have anti-tumour efficacy in humans. In the single dose study in mice, the 50 mg kg−1 dose level was also associated with maximum tissue concentrations in muscle and lung (low uptake tissues for SG-ASOs) of 13 and 19 µg g−1, respectively. Consequently, we inferred that average LY2181308 tumour concentrations of about 10 to 20 µg g−1 would be expected to exhibit pharmacologically relevant target inhibition (>50%) with anti-tumour effect. Furthermore, in vivo mouse data also showed that following a single dose of LY2181308 the peak tissue concentration and maximum target inhibition occurred at approximately the same time (about 24 h post dose) indicating an almost direct relationship between the tissue concentration and the effect on target. These data support the assumption of a rapid turn-over rate (i.e short half-lives) for survivin mRNA and survivin protein (see Table 3). This assumption is consistent with literature reports on PK/PD relationship of SG-ASOs [16].
Table 3.
Predicted LY2181308 pharmacodynamic parameters
| Parameters | Value | |
|---|---|---|
| EC50* on survivin mRNA | 20 µg g−1 | |
| KoutM, rate constant of degradation of survivin mRNA† | 1.386 h−1 | tkoutM = 30 min |
| KoutP, rate constant of degradation of survivin protein† | 0.347 h−1 | tkoutP = 2 h |
EC50: concentration leading to 50% inhibition of the target.
tkoutM, survivin mRNA half-life; tkoutP, survivin protein half-life.
Results
Monkey preclinical PK model
To build the predictive PK/PD model for the SG-ASO LY2181308, we used mainly the plasma and tissue PK data from cynomolgus monkeys. We first grouped the tissue PK data based on their degree of uptake, into low and high uptake tissues. The low uptake tissues included, muscle, lung, jejunum, lymphatic nodes, prostate, pancreas, spleen and skin. In these tissues LY2181308 concentrations ranged from 10 to 70 µg g−1 (single dose study). The high uptake tissues included liver and kidney medulla, where LY2181308 concentrations ranged from 100 to 400 µg µg−1 (single dose study). LY2181308 concentration in the kidney cortex was the highest (700 to 800 µg g−1 concentration range). Because a tumour model in non-human primates is extremely difficult to conduct, the low-uptake tissues lung and muscle were used as surrogates to describe possible tumour concentrations of LY2181308. We then combined the tissue and plasma concentrations of LY2181308 in a three-compartmental model (see Figure 3).
The left column of Table 5 presents the PK model parameter values in monkeys. As expected for SG-ASOs, the model predicted a moderate clearance and an extensive volume of distribution. It is important to remember that this model is not a physiological based PK (PBPK model), and hence the volume of distribution will not truly represent the tissue volume. The volumes of distribution indicate the extent of distribution in each compartment and make it possible to scale the amount of drug in each compartment to the corresponding concentrations. This model showed a good fit for both the tissue and plasma preclinical monkey PK data as illustrated by the posterior predictive check plot presented in Figure 4 which does not indicate any major bias of the model. The simulated LY2181308 plasma concentrations in monkey are consistent with the observed preclinical monkey PK data (multiple dose study).
Table 5.
Predicted and observed PK parameters (mean and standard error on the mean (SEE)in %)
| Plasma pharmacokinetics: distribution clearance from central to peripheral compartments central volume of distribution | |||
|---|---|---|---|
| Monkey | Humans | ||
| model A | scaled model A§ | model B | |
| Estimated | Predicted | Estimated | |
| Parameters | Mean (SEE%) | Mean (SEE%) | Mean (SEE%) |
| Central Vd (l) | 0.418 (12) | 6.27 | 4.09 (9) |
| Distribution clearance 1* (l h−1) | 0.167 (6) | 2.51 | 2.54 (4) |
| Distribution clearance 2* (l h−1) | 0.0043 (25) | 0.0648 | 0.0608 (30) |
| Distribution clearance 3* (l h−1) | 1.67 (24) | ||
| Peripheral (tissue) pharmacokinetics: elimination clearance and peripheral volumes of distribution | |||
|---|---|---|---|
| Monkey | Humans | ||
| model A | scaled model A§ | model B | |
| Estimated | Predicted | Estimated | |
| Parameters | Mean (SEE%) | Mean (SEE%) | Mean (SEE%) |
| Peripheral Vd 1† (l) | 1195 (22) | 17925 | 25900 (16) |
| Peripheral Vd 2† (l) | 0.13 (54) | 1.97 | 0.936 (23) |
| Peripheral Vd 3† (l) | 2.51 (12) | ||
| Elimination clearance (l h−1) | 3.45 (15) | 51.8 | 23.1 (48) |
| Terminal half-life‡ (days) | 9 | 9 | 32.7 (22–52) |
Distribution clearance 1, 2 and 3 = LY2181308 distribution from central compartment to first (low uptake tissue) and second/third (high uptake tissue) peripheral compartments, respectively.
Peripheral volumes of distribution.
derived from the model parameters (mean and range).
Assuming weight of 5 kg monkey and 75 kg human.
Figure 4.
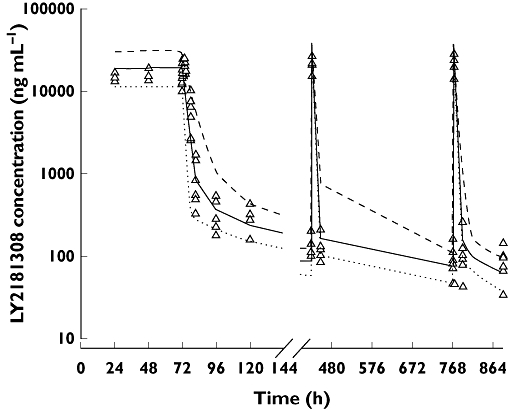
Posterior predictive check plot for the preclinical monkey PK model. Model simulated and observed LY2181308 concentrations following LY2181308 15 mg kg−1 day−1 (i.v. infusion over 72 h) followed by 4 mg kg−1 (i.v. infusion over 3 h) twice weekly from day 8. LY2181308 observation ( ); median model prediction (
); median model prediction ( ); 5th percentile prediction (
); 5th percentile prediction ( ); 95th percentile prediction (
); 95th percentile prediction ( )
)
Furthermore, this preclinical monkey PK model was tested using a separate single dose monkey PK study. In this study we observed, following a 20 mg kg−1 dose over 3 h, a mean LY2181308 plasma Cmax and AUC of 121 000 ng ml−1 and 500 000 ng ml−1 h, respectively. These values are consistent with the model predicted/simulated Cmax and AUC of 131 446 ng ml−1 and 573 371 ng ml−1 h, respectively.
Similarly, the model predictions of tissue concentrations are in agreement with the observed values. In the low uptake tissues, the observed LY2181308 concentrations showed a range of 10 to 70 µg g−1 24 h after a single 20 mg kg−1 dose of LY2181308. This compares favourably with the predicted or simulated range of 49 to 119 µg g−1 (median 70 µg g−1). This illustrate that the model slightly over-predicts the concentration in the low-uptake tissue. Observed LY2181308 concentrations in high uptake tissues (liver and kidney), 5 days after repeated dosing (multiple dose study) ranged from 335 to 688 µg g−1. These values were consistent with the observed value in the single dose study (100 to 400 µg g−1) and with the model predictions of 83–558 µg g−1 (median 220 µg g−1).
First in human study: dosing strategy and prediction in human
Using allometric scaling per bodyweight (assuming 5 kg and 75 kg for monkey and human bodyweight, respectively, and scaling coefficient of 1), the predicted parameters for humans were derived from the monkey PK model (left column of Table 5). This resulted in the predictions (mean value) for the human PK parameters presented in Table 5 (middle column).
One could criticize the choice of an allometric scaling coefficient of 1 for LY2181308 clearance as it is well known that for most small molecules which are renally and/or hepatically cleared the scaling coefficient is of the order of 0.67–0.75. The clearance of small molecules is dependent on the renal and hepatic blood flow, on the intrinsic hepatic clearance and fraction of unbound drug in plasma, whereas the clearance of SG-ASO is primarily dependent on the ability of the ASO to distribute into tissues. Distribution into tissues is the primary step to enable clearance of the SG-ASO by nucleases. Despite significant protein binding in plasma (about 96% in humans for LY2181308), SG-ASO such as LY2181308 extensively distribute into tissues, with evidence (immuno-histochemisry) of intracellular vesicles containing SG-ASOs suggesting that endocytosis may be a mechanism for cellular uptake [9]. After this cellular uptake, the drug is released from the vesicles within the cells and is available for activity and clearance by nucleases. This illustrates the difference between the factors determining the clearance of small molecules and SG-ASOs. Therefore, it is not unexpected that the allometric scaling coefficient for the clearance of SG-ASOs can be different from that for small molecules and shown by Geary et al. [9].
These results of the allometric scaling led to the design of a loading and maintenance dosing strategy paradigm for the FHD based on the predicted tissue half-life: three consecutive 3 h infusions every 24 h followed by a weekly 3 h infusion as maintenance doses. For each dose the amount of LY2181308 administered was the same. The aim was to rapidly achieve sufficient tissue concentrations of LY2181308 to maximally inhibit survivin protein in the tumour and to maintain that level of inhibition throughout the treatment course. Based on this dosing schedule it was anticipated that the steady state tissue concentration would be achieved following the loading dose (Table 4).
Table 4.
Predicted human LY2181308 pharmacokinetic parameters using the preclinical model
| Predictions (mean 5th–95th percentile) | |||
|---|---|---|---|
| Dose (mg) | Cmax plasma (ng ml−1) | AUC(0,24 h) plasma (ng ml−1 h) | Caverage at steady state in low uptake tissues (µg g−1) |
| 100 | 8 732 | 37 830 | 4.4 |
| 5 651–12 920 | 17 856–77 605 | 2.5–7.2 | |
| 200 | 17 464 | 75 660 | 8.8 |
| 11 302–25 839 | 35 713–155 211 | 5.0–14.4 | |
| 400 | 34 927 | 151 320 | 17.7 |
| 22 604–51 679 | 17 425–310 422 | 10.0–28.8 | |
| 600 | 52 391 | 226 980 | 26.5 |
| 33 907–77 518 | 107 137–465 633 | 15.0–43.2 | |
| 750 | 65 489 | 283 725 | 33.2 |
| 42 838–96 897 | 133 922–582 041 | 18.8–54.0 | |
Predictions from the model (Table 4) indicated that, for LY2181308 doses greater or equal to 400 mg, LY2181308 average concentrations in low uptake tissue should exceed, in some patients, the predicted efficacious concentration of 20 µg g−1 (EC50) determined from the mouse data [3]. Hence survivin protein inhibition should be observed for LY2181308 doses greater than 400 mg. In order to increase the probability of achieving the relevant tissue concentration in the majority of the patient population, the model indicated that a dose of 750 mg might be required. The GLP 1 month toxicology study supported a starting dose of 100 mg in humans. This study together with the single dose monkey study also informed on LY2181308 potential off-target Cmax related toxicities that could be observed in humans. The primary toxicities identified included transient peak related aPTT prolongation and complement activation related to LY2181308 Cmax concentrations greater than 100 000 ng ml−1. The model predicted that, at a clinical dose of 750 mg (3 h infusion), LY2181308 maximum plasma concentrations should remain below 100 000 ng ml−1.
Using the pharmacodynamic parameters (Table 3) derived from the mouse PK/PD data [3], survivin protein inhibition profiles were simulated (Figure 5). The model simulations indicated that significant inhibition of the target should be achieved in patients administered 750 mg of LY2181308.
Figure 5.
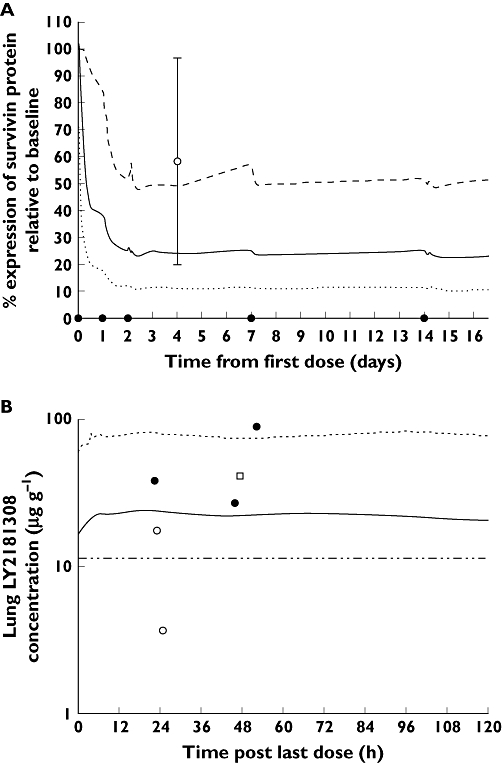
Following LY2181308 750 mg, survivin protein (expressed as % of baseline) in tumour biopsies of patients – model simulated profile compared with observed data. The dot on the time axis indicates LY2181308 dose (A). Following 750 mg LY2181308, steady state predicted LY2181308 concentrations (µg g−1) (in lung, a low uptake tissue) compared with observed clinical tumour biopsies in humans (B). (A) median simulated profile (PKPD model) ( ); 5th percentile simulated profile (PKPD model) (
); 5th percentile simulated profile (PKPD model) ( ); 95th percentile simulated profile (PKPD model) (
); 95th percentile simulated profile (PKPD model) ( ); observed median +/− standard deviation (n = 8) (
); observed median +/− standard deviation (n = 8) ( ); (B) 400 mg LY2181308 · Observation (
); (B) 400 mg LY2181308 · Observation ( ); 600 mg LY2181308 · Observation (
); 600 mg LY2181308 · Observation ( ); 750 mg LY2181308 · Observation (
); 750 mg LY2181308 · Observation ( ); median (
); median ( ); 95th percentile (
); 95th percentile ( ); preclinical EC25 for survivin mRNA inhibition predicted to lead to 50 % survivin protein inhibition (
); preclinical EC25 for survivin mRNA inhibition predicted to lead to 50 % survivin protein inhibition ( )
)
First in human clinical PK and PD results
Comparison of the predicted plasma exposure (using the preclinical model, Table 4) and observed clinical plasma exposure (Table 6) show that the predictions were accurate. The clinical tumour LY2181308 concentration data (n = 4 patients) and the survivin protein inhibition data (n = 16 patients) were too limited to build a clinical plasma–tissue PK model and a clinical PK/PD model. The modelling analysis of the clinical data was therefore aimed at the plasma LY2181308 concentration data only.
Table 6.
Observed LY2181308 plasma pharmacokinetic data in cancer patients
| Observations (mean (CV %)) Day 3 | |||
|---|---|---|---|
| Dose(mg) | n* | Cmax (ng ml−1) | AUC (0,24 h)(ng ml−1 h) |
| 100 | 1 | 7 060 | 27 161 |
| 200 | 1 | 12 815 | 69 733 |
| 400 | 4 | 31 370 (23.6) | 152 637 (21.1) |
| 600 | 3 | 45 654 (13.1) | 211 467 (23.9) |
| 750 | 24 | 69 120 (34.2) 39 923–155 514† | 342 794 (31.5) 187 344–603 944† |
| 900 | 1§ | 86 787 | 251 031 |
| 1 000 | 1‡ | 84 037 | 425 279 |
Number of patients.
Range.
Following a 4 h infusion, instead of 3 h, in order to prevent peak related toxicity.
On day 3, this patient was dosed over 1.17 h instead of the 4 h prescribed for the higher doses.
In contrast to the preclinical three-compartment PK model, a four-compartment PK model (Figure 6) was found to be the best fit for the clinical LY2181308 plasma concentration vs.time data. The addition of the fourth compartment improved the statistical criteria (the objective function dropped by 46 points) relative to a three compartmental model.
The LY2181308 clinical PK parameters for this four-compartment model are presented in Table 5 right column. The inter-individual variability, reported as coefficient of variation CV% (standard error on the estimate SEE%), was estimated for a) the central volume of distribution, 18% (134%), b) the plasma distribution clearance to the first peripheral compartment, 15.4% (44.2%) and c) the first and third peripheral volume of distributions 38.6% (31.6%) and 45.2% (51.9%), respectively. The residual variability was 28.8% (32.9% SEE).
The central volume of distribution, 4.09 l, is consistent with plasma volume. The lower volume of distribution 0.936 l into which the LY2181308 is estimated to be less readily distributed (distribution clearance of 0.0608 l h−1) may represent distribution into the red blood cells into which there is evidence that LY2181308 does not readily distribute [7]. The estimated two other volumes of distribution 25 900 and 2.5 l are consistent with predicted volumes of distribution in low (17 925 l) and high (1.97 l) uptake tissues, respectively (Table 5).
The posterior predictive check for the LY2181308 plasma clinical PK model are presented in Figure 7 and show that the model simulated median, 5th and 95th percentiles are consistent with the observed median, 5th and 95th percentiles profiles. This indicates that the four-compartment PK model described the clinical plasma PK data adequately and without bias.
Figure 7.
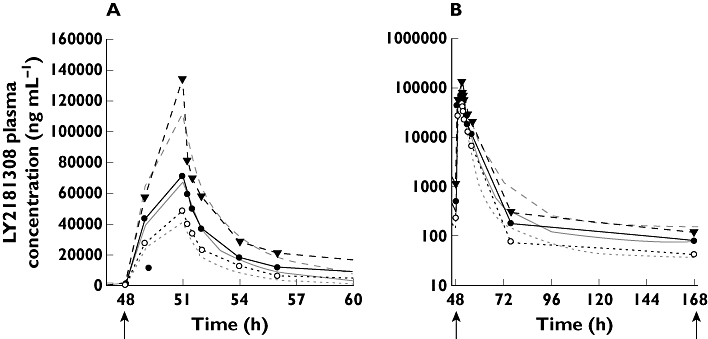
Posterior predictive check plot for the clinical PK model. Model simulated and observed LY2181308 concentration vs. time data following administration of LY2181308 750 mg i.v. infusion over 3 h in cancer patients (day 3 profile displayed). The arrows indicate start of LY2181308 infusion with linear plot (A) and semi-logarithmic plot (B). median simulate profile ( ); 5th percentile simulated profile (
); 5th percentile simulated profile ( ); 95th simulated profile (
); 95th simulated profile ( ); observed mean profile (
); observed mean profile ( ); observed 5th percentile profile (
); observed 5th percentile profile ( ); observed 95th percentile profile (
); observed 95th percentile profile ( )
)
The above mentioned clinical PK model parameters are in good agreement with the predicted human PK parameters based upon the monkey PK model (Table 5). Overall, the difference between the predicted clinical PK parameter values and the observed value remained reasonable (median 20%, ranging from 1.4 to 55%). The least well predicted parameter was the terminal half-life (32.7 days vs. 9 days).
Only a small percentage (less than 10%) of the plasma AUC was under this terminal half-life. Consequently the under-prediction of the half-life did not adversely affect the predictions of LY2181308 plasma AUC and Cmax which were adequately predicted (Table 4 compared with Table 6).
The terminal half-life was estimated from the model. Based on the standard error of the model estimated parameters and the posterior predictive check plot, the model was judged to be reliable and so was the estimated terminal half-life. It is important to note that given the weekly dosing interval, relative to the anticipated long terminal half-life, it was not possible to define an optimal sampling scheme to estimate reliably the terminal half-life using classical non-compartmental analysis. In order to do this, patients would have been required to come regularly after treatment discontinuation for PK sampling which is not easily ethically justified.
In tumour biopsies, LY2181308 concentrations (measured by ELISA) ranged from 3.64 to 87.4 µg g−1 (median 22.4 µg g−1; n = 5) following 750 mg [8, 9]. Furthermore, analysis of LY2181308 bio-distribution in tissues using PET technology in three patients indicated that LY2181308 does distribute into the tumour at concentrations ranging from 13.9–52.8 µg g−1 (median 32.5 µg g−1; n = 4) [7]. This was consistent with the predictions of 18.8 to 54 µg g−1 (5th–95th percentiles), using the preclinical monkey model (see Table 4 and Figure 5).
The patient population enrolled in this phase I study for LY2181308 was 40 patients which is still a modest size to identify reliably PK vs. covariate relationships. No clear relationship could be detected with bodyweight or BSA (Figure 8). The data may indicate a trend that patients with lower creatinine clearance may have higher exposure in tissue (using LY2181308 trough or pre-dose concentrations as the best correlated plasma PK parameter to the tissue exposure) (Figure 8). It is important to bear in mind that this trend is only based on data from four patients with the lowest creatinine clearance in the population enrolled in that study (<60 ml min−1). The case report [17] about a patient in this FHD study who has been treated for more than 1 year with LY2181308 indicated that the kidney likely plays an important role in the clearance of LY2181308 which would be consistent with a correlation between PK and creatinine clearance. More data are needed to determine further the relationships of PK vs. covariates.
Figure 8.
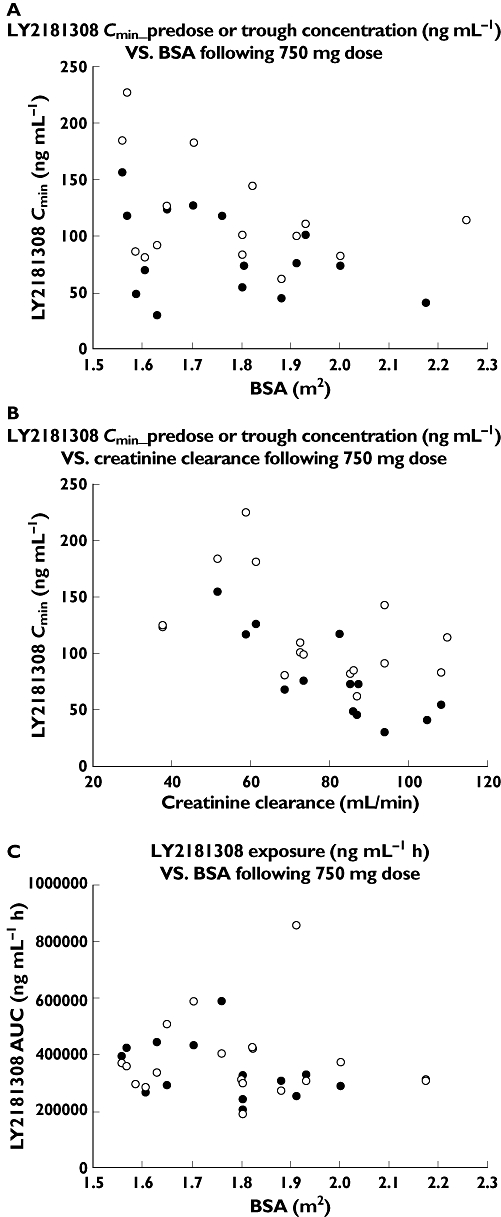
PK vs. covariate exploratory graphs. A) LY2181308 Cmin pre-dose or trough concentration vs. BSA following a 750 mg dose, B) LY2181308 Cmin pre-dose or trough concentration vs. creatinine clearance following a 750 mg dose and C) LY2181308 exposure vs. BSA following a 750 mg dose. (A) Day 15 ( ); Day 22(
); Day 22( ); (B) Day 15 (
); (B) Day 15 ( ); Day 22 (
); Day 22 ( ); (C) Day 3 (
); (C) Day 3 ( ); Day 15 (
); Day 15 ( )
)
With respect to target inhibition, at the LY2181308 750 mg dose level, we observed median inhibition of survivin mRNA and protein of 20% ± 34 (SD) (n = 9) and 23% ± 63 (SD) (n = 10), respectively. These median values of target inhibition were calculated using all available pre and post treatment biopsy data at 750 mg [9]. For some of these patients at 750 mg (n = 2 out of 9), little expression of survivin (gene or protein) in pre-treatment biopsies was observed. Hence for these two patients it was not unexpected to detect little or no reduction of survivin mRNA or protein expression following treatment with LY2181308 since the baseline pre-treatment expression levels were already low. When the analysis was rerun excluding those two patients, the median survivin mRNA and protein inhibition were 34% ± 20 (SD) (n = 7) and 41.1% ± 38.4 (SD) (n = 8), respectively, for 80% of the patients with evaluable data at the 750 mg dose. In comparison with these observations, the model predicted median survivin protein inhibition of 76% (51 to 89% 5th 95th percentile), was higher than the observed median level of inhibition as illustrated in Figure 5.
Discussion
We have developed a PK/PD model for the SG-ASO LY2181308 to predict the dose and dose regimen for the FHD study in cancer patients. Several assumptions were made when scaling the preclinical PK and PD data of LY2181308 to humans. Based on the preclinical monkey PK, human PK would have been adequately predicted based on allometric scaling per bodyweight with a coefficient of 1. This assumption was reasonable according to other reports on SG-ASO pharmacokinetics [6] and it was later confirmed in the FHD study.
However, predicting LY2181308 concentration in tumours posed a greater challenge. We used the low-uptake tissues, such as lung and muscle, as surrogates for predicting LY2181308 tumour concentration. The selection of low uptake tissues was based on the assumption that LY2181308 would not easily distribute into tumour tissue. The subsequent confirmatory study in patients suggested that this choice was reasonable as confirmed by tumour concentrations obtained by both the biodistribution study with carbon-11 radiolabled LY2181308 [7] and assessment of tumour biopsies using ELISA [8, 9].
In contrast to the PK parameters, there was a clear over-estimation of the PD effect. To assume that murine xenograft models accurately predict anti-tumour effect in patients is not without controversy [18, 19]. In addition, the xenograft data were not extensive enough to allow the inclusion of additional parameters, which were used for cytotoxic agents [1]. This would have required tumour growth delay experiments at different doses and dosing regimens. Also, the assumption that the LY2181308 concentration range leading to target inhibition and anti-tumour effects in mice should also lead to target inhibition and efficacy in humans is perhaps not valid. The LY2181308 concentrations and percentage of survivin inhibition required for anti-tumour efficacy in preclinical models may be different from those needed in humans.
Based on this PK and PD information, a multi-compartment PK/PD model was developed, which adequately described the multi-phasic PK profile of LY2181308 and over-predicted the associated PD effect. The human plasma exposure (AUC and Cmax) were accurately predicted (Table 4 compared with Table 6). However, the half-life (t1/2) of the terminal phase was under-predicted by the preclinical model (Table 5). This predicted mean value for t1/2 was 9 days whereas the observed mean value was 32.7 days (range 22 to 52 days) in humans. This is likely explained by the lack of preclinical data both during the initial rapid decreasing phase (after the end of infusion) and during the long terminal phase. It is relevant to note that the preclinical LY2181308 PK data were collected up to 5 days post-dose whereas the clinical PK data were collected over a longer period of time (over 22 days of treatment and up to 7 days post-dose). This sampling scheme enabled us to determine an additional fourth compartment in the clinical plasma pharmacokinetic profile (compared with the three compartments for the preclinical model). This fourth compartment corresponds to a small volume of distribution (2.51 l) and rapid equilibrium rate constants of 0.4 h-1 (K14) and 0.7 h-1 (K41). It enabled us to better describe the entire LY2181308 plasma vs. time curve particularly the initial rapid distribution phase and the terminal phase. Consistent with the under prediction of the terminal half-live (which is proportional to the ratio V : CL), LY2181308 tissue elimination clearance was over-predicted (predicted 51.8 l h−1vs. observed 23.1 l h−1) and LY2181308 tissue distribution volume was under-predicted (predicted 17925 l vs. observed 25900 l) by the preclinical model. Overall, the difference between the predicted PK parameter values and the observed values (Table 5), remained reasonable (median 20%, ranging from 1.4 to 55%). The terminal elimination phase accounted for only 9% of the overall plasma exposure, indicative of extensive distribution of the drug from plasma into tissue. This explains why LY2181308 plasma AUC and Cmax were still appropriately predicted (Table 6 compared with Table 4) despite the under-prediction of the terminal half-life (predicted 9 days vs. observed 32 days). Because of this long half-life, a loading and maintenance dosing paradigm was proposed to achieve quickly and maintain relevant pharmacological concentrations of SG-ASOs in the target tissue. The dosing interval for the maintenance regimen was based on the predicted half-life in humans, the proposed dose to achieve target inhibition and the appropriate margin of safety established by preclinical toxicology studies [20]. These three factors describe the therapeutic window in which novel therapeutic agents, such as LY2181308, can be safely evaluated in patients.
Because of these PK/PD assumptions and observations, the dosing strategy for LY2181308 is flat-fixed dosing. Historically, doses of anti-cancer drugs were adjusted per BSA according to the belief that patients with a lower BSA would have lower volume of distribution and lower clearance of the drug compared to patients with a higher BSA. This dose adjustment was made to decrease the variability in the drug exposure and aimed ultimately at preventing either toxicity (in patients with a low BSA) or sub-efficacious exposure (in patient with a high BSA). However, retrospective analysis [21] has shown that dose adjustment per BSA is not always adequate and does not necessarily lead to decreased variability in exposure. The patient population enrolled in this phase I study for LY2181308 was 40 patients which is still a modest size to identify reliably PK vs. covariate relationships. No clear correlation could be detected with bodyweight or BSA. The safety data, the variability in LY2181308 pharmacokinetics and the PK/PD data gathered from this study support the on-going investigation of LY2181308 with flat-fixed dosing in phase II studies.
The PK/PD model predicted that doses greater than 400 mg of LY2181308 could significantly inhibit the target sites in patients. The observed clinical data indicated that target inhibition was indeed observed in patients after receiving the 750 mg dose as a loading and maintenance regime despite the semiquantitative nature of the IHC measurements and the variability in the biopsy samples.
Although the observed level of survivin inhibition is lower than expected, it does translate into meaningful pharmacological changes in the tumour samples, including restoration of the apoptosis pathway as presented by Talbot et al. [9] and Saleem et al. [7].
In conclusion, the integration of LY2181308 PK and PD preclinical data helped design the first clinical trial of LY2181308. In the case of targeted therapies, such as SG-ASOs, toxicity is not correlated with the degree of target inhibition. Therefore, it is critically important to determine the dose and dose schedule not solely on the safety/toxicity profile but also on pharmacological activity. Lack of integration of the pharmacological activity data within the overall data package (toxicology and PK) during early clinical development may lead to the selection of an unsafe and ineffective dose for the drug candidate. During the preclinical development of LY2181308 we gathered data on both survivin gene and protein inhibition. This enabled us to determine the relevant LY2181308 dose ranges (100 to 1000 mg) for clinical investigation. The data from the FHD study confirmed that LY2181308 at the 750 mg dose was safe and induced significant target inhibition and pharmacological activity [7–9]. The dosing regimen consisting of a 3 h infusion administered on three consecutive days (on day 1, day 2 and day 3) as a loading dose followed by a weekly maintenance dose starting on day 8, led to relevant concentrations in tumours and significant target inhibition. This dosing regimen is currently being applied in clinical trials with other SG-ASOs [22, 23].
Appendix 1
PK/PD models differential equations
Model A: Preclinical monkey LY2181308 plasma PK model built using monkey PK data
Model A estimated parameters were scaled to human using allometric (refer to Table 2 for allometric scaling equation)
DADT(C)=K1c×A(D1)+K2c×A(D2)−(Kc1+Kc2)×A(C); Change over time of LY2181308 drug amount A in central plasma compartment C.
DADT(D1) = Kc1 × A(C) − (K1c + K10) × A(D1); Change over time of LY2181308 drug amount A in distribution compartment D1 low uptake tissues.
DADT(D2) = Kc2 × A(C) − (K2c + K20) × A(D2); Change over time of LY2181308 drug amount A in distribution compartment D2 high uptake tissues.
Model B: Clinical LY2181308 plasma PK model built using human PK data
DADT(C) = K1c × A(D1) + K2c × A(D2) + K3c × A(D3) − (Kc1 + Kc2 + Kc3) × A(C); Change over time of LY2181308 drug amount A in central plasma compartment C.
DADT(D1) = Kc1 × A(C) − (K1c + K10) × A(D1); Change over time of LY2181308 drug amount A in distribution compartment D1 surrogate for low uptake tissues.
DADT(D2) = Kc2 × A(C) − (K2c + K20) × A(D2); Change over time of LY2181308 drug amount A in distribution compartment D2 high uptake tissues.
DADT(D3) = Kc3 × A(C) − (K3c + K30) × A(D3); Change over time of LY2181308 drug amount A in distribution compartment D3 high uptake tissues.
Relationship between model parameters
Distribution clearance 1 = Kc1 × central Vd = K1c × Peripheral Vd 1
Distribution clearance 2 = Kc2 × central Vd = K2c × Peripheral Vd 2
Distribution clearance 3 = Kc3 × central Vd = K3c × Peripheral Vd 3
K10 = Elimination clearance/Peripheral Vd 1; K20 = K30 = K10
Model C: PK/PD model.
PD parameters
Emax = 1
GAM = 1.92
GAM1 = 1
EC50 = 20
KinM = 1.386
KoutM = KinM
B50 = 0.248
KoutP = 0.347
KinP = KoutP
Effect of drug concentration on rate of formation of survivin mRNA:
 |
Change over time of survivin mRNA amount:
Effect of survivin mRNA on the survivin protein expression:
Change over time of survivin protein amount:
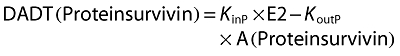 |
Competing interests
All authors were employees of Eli Lilly and Company which sponsored the study at the time the work was performed. All authors except M B are still employed by Eli Lilly and Company at the time of submission of this paper.
The authors would like to express their gratitude to the clinical investigators, Drs Talbot, Ranson and de Bono, to the sites nurses and clinical study managers and to the patients who participated in this first in man clinical trial. The authors thank the Charles Rivers Laboratories in Montreal for the animal experiments in monkeys.
REFERENCES
- 1.Simeoni M, Magni P, Cammia C, De Nicolao G, Croci V, Pesenti E, Germani M, Poggesi I, Maurizio Rocchetti M. Predictive pharmacokinetic-pharmacodynamic modeling of tumour growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004;64:1094–101. doi: 10.1158/0008-5472.can-03-2524. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, Sinha V, Forgue ST, Callies S, Ni L, Peck R, Allerheiligen SRB. Model-based drug development: the road to quantitative pharmacology. J Pharmacokinet Pharmacodyn. 2006;33:369–93. doi: 10.1007/s10928-006-9010-8. [DOI] [PubMed] [Google Scholar]
- 3.Patel BKR, Carrasco RA, Stamm NB, Marcusson E, Sandusky G, Iversen P. Antisense inhibition of survivin expression as a cancer therapeutic. Submitted Cancer Research. [DOI] [PubMed]
- 4.Li F, Brattain MG. Role of the survivin gene in pathophysiology. Am J Pathol. 2006;169:1–11. doi: 10.2353/ajpath.2006.060121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 6.Geary RS, Yu RZ, Watanabe T, Henry SP, Hardee GE, Chappell A, Matson J, Sasmor H, Cummins L, Levin AA. Pharmacokinetics of a tumour necrosis factor-alpha phosphorothioate 2′-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug Metab Dispos. 2003;31:1419–28. doi: 10.1124/dmd.31.11.1419. [DOI] [PubMed] [Google Scholar]
- 7.Saleem A, Ranson M, Callies S, Lahn M, Prenant C, Brown G, Matthews JC, Dence CS, McMahon A, Price P. Microdosing imaging pharmacokinetic (PK) study of the antisense oligonucleotide (ASO) to survivin (LY2181308) using positron emission tomography (PET): a novel paradigm in clinical drug development. J Clin Oncol. 2009;27:15S. [Google Scholar]
- 8.Talbot DC, Davies J, Callies S, Andre V, Lahn M, Ang J, De Bono JS, Ranson M. First human dose study evaluating safety and pharmacokinetics of LY2181308, an antisense oligonucleotide designed to inhibit survivin. J Clin Oncol. 2008;26:20S. [Google Scholar]
- 9.Talbot DC, Davies J, Olsen A, Andre V, Lahn M, Powell E, Kadam S, de Bono J, McHugh P, Ranson M. Pharmacodynamic (PD) evaluation of LY2181308 in patients with metastatic malignancies. J Clin Oncol. 2009;27:15S. [Google Scholar]
- 10.Knudsen BS. Evaluation of the branched-chain DNA assay for measurement of RNA in formalin-fixed tissues. J Mol Diagn. 2008;10:169–76. doi: 10.2353/jmoldx.2008.070127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacus S, Flowers JL, Press MF, Bacus JW, McCarty KS., Jr The evaluation of estrogen receptors in primary breast carcinoma by computer-assisted image analysis. Am J Clin Pathol. 1988;90:233–9. doi: 10.1093/ajcp/90.3.233. [DOI] [PubMed] [Google Scholar]
- 12.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM Users Guide. NONMEM Project Group. San Francisco CA: University of California; 1994. [Google Scholar]
- 13.Sheiner LB. Population pharmacokinetics: the population approach to pharmacokinetic data analysis: rational and standard data analysis methods. Drug Metab Rev. 1984;15:153–71. doi: 10.3109/03602538409015063. [DOI] [PubMed] [Google Scholar]
- 14.Sun H, Fadiran EO, Jones CD, Lesko L. Population pharmacokinetics – a regulatory perspective. Clin Pharmacokinet. 1999;37:41–51. doi: 10.2165/00003088-199937010-00003. [DOI] [PubMed] [Google Scholar]
- 15.Wade JR, Beal SL, Sambol NC. Interaction between structural, statistical and covariate models in population pharmacokinetic analysis. J Pharmacokinet Biopharm. 1994;22:165–77. doi: 10.1007/BF02353542. [DOI] [PubMed] [Google Scholar]
- 16.Yu RZ, Geary RS, Leeds JM, Watanabe T. Comparison of pharmacokinetics and tissue disposition of an antisense phosphorothioate oligonucleotide targeting human Ha-ras mRNA in mouse and monkey. J Pharm Sci. 2001;90:182–93. doi: 10.1002/1520-6017(200102)90:2<182::aid-jps9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 17.Herrington WG, Talbot D, Lahn MM, Brandt JT, Callies S, Nagle R, Winearls CG, Roberts ISD. Long-term administration of the survivin mRNA-targeted antisense oligonucleotide LY2181308 causing renal injury. Am J Kidney Dis. 2010 doi: 10.1053/j.ajkd.2010.09.024. in press. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–31. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamp A. What's wrong with our cancer models? Nat Rev Drug Discov. 2005;4:161–5. doi: 10.1038/nrd1635. [DOI] [PubMed] [Google Scholar]
- 20.Jason TL, Koropatnick J, Berg RW. Toxicology of antisense therapeutics. Toxicol Appl Pharmacol. 2004;201:66–83. doi: 10.1016/j.taap.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Mathijssen Ron HJ, De Jong FA, Loos WJ, Van Der Bol JM, Verweij J, Sparreboom A. Flat-fixed dosing versus body surface area based dosing of anticancer drugs in adults: does it make a difference? Oncologist. 2007;12:913–23. doi: 10.1634/theoncologist.12-8-913. [DOI] [PubMed] [Google Scholar]
- 22.Chi KN, Eisenhauer E, Fazli L, Jones EC, Goldenberg SL, Powers J, Tu D, Gleave ME. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J Natl Cancer Inst. 2005;97:1287–96. doi: 10.1093/jnci/dji252. [DOI] [PubMed] [Google Scholar]
- 23.Dean E, Jodrell D, Connolly K, Danson S, Jolivet J, Durkin J, Morris S, Jowle D, Ward T, Cummings J, Dickinson G, Aarons L, Lacasse E, Robson L, Dive C, Ranson M. Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion in patients with advanced refractory cancer. J Clin Oncol. 2009;27:1660–6. doi: 10.1200/JCO.2008.19.5677. [DOI] [PubMed] [Google Scholar]