

Abstract
AIMS
Sitagliptin is a selective inhibitor of dipeptidyl peptidase-4 (DPP-4) used to treat type 2 diabetes. The present aim was to evaluate pharmacokinetic (PK), pharmacodynamic (PD) and safety characteristics of sitagliptin following single doses in healthy, young Japanese males.
METHODS
In this alternating two-panel, randomized, controlled double-blind study, six healthy Japanese male subjects (aged 20–46 years) in each panel received single oral doses of 5–400 mg sitagliptin and two received placebo. Plasma and urine drug concentrations were measured from 0–48 h post dose and plasma DPP-4 inhibition from 0–24 h post dose. The results were compared with historical data from young, healthy non-Japanese males.
RESULTS
Plasma concentrations of sitagliptin increased approximately in proportion to dose; maximum concentrations occurred 2–6 h post-dose. The mean apparent terminal half-life for plasma sitagliptin was 9–14 h, with the half-life slightly decreasing as the dose increased. The mean dose fraction excreted unchanged in the urine was 0.73–1.00. Ingestion of a traditional Japanese breakfast prior to dosing had only a minor effect on PK parameters. After correction for dilution and competition effects during assay, doses of sitagliptin ≥50 mg resulted in weighted average DPP-4 inhibition from 0–24 h post-dose >94% (without correction, >78%). No clinically meaningful differences in PK and DPP-4 inhibition parameters were found between Japanese and non-Japanese subjects. Sitagliptin was generally well tolerated and there were no serious adverse experiences or episodes of hypoglycaemia.
CONCLUSIONS
The PK and PD findings from this study are consistent with once daily dosing of sitagliptin in Japanese patients with type 2 diabetes.
Keywords: DPP-4 inhibitor, MK-0431, race
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Sitagliptin is an oral antihyperglycaemic agent that improves glycaemic control by inhibiting dipeptidyl peptidase-4 (DPP-4), the enzyme that is principally responsible for inactivation of incretins. Incretins are endogenous peptide hormones that support glycaemic homeostasis through glucose-dependent stimulation of insulin secretion by pancreatic β-cells and suppression of glucagon secretion by α-cells.
Pharmacokinetic properties of sitagliptin and inhibition of plasma DDP-4 by sitagliptin have been characterized previously in young, normoglycaemic, non-Japanese adult males and in other non-Japanese subjects. It was found in these studies that doses of at least 100 mg once daily produced nearly complete inhibition of DPP-4 over 24 h.
WHAT THIS STUDY ADDS
The findings from this study suggest that the pharmacokinetic and pharmacodynamic (i.e. plasma DPP-4 inhibition) properties of sitagliptin in Japanese subjects are not substantially different from responses in non-Japanese subjects.
Consumption of a standardized traditional Japanese breakfast prior to dose ingestion did not alter plasma sitagliptin concentrations to a clinically meaningful extent.
Oral administration of sitagliptin at approved doses provided nearly complete inhibition of DPP-4 over an interval of 24 h. The findings support once daily dosing with sitagliptin in Japanese patients with type 2 diabetes.
Introduction
Sitagliptin is an oral antihyperglycaemic agent that acts via selective inhibition of dipeptidyl peptidase-4 (DPP-4), the enzyme principally responsible for inactivation of the endogenous incretin hormones glucagon-like-1 peptide (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) [1]. By prolonging the activity of GLP-1 and GIP, sitagliptin augments two key incretion functions: glucose-dependent stimulation of insulin secretion by pancreatic β-cells and suppression of glucagon release from pancreatic α-cells [2, 3]. In large, randomized, placebo-controlled clinical trials, sitagliptin has been found to improve glycaemic control, measures of β-cell function and to be generally well tolerated in patients with type 2 diabetes, when given as monotherapy and when administered in combination with other oral antihyperglycaemic agents [4, 5].
Pharmacokinetic (PK), pharmacodynamic (PD) and safety characteristics of sitagliptin were initially characterized in non-Asian subjects who resided in Europe and the United States [6–9]. These studies found that orally administered sitagliptin was well absorbed and approximately 80% was excreted unchanged in the urine. The pharmacokinetic behaviour of sitagliptin was not altered when doses were repeated daily for 10 days and plasma concentrations varied approximately in proportion to dose over a wide range of doses (1.5–600 mg). Except in patients with renal insufficiency, the apparent terminal half-life (t1/2) of sitagliptin was in the range of 8–14 h. In patients with renal insufficiency, it was found that sitagliptin clearance varied approximately in proportion with creatinine clearance [10].
Urinary excretion of sitagliptin is actively mediated by human organic anion transporter (hOAT3), organic anion transporting polypeptide (OATP4C1) and multidrug resistance P-glycoprotein [11]. Many drug transporter genetic polymorphisms have been described and specific variants have been shown to differ in frequency across different ethnic populations [12, 13]. Hypothetically, these differences could contribute to inter-ethnic variability in the pharmacokinetic properties of sitagliptin. The present study was conducted in preparation for the use of sitagliptin to treat type 2 diabetes in Japan (where it is now approved) and in other Asian patient populations. The specific purpose of this study was to evaluate PK, PD and safety characteristics of sitagliptin in young, healthy male Japanese subjects and to compare these with historical data from a prior study conducted in Belgium in young, healthy non-Japanese males [6].
Methods
Study participants
Eligible subjects were healthy, young (18–45 years), normoglycaemic males. For the purposes of this study conducted in Hawaii, a subject was considered to be Japanese if all four of his biological grandparents were of Japanese descent and had been born in Japan. In the European study conducted previously, the race of all subjects was reported as ‘White’. The eligibility of subjects was confirmed based upon medical history, physical examination, vital signs, 12-lead electrocardiograms (ECGs) and laboratory tests (haematology, blood chemistry, and urinalysis). Subjects were required to be within 15% of ideal bodyweight based on the Metropolitan Life Height and Weight Table. Criteria for exclusion included pre-existing hepatic, cardiovascular or neurological disease, diabetes, renal disease or renal inadequacy (creatinine clearance ≤80 ml min−1), the habit of smoking ≥10 cigarettes/day, and the use or anticipated use of prescription or non-prescription drugs within 2 weeks prior to or during the study.
This study was performed at a single clinical research centre in Honolulu, HI, USA (Radiant Research). All procedures conformed to the guidelines for good clinical practice and ethical standards for human experimentation established by the Declaration of Helsinki. The study protocol was approved by an independent institutional review board (Quorum Review, Inc., Seattle, WA, USA), and all subjects provided written informed consent prior to participation. Subjects agreed to refrain from consumption of grapefruit and grapefruit juice from 2 weeks prior to the study until its completion and to refrain from consumption of any fruit juice on treatment days.
Study design
This was a double-blind, randomized, placebo-controlled, alternating panel, rising dose study. There were two panels (A and B) of eight subjects each and four dosing periods. In each dosing period, six subjects in each panel received sitagliptin and two received an image-matched placebo. Treatment allocations were randomized independently in each dosing period. Doses given to any individual subject were separated by at least 1 week of washout. The sequence of doses was 5, 25, 100 and 25 mg for subjects in panel A and 12.5, 50, 200 and 400 mg for subjects in panel B. Safety and tolerability data were reviewed at each dose before proceeding to the next higher level.
Subjects fasted overnight (for at least 8 h) prior to receiving doses, except in Period 4, in which subjects in panel A ingested a standardized traditional Japanese breakfast prior to dosing. This meal consisted of rice, dried plum, dried bonito shavings, seasoned laver (seaweed), salmon, Japanese radish, soy sauce, spinach, tofu, green leeks, red miso and seasonings. It contained approximately 27 g of protein, 9 g of fat and 53 g of carbohydrates, with a total content of approximately 411 kcal.
Subjects were required to remain in the research unit for 48 h after dosing. They were allowed unlimited water but no food for the first 4 h post-dose, after which they received a standardized lunch and later, at approximately 10 h post-dose, a standardized dinner.
Pharmacokinetic assessments
Blood samples (6 ml) were collected prior to each dose and at 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 36 and 48 h post-dose. The samples were placed on ice immediately and centrifuged within 30 min to obtain plasma. Two aliquots of 200 µl each were taken from this plasma and stored at −70°C for subsequent assays of sitagliptin concentration and DPP-4 activity. Urine was collected immediately prior to dosing (at which time subjects were instructed to void completely) and over the following post-dose intervals (each terminated by complete voiding): 0–2, 2–4, 4–8, 8–12, 12–24, 24–36 and 36–48 h. Within each collection interval, urine voids were consolidated and stored at 0–4°C. At the end of the interval, the total volume was recorded and a 4 ml aliquot was stored at −70°C for subsequent assay of sitagliptin concentration.
Sitagliptin concentrations were measured using turbulent flow online extraction liquid chromatography and tandem mass spectrometry [14, 15]. The lower limits of quantitation were 0.5 ng ml−1 and 0.1 µg ml−1 in plasma and urine, respectively, and the linear calibration ranges were 0.5–1000 ng ml−1 and 0.1–50 µg ml−1, respectively. The within-day precision (relative SD%, n = 5) for plasma quality control samples varied from 2.0% to 5.3% and accuracy from 103% to 105% of the nominal value. The between-day precision for 100 sets of plasma quality-control samples in 29 analytical runs varied from 6.3% to 9.0% and the accuracy from 98.8% to 104% of the nominal value. For the urine quality control samples, the within-day precision (n = 5) and accuracy were 1.8% to 2.6% and 96.2% to 106% of the nominal value, respectively. The between-day precision for 56 sets of urine quality-control samples over a 6 month period varied from 3.8% to 5.5% and the accuracy from 102% to 105% of the nominal value. The assays of sitagliptin were performed by Merck Research Laboratories in West Point, PA, USA.
The following PK parameters were determined as previously described [6]: area under the curve (AUC) for plasma concentration over time extrapolated out to infinity AUC0–∞, maximal plasma concentration (Cmax), the post-dose time of plasma Cmax (tmax), the apparent terminal half-life of sitagliptin in the plasma (t1/2), the dose fraction that was excreted unchanged [fe,(0,∞)], and the renal clearance of sitagliptin (CLR).
Pharmacodynamic assessment
Plasma DPP-4 inhibition was assayed as previously described [6] by Merck Research Laboratories in Rahway, NJ, USA. Plasma samples were diluted 2.5-fold with buffer and fluorogenic substrate prior to analysis [6]. Given that binding of sitagliptin to the DPP-4 enzyme is competitive and rapidly reversible [16], this assay is expected to underestimate actual levels of inhibition in vivo unless the raw velocities measured (i.e. uncorrected percent DPP-4 inhibition values) are corrected to account for the dilution of sitagliptin and competition between sitagliptin and the fluorogenic substrate for binding to the enzyme active site [17]. A recently described statistical method corrects for these effects of dilution and competition [18]. This correction method was applied to data in this study. However, uncorrected inhibition values are also reported herein to facilitate comparison with previously published results. The summary measure of DPP-4 inhibition in any individual or group at a given sitagliptin dose was the weighted average inhibition observed from 0–24 h post-dose [WAI(0,24 h].
Safety and tolerability assessment
Safety and tolerability were assessed by physical examination, laboratory analyses of blood and urine, and collection of adverse experiences (AEs). Physical examination and laboratory analyses were performed pre-dose, 24 h post-dose and 10–14 days post-study. Physical examination included recording of vital signs and 12-lead ECGs. Laboratory analyses included routine haematology, serum chemistry (with measurements of fasting glucose, liver transaminases and muscle creatinine phosphokinase), and urinalysis. Vital signs were also recorded 2, 4, and 8 h post-dose and fasting serum glucose (FSG) was measured at 4 and 10 h post-dose (before lunch and dinner). Clinical AEs were collected through 14 days post-study and were evaluated for intensity (mild, moderate or severe), duration, outcome (recovered or not recovered), and relationship to study drug (definitely not, probably not, possibly, probably or definitely related, as rated by the investigator).
Statistical analysis
The plasma sitagliptin PK parameters AUC0–∞, Cmax, tmax, and apparent terminal t1/2 were analysed using a mixed-effects analysis of variance (anova) model appropriate for application to studies with an alternating panel, rising-dose design. Panel and dose within panel were included as fixed-effect factors in this model and subject within panel was included as a random-effect factor; period and sequence effects were assumed to be negligible. Summary statistics were computed for urinary PK parameters.
The same mixed-effects anova model was used to assess the effects of food ingestion on PK parameters. In this analysis, appropriate weighted linear contrasts were used to estimate fed vs. fasted geometric mean ratios (GMRs) and their 95% confidence intervals (CIs) for plasma sitagliptin AUC0–∞ and Cmax measured on a log-scale. It was assumed that data would be recorded from six subjects and that within-subject standard deviations for AUC0–∞ and Cmax would be 0.106 ln (nm·h) and 0.161 ln (nm), respectively. Under these conditions, and with observed GMRs of 1.00, the 95% CIs for AUC0–∞ and Cmax would be (0.87, 1.15) and (0.81, 1.23), respectively, after backtransformation from the log scale.
The same anova model was also used to analyse placebo-adjusted sitagliptin effects on WAI(0,24 h). For this analysis, data from placebo-treated subjects within a panel were pooled across all four of the dosing periods and the percent inhibition of DPP-4 in each subject at each post-dose timepoint was calculated using that individual's baseline (pre-dose) DPP-4 activity as a reference. Values for DPP-4 WAI(0,24 h) were then calculated by dividing the AUC for DPP-4 inhibition over 0–24 h post-dose by 24. All analyses were done on a log-percent scale with subsequent back-transformation of results to a linear scale. To estimate EC50 (defined as the plasma concentration of sitagliptin that achieved 50% inhibition of DPP-4), data were pooled across all doses, subjects and timepoints and fitted to a simple Emax (maximum drug effect) model by use of the Gauss-Newton method [19].
Present findings were compared with historical data from non-Japanese subjects [6] by using appropriate linear contrasts to estimate GMRs (Japanese/non-Japanese) and their corresponding 90% CIs for AUC0–∞ and Cmax. For DPP-4 WAI(0,24 h), the differences between means at each dose were compared between Japanese and non-Japanese subjects.
The assumption of normality was confirmed in the anova models by applying the Shapiro-Wilk test. Homogeneity of variances was confirmed by Levene's test.
Results
Subject demographics and disposition
Out of the 16 subjects who were initially enrolled, two discontinued while the study was in progress. Details regarding these discontinuations are presented with the safety results. The discontinued subjects were replaced as needed so that pharmacokinetic and pharmacodynamic data were available in each period from complete panels of eight subjects each. All enrolled subjects were included in the safety analyses. The mean age of subjects was 33 years (range 20–46 years), mean weight was 69 kg (range 48–95 kg) and mean height was 171 cm (range 156–195 cm).
Pharmacokinetics
Plasma sitagliptin concentrations increased in a dose-dependent manner throughout the range of doses studied and concentration-time profiles were similarly shaped regardless of dose (Figure 1). Plasma sitagliptin AUC0–∞ increased approximately in proportion to dose whereas Cmax increased at a rate greater than dose (Table 1). Pooling data across the two subject panels and seven doses tested, a regression analysis of log-transformed AUC0–∞ and log-transformed dose resulted in a best-fit slope coefficient (95% CI) of 0.995 (0.973, 1.017). (A coefficient of 1.00 would indicate exact dose-proportionality.) For log-transformed Cmaxvs. log-transformed dose, the best-fit slope coefficient was 1.207 (1.164, 1.251). The mean apparent terminal half-life for plasma sitagliptin was 9–14 h, with a significant trend towards decreasing half-life with increasing dose (P < 0.001). The AUC0–∞ and Cmax values observed in the present study, as well as all other PK parameters, were generally comparable with those previously observed in healthy, young non-Japanese males (Table 2). At the lowest dose tested (5 mg), mean Cmax was 36% greater in Japanese subjects than in non-Japanese. At the clinically relevant dose of 100 mg, the difference was 30%. The difference between Japanese and non-Japanese was ≤15% in all other comparisons.
Figure 1.
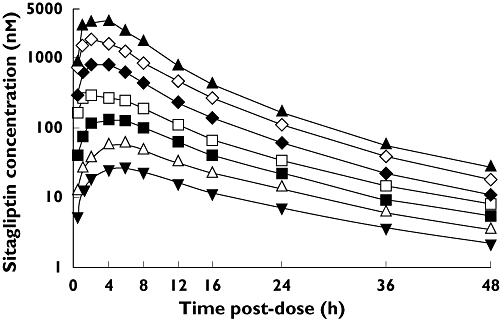
Mean plasma concentrations of sitagliptin over time in healthy, young Japanese males given the following single oral doses while in the fasted state: 5 ( ), 12.5 (
), 12.5 ( ), 25 (
), 25 ( ), 50 (
), 50 ( ), 100 (
), 100 ( ), 200 (
), 200 ( ) and 400 (
) and 400 ( ) mg
) mg
Table 1.
Summary of pharmacokinetic parameters following administration of single oral sitagliptin doses to fasted (except as indicated), healthy, young Japanese males
| Dose (all n = 6) | 5 mg | 12.5 mg | 25 mg | 25 mg – fed | 50 mg | 100 mg | 200 mg | 400 mg |
|---|---|---|---|---|---|---|---|---|
| AUC0–∞ (µm h)* | 0.50 ± 0.10 | 0.96 ± 0.15 | 2.04 ± 0.35 | 2.23 ± 0.23 | 3.76 ± 0.63 | 8.65 ± 1.64 | 16.5 ± 3.85 | 32.0 ± 4.73 |
| Cmax (nm)* | 27 ± 5 | 59 ± 7 | 149 ± 33 | 171 ± 24 | 309 ± 83 | 959 ± 307 | 1970 ± 715 | 3950 ± 541 |
| tmax (h)† | 6 | 4 | 5 | 5 | 2 | 2 | 3 | 2 |
| Apparent t1/2 (h)‡ | 13.8 ± 1.6 | 12.3 ± 0.8 | 11.6 ± 1.8 | 11.8 ± 1.5 | 11.4 ± 2.4 | 9.6 ± 0.9 | 9.1 ± 1.2 | 9.1 ± 0.9 |
| fe(0,∞)§ | 0.73 ± 0.06 | 0.85 ± .10 | 0.79 ± 0.09 | 0.86 ± 0.08 | 0.85 ± 0.09 | 0.88 ± 0.08 | 0.95 ± 0.05 | 1.00 ± 0.10 |
| CLR (ml min−1)* | 299 ± 57 | 445 ± 79 | 397 ± 55 | 391 ± 53 | 464 ± 69 | 415 ± 74 | 475 ± 88 | 513 ± 90 |
Geometric least-squares mean ± SD;
Median;
Harmonic mean ± between-subject Jackknife SD;
arithmetic least-squares mean ± SD.
AUC0–∞, post-dose area under the curve for plasma sitagliptin concentration over time extrapolated to infinity; CLR, renal clearance of sitagliptin; Cmax, maximum plasma concentration of sitagliptin; fe (0,∞), total fraction of sitagliptin dose excreted unchanged in urine with time extrapolated to infinity; SD, standard deviation; tmax, time to reach Cmax; t1/2, plasma half-life.
Table 2.
Comparison of plasma sitagliptin AUC0–∞ and Cmax between fasted, healthy, young Japanese males in the present study (Table 1) and historical data from fasted, healthy, young non-Japanese males [6]
| Dose (mg) | n | GMR for AUC0–∞(90% CI) | GMR for Cmax(90% CI) |
|---|---|---|---|
| 5 | 6 | 1.14 (1.02, 1.27) | 1.36 (1.11, 1.67) |
| 12.5 | 6 | 1.02 (0.91, 1.14) | 1.14 (0.92, 1.40) |
| 25 | 6 | 1.05 (0.94, 1.17) | 1.11 (0.90, 1.36) |
| 50 | 6 | 0.91 (0.82, 1.02) | 0.95 (0.77, 1.18) |
| 100 | 6 | 1.11 (1.00, 1.23) | 1.30 (1.07, 1.57) |
| 200 | 6 | 1.06 (0.97, 1.17) | 1.02 (0.85, 1.21) |
| 400 | 6 | 0.87 (0.78, 0.97) | 0.85 (0.69, 1.03) |
AUC0–∞, post-dose area under the curve for plasma sitagliptin concentration over time extrapolated to infinity; CI, confidence interval; Cmax, maximum plasma concentration of sitagliptin; GMR, ratio of geometric least-square means (Japanese/non-Japanese).
Mean fe(0,∞) increased from 0.73 to 1.00 between the lowest and highest doses tested. There was a weak trend towards increasing CLR with increasing dose (P < 0.02), but after exclusion of the lowest and highest test doses (i.e. including doses 12.5–200 mg), the interaction was not significant (P > 0.200). The mean value of CLR observed within this more restricted dose range was 430 ml min−1 (range 397–475 ml min−1).
Time course profiles for mean plasma sitagliptin concentration in fasted and fed subjects are compared in Figure 2. Slightly higher values for AUC0–∞ and Cmax were observed in fed subjects compared with fasted, as was observed previously in young, healthy non-Japanese males [6]. The GMRs (fed/fasted) and 95% CIs for the AUC0–∞ and Cmax were 1.12 (1.05, 1.19) and 1.18 (1.00, 1.39), respectively. tmax and apparent terminal t1/2 were not altered significantly by food (P = 0.693 and 0.711, respectively).
Figure 2.
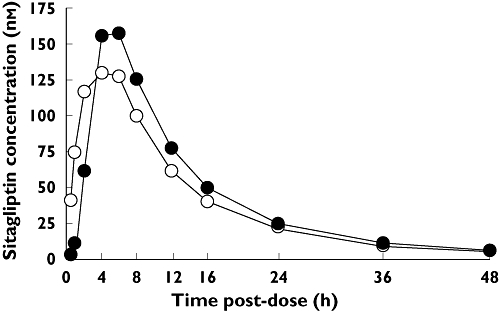
Mean plasma sitagliptin concentrations over time in healthy, young Japanese males given a single oral dose of 25 mg sitagliptin while in either the fed ( ) or fasted (
) or fasted ( ) state
) state
Pharmacodynamics
Profiles for plasma DPP-4 inhibition vs. time are shown in Figure 3. When low doses were given, these profiles had a shape similar to their corresponding plasma concentration-time profiles. At doses greater than 25 mg, however, the time course profiles trended towards saturation, especially during the first 12 h post-dose. After correction for underestimation of inhibition due to sample dilution and substrate competition, WAI(0,24 h) exceeded 94% at all doses ≥50 mg (Table 3). The corrected level of DPP-4 inhibition at 24 h post-dose (a time that would correspond to the trough concentration when sitagliptin is taken once daily) exceeded 88% at all doses ≥50 mg (Figure 3B). The relationship between dose and DPP-4 inhibition observed in this present study in Japanese subjects was very similar to that observed in non-Japanese subjects (Table 3 and Figure 3).
Figure 3.
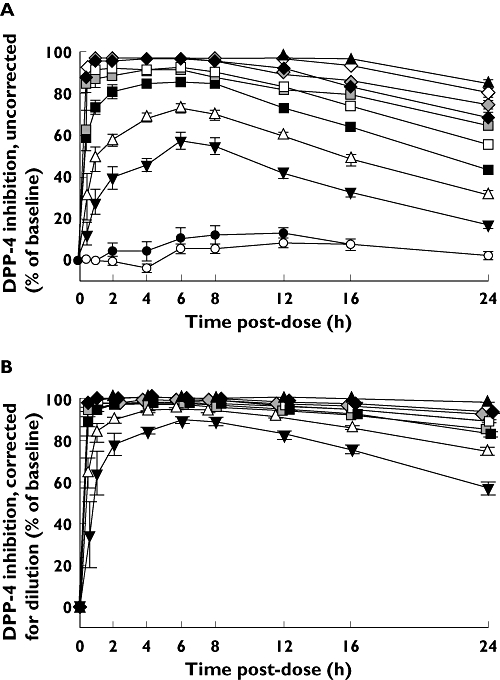
Mean levels of DPP-4 inhibition in the plasma of young, healthy, fasted, male Japanese subjects (except as noted) given single oral doses of sitagliptin. The values in (A) were not corrected for dilution of plasma samples during the assay procedure. The values in (B) were corrected for this artifact following a previously described statistical procedure [14]. Japanese subjects received either placebo ( and
and  in panels A and B) or doses of 5 (
in panels A and B) or doses of 5 ( ), 12.5 (
), 12.5 ( ), 25 (
), 25 ( ), 50 (
), 50 ( ), 100 (
), 100 ( ), 200 (
), 200 ( ) and 400 (
) and 400 ( ) mg sitagliptin. Historical data are included from young, healthy, fasted, non-Japanese male subjects given single oral doses of 50 (
) mg sitagliptin. Historical data are included from young, healthy, fasted, non-Japanese male subjects given single oral doses of 50 ( ) and 100 (
) and 100 ( ) mg sitagliptin
) mg sitagliptin
Table 3.
Weighted average percent inhibition of plasma DPP-4 activity from 0–24 h post-dose [WAI(0,24 h)] following administration of single oral sitagliptin doses to fasted, healthy, young Japanese and non-Japanese males
| A) Mean WAI(0,24 h)*†of plasma DPP-4 activity (uncorrected for sample dilution during assay) | |||||||
|---|---|---|---|---|---|---|---|
| Dose (all n = 6) | 5 mg | 12.5 mg | 25 mg | 50 mg | 100 mg | 200 mg | 400 mg |
| Japanese | 36.8 | 53.3 | 68.4 | 78.1 | 85.9 | 91.5 | 93.1 |
| Non-Japanese‡ | 32.6 | 53.6 | 67.2 | 79.3 | 86.9 | 91.1 | 93.4 |
| Mean difference | 4.3 | −0.3 | 1.2 | −1.2 | −1.0 | −0.5 | −0.3 |
| (90% CI) | (−2.3, 10.8) | (−5.0, 4.4) | (−2.0, 4.5) | (−3.3, 1.0) | (−2.3, 0.4) | (−1.3, 0.4) | (−1.0, 0.4) |
| B)Mean WAI(0,24 h)*†of plasma DPP-4 (corrected for sample dilution during assay) | |||||||
|---|---|---|---|---|---|---|---|
| Dose (all n = 6) | 5 mg | 12.5 mg | 25 mg | 50 mg | 100 mg | 200 mg | 400 mg |
| Japanese | 74.4 | 85.5 | 91.4 | 94.3 | 96.4 | 97.6 | 97.7 |
| Non-Japanese‡ | 72.6 | 86.2 | 91.4 | 95.0 | 96.8 | 97.6 | 98.0 |
| Mean difference | 1.8 | −0.7 | −0.0 | −0.7 | −0.4 | −0.1 | −0.3 |
| (90% CI) | (−9.5, 13.2) | (−12.0, 10.6) | (−11.4, 11.3) | (−12.0, 10.7) | (−11.8, 10.9) | (−19.7, 19.6) | (−11.7, 11.0) |
Calculated as (1 − GMR) × 100, where GMR is geometric mean ratio of weighted average DPP-4 enzyme activity from 0–24 h post-dose : DPP-4 enzyme activity at pre-dose.
Back-transformed from log scale.
Non-Japanese data from reference 6.
CI, confidence interval; DPP-4, dipeptidyl-peptidase-4.
The best-fit estimate (±SE) for EC50 in the present study was 26.2 ± 0.8 nm (based on DPP-4 inhibition data not corrected for dilution and competition effects). This value was very close to the estimate of 25.7 ± 0.5 nm previously derived by analysis of DPP-4 inhibition data (also uncorrected) from non-Japanese subjects [6].
Safety and tolerability
Sitagliptin was generally well tolerated in this study. Twelve subjects reported a total of 26 clinical AEs. None of these was serious and the incidence of these AEs appeared to be unrelated to the timing of treatments. During the course of the four dosing periods, there were 48 instances (as per protocol) in which subjects were given sitagliptin and 16 in which subjects were given placebo. The 48 administrations of sitagliptin were followed in the same dose period by 16 AEs reported for nine subjects, and the 16 administrations of placebo were followed in the same dose period by nine AEs reported for six subjects. There was also one pre-study AE reported.
All AEs were considered to be mild except one, an AE of headache reported to be of moderate intensity. This AE began in Period 4, 6 days after administration of 400 mg sitagliptin, and it had a duration of 3 days. The investigator considered it to be probably not drug-related. Among the other AEs, three were considered by the investigator as possibly drug-related. These included AEs of drowsiness and headache reported in one subject and somnolence in another. All occurred within 1 day of dosing with 50 mg of sitagliptin.
One subject withdrew from the study for a non-medical reason after receiving placebo in Period 1. A different subject discontinued the study in Period 2 after receiving a 5 mg dose of sitagliptin in Period 1 and placebo in Period 2. Elevated levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were observed in this subject beginning from 24 h post-dose in Period 1 (levels 1.2 and 1.5 times, respectively, the upper limits of normal for these enzymes) and continuing into Period 2, when elevations in serum alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT) were also observed. These enzymes reached levels from 2.1 to 6.5 times their respective upper limits of normal on the second and third days of Period 2, declining thereafter. Examination by upper abdominal ultrasound revealed the presence of gallbladder cholesterol polyps with small adherent stones. The increased enzyme levels and polyps were reported as non-serious AEs in Period 2 and discontinuation of the subject was attributed to these AEs conjointly. The investigator considered the elevated enzymes as probably not related to the study drug and the polyps to be definitely not related to the study drug, given that cholesterol polyps generally require years to develop and thus would have been a subclinical condition present but undetected at the time of enrolment. Serum levels of the liver enzymes were followed through a period of 30 days subsequent to day 2 of Period 1 (the day on which elevated levels were first present). The enzymes AST, ALT and ASP returned to within normal limits during this time; GGT was still slightly above the upper limit of normal when last observed (61 IU l−1, with 51 IU l−1 being the upper limit of normal).
No AEs of hypoglycaemia were reported. Baseline (pre-dose) values for mean FSG varied between 4.7 and 5.2 mmol l−1 (85.3–94.3 mg dl−1). Mean FSG values observed 4, 10 and 24 h after dosing (prior to lunch, dinner and the following day's breakfast) did not differ meaningfully from baseline values. There were no changes in vital signs or ECG parameters that appeared to be related to dose or clinically meaningful.
Discussion
Initial studies of the PK, PD and safety characteristics of sitagliptin were performed in Europe and North America, which is where this medication was also initially introduced into clinical use. More recently, sitagliptin has been approved for prescribing in several Asian countries, including Japan, China and Korea. This study addressed the question whether young, healthy, male Japanese subjects are similar to non-Japanese subjects in their responses to single oral doses of sitagliptin. The present findings suggest that there are no clinically important differences between Japanese and non-Japanese subjects with respect to the PK, PD and safety characteristics of sitagliptin.
Clinical experience with sitagliptin supports a conclusion that this medication has a wide therapeutic index. Significant glycaemic improvement has been observed with doses of 50, 100 and 200 mg day−1[20–23], and sitagliptin has been generally well tolerated after single oral doses up to 800 mg [7], daily doses of 400 mg for 28 days [8] and daily doses of 200 mg for 24 weeks [23, 24]. Against this background of efficacy and safety observations made over a wide range of sitagliptin doses, none of the findings from the present study suggest differences between Japanese and non-Japanese responses to sitagliptin that would be of clinical consequence.
Sitagliptin improves glycaemic control through selective inhibition of DPP-4, an action that increases active levels of the endogenous incretins GLP-1 and GIP [1, 2]. Previous studies have shown that maximum glucose-lowering efficacy is observed with doses that are sufficient to inhibit DPP-4 by >80% (uncorrected for dilution and competition effects) throughout an entire dose cycle [6, 16]. With correction for dilution and substrate competition effects during in vitro assay of plasma samples, 80% inhibition in vitro corresponds to approximately 95% inhibition in vivo[18]. This level of DPP-4 inhibition is achieved by plasma sitagliptin concentrations ≥100 nm, and at this level of inhibition, plasma concentrations of intact GLP-1 are increased approximately two-fold [6–8]. The present findings suggest that DPP-4 inhibition can be sustained at this level by single oral doses of sitagliptin in young, healthy Japanese males, as has previously been found in non-Japanese young, healthy males and other demographic groups.
In previous studies, sitagliptin has generally exhibited a favourable safety and tolerability profile [24] and the present findings, including the absence of hypoglycaemia, were consistent with prior experience. Incretins support glucose homeostasis by stimulating insulin release and suppressing glucagon release only when blood glucose is elevated; during euglycaemia, these actions are limited [3, 25]. This explains why sitagliptin did not elicit any large or clinically meaningful adverse reductions in blood glucose in the normoglycaemic subjects in this study.
Cholesterol polyps are the most common form of gallbladder polyp (which have a prevalence of approximately 5% in the general population) and they are generally asymptomatic [26]. It is possible, nonetheless, that the polyps and accompanying gallstones found in the subject in this study may have played a role in his elevated liver enzymes through transient obstruction of the flow of bile or injury to the bile duct epithelium [27]. In a large pooled analysis of safety data from 19 controlled clinical trials with sitagliptin, no significant differences were found between sitagliptin-exposed and non-exposed patients for incidence rates of AEs in the system organ class of hepatobiliary disorders (counted together as a class); neither were there significant differences in the incidence rates for the AEs of increased ALT and increased AST [24].
In conclusion, this study found no clinically relevant differences between Japanese and non-Japanese subjects with respect to PK and PD parameters observed after single oral doses of sitagliptin. The present data are supportive of once daily dosing with sitagliptin in Japanese patients with type 2 diabetes.
Competing interests
All of the authors, except for J L R, are present or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., and may also hold stock or stock options in this company. This study was funded by Merck & Co., Inc.
REFERENCES
- 1.Herman GA, Stein PP, Thornberry NA, Wagner JA. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: focus on sitagliptin. Clin Pharmacol Ther. 2007;81:761–7. doi: 10.1038/sj.clpt.6100167. [DOI] [PubMed] [Google Scholar]
- 2.Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P, Dietrich B, Golor G, Schrodter A, Keymeulen B, Lasseter KC, Kipnes MS, Snyder K, Hilliard D, Tanen M, Cilissen C, De Smet M, de Lepeleire I, Van Dyck K, Wang AQ, Zeng W, Davies MJ, Tanaka W, Holst JJ, Deacon CF, Gottesdiener KM, Wagner JA. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels following an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:4612–9. doi: 10.1210/jc.2006-1009. [DOI] [PubMed] [Google Scholar]
- 3.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 4.Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP. Sitagliptin, a DPP-4 inhibitor for the treatment of patients with type 2 diabetes: a review of recent clinical trials. Curr Med Res Opin. 2008;24:489–96. doi: 10.1185/030079908x261069. [DOI] [PubMed] [Google Scholar]
- 5.Lyseng-Williamson KA. Sitagliptin. Drugs. 2007;67:587–97. doi: 10.2165/00003495-200767040-00007. [DOI] [PubMed] [Google Scholar]
- 6.Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, Snyder K, Hilliard D, Tanen M, Tanaka W, Wang AQ, Zeng W, Musson D, Winchell G, Davies MJ, Ramael S, Gottesdiener KM, Wagner JA. Pharmacokinetics and pharmacodynamics of single doses of sitagliptin, an inhibitor of dipeptidyl peptidase-IV, in healthy subjects. Clin Pharmacol Ther. 2005;78:675–88. doi: 10.1016/j.clpt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Bergman AJ, Stevens C, Zhou YY, Yi B, Laethem M, De Smet M, Snyder K, Hilliard D, Tanaka W, Zeng W, Tanen M, Wang AQ, Chen L, Winchell G, Davies MJ, Ramael S, Wagner JA, Herman GA. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Ther. 2006;28:55–72. doi: 10.1016/j.clinthera.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 8.Herman GA, Bergman A, Liu F, Stevens C, Wang AQ, Zeng W, Chen L, Snyder K, Hilliard D, Tanen M, Tanaka W, Meehan AG, Lasseter K, Dilzer S, Blum R, Wagner JA. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol. 2006;46:876–86. doi: 10.1177/0091270006289850. [DOI] [PubMed] [Google Scholar]
- 9.Vincent SH, Reed JR, Bergman AJ, Elmore CS, Zhu B, Xu S, Ebel D, Larson P, Zeng W, Chen L, Dilzer S, Lasseter K, Gottesdiener K, Wagner JA, Herman GA. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos. 2007;35:533–8. doi: 10.1124/dmd.106.013136. [DOI] [PubMed] [Google Scholar]
- 10.Bergman AJ, Cote J, Yi B, Marbury T, Swan SK, Smith W, Gottesdiener K, Wagner J, Herman GA. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care. 2007;30:1862–4. doi: 10.2337/dc06-2545. [DOI] [PubMed] [Google Scholar]
- 11.Chu XY, Bleasby K, Yabut J, Cai X, Chan GH, Hafey MJ, Xu S, Bergman AJ, Braun MP, Dean DC, Evers R. Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein. J Pharmacol Exp Ther. 2007;321:673–83. doi: 10.1124/jpet.106.116517. [DOI] [PubMed] [Google Scholar]
- 12.Ieiri I, Takane H, Hirota T, Otsubo K, Higuchi S. Genetic polymorphisms of drug transporters: pharmacokinetic and pharmacodynamic consequences in pharmacotherapy. Expert Opin Drug Metab Toxicol. 2006;2:651–74. doi: 10.1517/17425255.2.5.651. [DOI] [PubMed] [Google Scholar]
- 13.Sissung TM, Baum CE, Kirkland CT, Gao R, Gardner ER, Figg WD. Pharmacogenetics of membrane transporters: an update on current approaches. Mol Biotechnol. 2010;44:152–67. doi: 10.1007/s12033-009-9220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng W, Musson DG, Fisher AL, Wang AQ. Determination of MK-0431 in human plasma using high turbulence liquid chromatography online extraction and tandem mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:1169–75. doi: 10.1002/rcm.2426. [DOI] [PubMed] [Google Scholar]
- 15.Zeng W, Musson DG, Fisher AL, Chen L, Schwartz MS, Woolf EJ, Wang AQ. Determination of sitagliptin in human urine and hemodialysate using turbulent flow online extraction and tandem mass spectrometry. J Pharm Biomed Anal. 2008;46:534–42. doi: 10.1016/j.jpba.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, Marsilio F, McCann ME, Patel RA, Petrov A, Scapin G, Patel SB, Roy RS, Wu JK, Wyvratt MJ, Zhang BB, Zhu L, Thornberry NA, Weber AE. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:141–51. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 17.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: Wiley-Interscience; 1975. [Google Scholar]
- 18.Alba M, Sheng D, Guan Y, Williams-Herman D, Larson P, Sachs JR, Thornberry N, Herman G, Kaufman KD, Goldstein BJ. Sitagliptin 100 mg daily effect on DPP-4 inhibition and compound-specific glycemic improvement. Curr Med Res Opin. 2009;25:2507–14. doi: 10.1185/03007990903209514. [DOI] [PubMed] [Google Scholar]
- 19.Hartley HO. The modified Gauss-Newton method for the fitting of non-linear regression functions by least squares. Technometrics. 1961;3:269–28. [Google Scholar]
- 20.Scott R, Wu M, Sanchez M, Stein PP. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract. 2007;61:171–80. doi: 10.1111/j.1742-1241.2006.01246.x. [DOI] [PubMed] [Google Scholar]
- 21.Hanefeld M, Herman GA, Wu M, Mickel C, Sanchez M, Stein PP. Once-daily sitagliptin, a dipeptidyl peptidase-4 inhibitor, for the treatment of patients with type 2 diabetes. Curr Med Res Opin. 2007;23:1329–39. doi: 10.1185/030079907X188152. [DOI] [PubMed] [Google Scholar]
- 22.Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006;49:2564–71. doi: 10.1007/s00125-006-0416-z. [DOI] [PubMed] [Google Scholar]
- 23.Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7. doi: 10.2337/dc06-0703. [DOI] [PubMed] [Google Scholar]
- 24.Williams-Herman D, Engel SS, Round E, Johnson J, Golm GT, Guo H, Musser B, Davies MJ, Kaufman KD, Goldstein BJ. Safety and tolerability of sitagliptin in clinical studies: a pooled analysis of data from 10 246 patients with type 2 diabetes. BMC Endocr Disord. 2010;10:7. doi: 10.1186/1472-6823-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 26.Myers RP, Shaffer EA, Beck PL. Gallbladder polyps: epidemiology, natural history and management. Can J Gastroenterol. 2002;16:187–94. doi: 10.1155/2002/787598. [DOI] [PubMed] [Google Scholar]
- 27.Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med. 2010;77:195–204. doi: 10.3949/ccjm.77a.09064. [DOI] [PubMed] [Google Scholar]