

Abstract
AIM
Gemcitabine (GEM) enters normal and tumour cells via concentrative (CNT) and equilibrative nucleoside transporters (ENT) and is subsequently deaminated to the inactive difluorodeoxyurine (dFdU) by cytidine deaminase (CDA). The aim of our study was to ascertain whether the nucleoside transporter genotype and the CDA activity phenotype can predict total GEM plasma clearance.
METHODS
Forty-seven patients received GEM 1000–1250 mg m−2 i.v. over 30 min. Plasma concentrations of GEM and dFdU were measured and individual pharmacokinetic profiles were determined. CDA activity was measured ex vivo in plasma samples. The two most common hENT1 and hCNT1 polymorphisms were determined from genomic DNA.
RESULTS
Multivariate analysis revealed that GEM plasma clearance (CL) was positively correlated with the end of infusion dFdU : GEM ratio (P < 0.0001), which is a marker of in vivo CDA activity. The ENT1 genotype characterized by high transport capacity (G/G) and age were inversely correlated with CL (P= 0.027 and 0.048, respectively). A strong correlation was found between end of infusion GEM concentration and area under the concentration–time curve from time 0 to infinity (AUC(0,∞)) (r2= 0.77).
CONCLUSIONS
Our results confirm the role of CDA and age on the interindividual variability of GEM CL and show the contribution of the hENT1 genotype for the first time.
Keywords: cytidine deaminase, deoxycytidine kinase, gemcitabine, nucleoside transporter, pharmacogenetics, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Gemcitacine is taken up by the cell through various nucleoside transporters of either the concentrative (CNT) or equilibrative type (ENT) and is then transformed into the inactive metabolite, dFdU, by cytidine deaminase (CDA) and into the active metabolite, dFdCMP, by deoxycytidine kinase (dCK).
While the major contribution of CDA to gemcitabine elimination is well recognized no data about the role of CNT and ENT activities have yet been reported. Both nucleoside transporters exhibit genetic polymorphisms characterized by different expression levels or nucleoside affinity.
WHAT THIS STUDY ADDS
The plasma clearance (CL) of gemcitabine has been determined following the standard 30 min infusion of 1000–1250 mg m−2. The in vivo CDA activity was measured as end of infusion metabolic ratio (MR = dFdU : gembitabine) and the variant hCNT-1 and hENT-1 alleles were genotyped.
Our results confirmed that gemcitabine CL is directly correlated with CDA activity and inversely correlated with age and, for the first time, show that patients heterozygous for the –706 G > C hENT-1 mutation have a lower CL as compared with wild type patients.
Introduction
Gemcitabine (GEM) is a prodrug with anticancer activity in a variety of solid tumours. As a hydrophilic compound, it crosses the plasma membrane primarily via various nucleoside transporters (NT) of either the concentrative (CNT) or equilibrative type (ENT). Once inside the cell, GEM is phosphorylated to the monophosphate metabolite, 2′, 2′- difluorodeoxycytidine monophosphate (dFdCMP), by deoxycytidine kinase (dCK) [1]. At the same time, GEM undergoes extensive deamination in the liver and several other tissues by cytidine deaminase (CDA) to form the inactive metabolite 2′,2′-difluorodeoxyuridine (dFdU), with renal excretion of the unchanged drug accounting for only 5% of the dose [2]. Thus, GEM elimination is expected to depend on the sequential actions of NTs, which make the drug available for intracellular metabolism, and of CDA and dCK, which transform it into deaminated and phosphorylated metabolites, respectively.
The concentrative nucleoside transporter 1 (CNT1) is predominantly expressed in liver, kidney and small intestine, whereas the equilibrative nucleoside transporter 1 (ENT1) is ubiquitously distributed [3]. Both NTs exhibit genetic polymorphisms associated with different transporter expression or nucleoside affinity [1]. Three single nucleotide polymorphisms (SNPs) have been identified in the promoter region of hENT1, at positions –1345C > G, –1050G > A and –706G > C, which are associated with increased function. The two most frequent hENT1 haplotypes in Caucasians are CGG/CGG (wild type: 79%) and CGG/CGC (heterozygous: 21%), and heterozygous individuals (CGG/CGC) have a 37% higher RNA expression compared with homozygous wild-type (CGG/CGG) [4]. In addition, hCNT1 gene shows several non-synonymous SNPs in the coding region, one of which (G > A substitution at position 565) is present in 28% of Caucasians and is characterized by reduced affinity for GEM [5].
Plasma and tissue CDA activity is highly variable between individuals [6–9]. Although several SNPs have been identified in the CDA gene, only the CDA208G > A variant is associated with reduced GEM plasma clearance [10] and enhanced risk of severe myelosuppression [11]. However, given the low frequency of the CDA208G>A allele (12.5% in Africans, 3.7% in Japanese, and 0% in Caucasians) [1], non-genetic factors may play a predominant role in explaining the variability in CDA activity. In this regard, increased CDA serum activity has been described in pregnancy and inflammatory diseases [12].
In contrast to the wide interpatient variability in GEM metabolic clearance, optimal GEM plasma concentrations should be within the narrow range of 10–20 µm (3–6 µg ml−1) at which the enzyme dCK, responsible for GEM intracellular activation, becomes saturated [13]. A correlation has also been reported between end of infusion GEM concentration and platelet count decrease, with a 50% reduction at 14.3 µg ml−1 (≈48 µm) [14]. Accordingly, methods able to predict GEM pharmacokinetics in the individual patient would be helpful in optimizing dose schedules.
The primary aim of our study was to verify whether, and to what extent, the two most common NT polymorphisms (hCNT1 565 G > A and hENT1–706 G > C) and the CDA metabolic activity (measured in vivo and in vitro) can predict GEM clearance (CL).
Methods
Patients and treatments
Forty-seven consecutive Caucasian patients (36 males and 11 females) with tumours of the lung (n= 30), pancreas (n= 6), kidney/bladder (n= 5), cholecystis (n= 4), ovary (n= 1) or pleura (n= 1) requiring GEM treatment were the study population. They were aged between 51 and 82 years (median 71, mean 69 years) and weighed 50–120 kg (mean 68.5 kg). GEM was infused intravenously over 30 min at doses of 1000–1250 mg m−2. The programmed chemotherapeutic regimen consisted of GEM alone in 15 patients and of the combination of GEM with platinum compounds in the others. Only 22 of 32 patients assigned to the combined therapy had already received at least one dose of cisplatin (n= 16) or carboplatin (n= 6) before GEM administration.
The study was approved by the Ethics Committee of the General Hospital of Rovigo, and all patients gave their written informed consent.
Pharmacokinetic study and drug assay
On the day of the study, no other antitumour agents were administered. Blood samples (2 ml) were collected before GEM infusion, at the end of infusion, and at 10, 20, 30, 45 and 60 min thereafter. Tetrahydrouridine (20 µl of 10 mg ml−1) was immediately added to the post-infusion samples to inhibit CDA and prevent ex vivo GEM deamination. Plasma was then separated and stored at −20°C until analysis. GEM and dFdU plasma concentrations were determined by an HPLC method modified after Lanz et al. [15]. The lowest detection limit was 0.02–0.025 mg l−1; intra-day and inter-day variability values were 2.1–3.4% and 5.2–7.5 %, respectively.
Pharmacokinetic analysis
The area under the concentration–time curve (AUC(0,∞)) was calculated using the trapezoidal rule from the start of infusion to the last sampling time, and was thereafter extrapolated to infinity using the slope (k) of the final exponential phase. AUC and k were calculated using the GraphPad PRISM software ver. 4 (San Diego, CA). Systemic clearance (CL) and apparent volume of distribution (Vz) were calculated using the standard formulas: CL = dose/AUC(0,∞); Vz= CL/k.
The metabolic ratio (MR) between the concentrations of dFdU and GEM at the end of infusion was taken as an index of in vivo CDA activity. The choice of end of infusion MR was dictated by Abbruzzese's results [2] showing that GEM plasma concentrations generally attain a plateau after 15 min of infusion.
Ex vivo determination of plasma CDA activity
Drug-free plasma (200 µl) was spiked with 2 µg GEM (= 10 mg l−1) and incubated for 2 h at 37°C. The reaction was stopped after 0.5, 1 and 2 h by adding 500 µl acetonitrile, and the corresponding GEM and dFdU concentrations were then determined as described above. The time-course of GEM and dFdU concentrations followed a one-compartment model with first-order kinetics, so that GEM elimination (KelGEM) and dFdU formation (KfodFdU) rates were calculated. CDA activity was quantified by measuring the rate constant (h−1) of dFdU formation (KfodFdU). The in vitro plasma CL was also calculated as for systemic CL (dose/AUC).
Genetic analysis
Genomic DNA was extracted from peripheral blood white cells with a commercial kit (Promega). Since the only variant hENT1 haplotype known in Caucasians differs from the wild type for the –706 G > C substitution (CGG/CGC vs. CGG/CGG), the analysis of hENT1 polymorphisms was restricted to the -706 G > C SNP. hENT1 PCR reaction was performed in a final solution of 50 µl containing 8% DMSO with the following primers:
Forward: 5′GGGAGCAGGAGAGGGACGCT3′;
Reverse: 5′AGGCAGGCAAGAGGACGAGAGG3′.
It consisted of 35 cycles with an annealing temperature of 70°C; genotyping was performed through RFLP analysis with AluI enzyme, which cuts the variant –706C allele into three fragments of 261, 165 and 129 bps, and the –706G allele into two fragments of 426 and 129 bps.
hCNT1 amplification was performed with the following primers:
Forward: 5′GGTGCTATTGTGTGTGGGTG3′;
Reverse: 5′.GCAGGCAAAGAGGAGAGCCA3′, modified at the 18th nucleotide to introduce a restriction site (G to C substitution).
Amplification involved 35 cycles at an annealing temperature of 62°C. Genotyping was performed through RFLP analysis with NlaIII, which cuts the variant 565A allele into two fragments of 20 and 174 bps.
Classification of ENT1 and CNT1 activity based on hENT1 and hCNT1 genotypes
According to in vitro functional data [4, 5], the hENT1 genotypes were classified as high activity (G/C and C/C) and low activity (G/G) genotypes. Similarly, the hCNT1 genotypes were classified as high (G/G) and low activity (G/A and A/A) genotypes. hENT1 and hCNT1 genotypes were further combined in order to obtain a total NT activity score as follows: high activity haplotype (high activity hENT1+ high activity hCNT1); intermediate activity haplotype (high activity hENT1+ low activity hCNT1, or low activity hENT1+ high activity hCNT1) and low activity haplotype (low activity hENT1+ low activity hCNT1).
Statistical analysis
Pharmacokinetic parameters are reported as mean values ± SD. Differences between groups were evaluated by means of the two-tailed t-test for unpaired data, after testing for normality of distribution (significance level P < 0.05). The correlation between variables was assessed by means of linear regression analysis. In an attempt to identify the determinants of CL m−2, multivariate linear regression analysis was carried out with age and MR (a marker of in vivo CDA activity) as continuous variables and gender, previous platinum therapy and NT genotypes as categorical variables. Females were labelled 1 and males 2. High and low activity hENT1 and hCNT1 genotypes were labelled 1 and 2, respectively, and the three activity levels of the combined genotypes were labelled 1, 2 and 3. The determination coefficient r2 and adjusted r2 were calculated. The ability of the linear equation to predict the dependent variable from the regression parameters was also evaluated by calculating the mean prediction error (MPE) and mean absolute error (MAE):
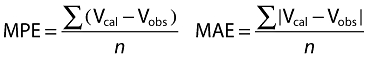 |
where Vobs is the observed value and Vcal the calculated value. MPE and MAE were then normalized by Vobs and expressed as percentual errors: MPE% and MAE%.
The statistical package STATISTICA ver. 7.1 (StatSoft Italia srl, Vigonza, Italy) was used to run all analyses.
Results
Pharmacokinetic data
The time courses of the mean GEM and dFdU plasma concentrations are shown in Figure 1A.
Figure 1.
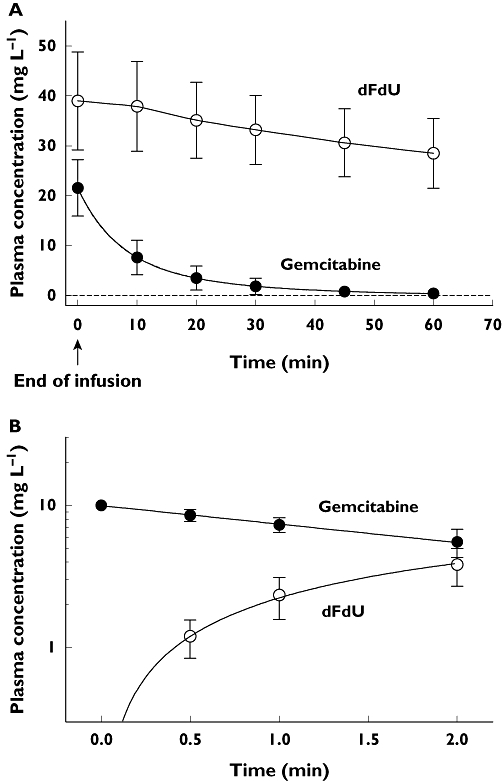
A) the time courses of the mean GEM and dFdU plasma concentrations in our patients (n= 47) after stopping GEM infusion. B) the time courses of the mean GEM and dFdU concentrations in patients' plasma incubated with GEM for 2 h at 37°C. Bars indicate SD
End of infusion GEM plasma concentration (GEMend), metabolic ratio, AUC(0.∞) and CL were widely variable between patients. Ranges and mean (± SD) values of the pharmacokinetic parameters are shown in Table 1.
Table 1.
Mean values (± SD), coefficient of variation (CV%) and ranges of the main pharmacokinetic parameters
| Mean (±SD) | CV% | Range | |
|---|---|---|---|
| GEMend (mg l−1) | 21.6 ± 5.6 | 25.9 | 9.7–35.6 |
| MR | 1.85 ± 0.53 | 28.6 | 0.76–4.2 |
| AUC(0,∞) (mg l−1 h) | 10.8 ± 3.2 | 29.6 | 4.8–19.3 |
| CL (l h−1 m−2) | 105.9 ± 28.3 | 26.7 | 55.3–209.8 |
| Vz (l m2−1) | 37.0 ± 30.4 | 82.2 | 10.0–129.3 |
GEMend was closely correlated with GEM AUC(0,∞) (Figure 2C, r2= 0.77, P < 0.0001). MPE% and MAE% were 1.96% ± 12.71% and 10.45% ± 7.34%, respectively. CL also showed a highly significant direct correlation with MR (Figure 2A, r2= 0.62, P < 0.0001) and a significant inverse correlation with age (Figure 2B, r2= 0.088, P= 0.043). Although MR was not linearly correlated with age, MR was slightly but significantly lower in patients aged ≥ 70 years (1.73 ± 0.62) than those <70 years (2.04 ± 0.42, P= 0.044). Gender did not influence CL variability, while Vz was significantly lower in females (20.9 ± 9.6 l m−2) than in males (42.7 ± 33.0 l m−2, P= 0.036).
Figure 2.
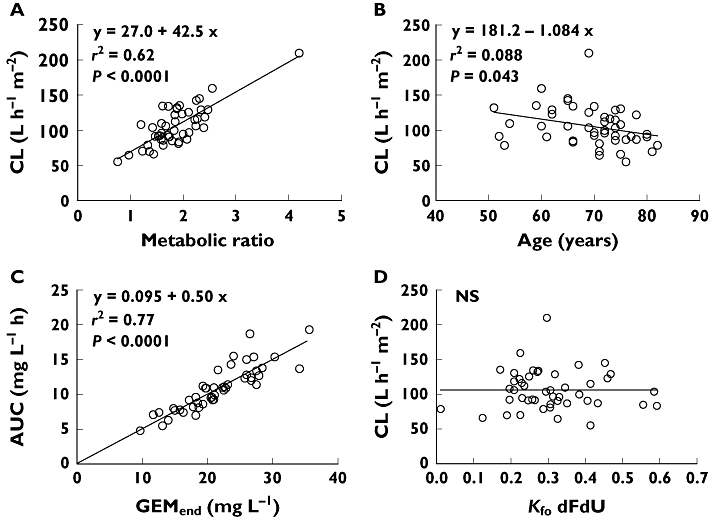
Linear regression analysis results (see Results for explanations). GEM CL is directly correlated with the metabolic ratio (A) and inversely with age (B). Gembitabine end infusion concentration (GEMend) is highly correlated with AUC(0,∞) (C), while no correlation was found between in vitro dFdU formation rate (KfodFdU) and in vivo CL (D)
As regards ex vivo CDA plasma activity data (Figure 1B), the mean KelGEM and KfodFdU, were 0.306 ± 0.117 h−1 (range 0.010–0.603 h−1) and 0.308 ± 0.117 h−1 (range 0.011–0.592 h−1), respectively, and were virtually coincident (linear correlation slope = 0.98, r2= 0.998), thus indicating that CDA is the only enzyme responsible for GEM inactivation in plasma. In contrast, absolutely no correlation was found between metabolite formation rate KfodFdU and CL (Figure 1D, P= 0.97). The GEM CL calculated in vitro was negligible (1.1 ± 2.8 ml h−1) as compared with the corresponding CL in vivo (192 ± 63 l h-1).
Genetic data
The frequencies of hENT1–706 and hCNT1 565 genotypes were the following: hENT 1 G/G (wild type) 62% (n= 29), hENT1 G/C 38% (n= 18), hENT1 C/C 0%; hCNT1 GG (wild type) 66% (n= 31), hCNT1 G/A 13% (n= 6), hCNT1 A/A 21% (n= 10). hENT1 allele distribution was in Hardy-Weinberg equilibrium. No association was found between the two polymorphisms (P= 0.4)
Following univariate analysis, none of the pharmacokinetic parameters (GEMend, MR, CL) differed significantly between the various hENT1 and hCNT1 genotypes/haplotypes (data not shown).
Multivariate linear regression analysis
The results showed that the only variables related with CL m−2 were, in decreasing order of significance, MR, hENT1 genotype and age (details in Table 2). Gender, previous platinum therapy, hCNT1 genotypes and combined hENT1 and hCNT1 haplotypes were not related with CL m−2. The multiple regression equation was the following:
Table 2.
Multiple linear regression analysis: dependent variable CL m-2. B values of the independent variables indicate the coefficients of the respective regression lines; t is the result of the statistical test
| Dependent variable: CL m−2, r2= 0,70; adjusted r2= 0.67, P < 0.000001 | ||||
|---|---|---|---|---|
| n= 47 | B | SE of B | t | P |
| Intercept | 87.7 | 24.7 | 3.54 | 0.00096 |
| dFdU : GEMend (MR) | 41.9 | 4.6 | 9.13 | <0.0000001 |
| hENT genotype | −11.1* | 4.9 | −2.04 | 0.027 |
| Age | −0.63 | 0.31 | −2.29 | 0.048 |
The B value refers to the low activity genotype (G/C); it must be multiplied by 2 for the high activity genotype (G/G).
where the value of G is 1 for the low activity genotype (G/G) and 2 for the high activity genotype (G/C). MPE% and MAE% were 2.63% ± 14.97% and 12.60% ± 8.31%, respectively.
The correlations of CL with MR and age confirmed the results of the univariate analysis, whereas the determinant role of hENT1 genotypes was only revealed by multivariate analysis. It appears that genotypes with higher NT activity are associated with a lower plasma CL.
Vz m−2 variability was explained only by gender (P= 0.043), females having a smaller Vz m−2 than males, thus confirming the results of the univariate analysis.
Discussion
The principal result of our study was the significant correlation of GEM CL with (i) hENT1 genotype, (ii) CDA activity in vivo (i.e. MR) and (iii) age.
The most novel finding of our study was the contribution of NT polymorphisms to GEM CL in vivo. According to the multiple regression equation derived from our data (see Table 1), patients with a high activity genotype have reduced GEM CL. In our series, an ‘average’ patient (i.e. aged 69 years, with an MR of 1.85) carrying the high activity hENT1 G/C genotype, would have a CL 10% lower than that of a patient of the same age and with the same metabolic capacity but with the low activity G/G genotype. It is worth noting that the role of the hENT1 genotype is even more important in the subset of patients with low CDA activity. Based on our equation, the effect of hENT1 polymorphism on total GEM CL consists of a variation of about 5% among patients with high CDA activity (MR = 4.25) but rises to 17% among those with low CDA activity (MR = 0.75). As a consequence, slow metabolizers carrying the hENT1 G/G genotype should be given 17% more drug to attain the same AUC as that of slow metabolizers with the G/C genotype. The association between high hENT1 activity and reduced GEM CL is apparently surprising, since one would expect that greater GEM cell uptake would lead to faster metabolism via CDA and dCK.
A possible explanation is that intracellular GEM metabolism undergoes partial saturation. It is known that dCK activity is fully saturated at low plasma concentrations (10–20 µm) [13, 16], which are easily attained at standard doses, whatever infusion rate is applied. Conversely, CDA activity is likely to become saturated for doses greater that 1750 mg m−2[14]. Knowing that the metabolic CL of a drug is equal to Vmax/(Km± drug concentration) and assuming that GEM metabolic CL is the sum of dCK and CDA activity, we calculated CL at different GEM concentrations using the Vmax and Km values of human dCK and CDA published by Bouffard et al. [17] (Table 3). CL was calculated at 9.7 mg l−1 (∼37 µm) and 35.6 mg l−1 (∼135 µm), which are the lowest and the highest GEM Cmax found in our patients, and at 13.3 mg l−1 (∼51 µm) and 48.6 mg l−1 (∼185 µm), which are the lowest and the highest GEM Cmax increased by 37%. The 37% increase was tentatively chosen assuming that the increase in intracellular GEM concentration in patients with a high activity hENT1 genotype would be similar to the increase in mRNA reported in human blood cells [4]. The results showed that a 37% increase in the lowest and highest Cmax yielded a 10% and 18% decline in CL, respectively. These theoretical percentages are similar to those obtained from our experimental data (5–17%), supporting our hypothesis.
Table 3.
Mean kinetic parameters (± SD) of purified human CDA and dCK, using gemcitabine as substrate [17]
| Enzyme | Km (µm) | Vmax (pmol min−1) |
|---|---|---|
| CDA (n= 3) | 95.7 ± 8.4 | 1200 ± 100 |
| dCK (n= 3) | 4.6 ± 0.6 | 14.9 ± 0.9 |
An additional, but less important, mechanism to explain the dependence of CL on NT may be increased GEM reabsorption at the renal tubule level. It is known that NT systems are well represented in both the proximal and distal tubules [18]. By means of in situ hybridization and immunohistochemical techniques, hENT1s have mainly been located in the basolateral membrane of the distal tubule [19], i.e. in that tract of nephron where passive drug reabsorption occurs. It is therefore possible that increased hENT1 activity in distal tubular cells produces greater GEM reabsorption and, consequently, a lower plasma CL. However, since only 1–10% of the GEM dose is excreted by the kidney as unchanged drug [2], this mechanism alone cannot account for the estimated decrease in GEM CL.
The influence of hENT1 polymorphisms on intracellular GEM biotransformations in normal tissues cannot be directly applied to the cancer setting. Pennycooke et al. [20] reported marked differences in hENT1 expression between tumoral and matched normal tissues in several human cancer types. In vitro studies showed that hENT1-deficient tumour cells are highly resistant to cytotoxicity by GEM [21, 22] and that inhibitors of NTs reduce sensitivity to the drug [23]. In addition, clinical studies in patients with pancreatic cancer treated with GEM have consistently shown that low expression of hENT1 in tumour tissue is associated with reduced overall survival [24–27]. On the whole, it seems that reduced drug tumour uptake by ENT1 limits intracellular GEM activation via dCK metabolism. It is therefore conceivable that tumours with low hENT1 expression (and low GEM uptake), unlike normal cells, are unable to saturate dCK, producing suboptimal amounts of the GEM active metabolite.
In summary, low hENT1 expression in normal tissues due to genetic polymorphisms increases GEM CL and reduces systemic drug exposure, whereas low hENT1 expression in tumoral tissues due to multiple pathway perturbation induces drug resistance. The simultaneous occurrence of these two events is probably associated with worse prognosis. This hypothesis is worth verifying in a prospective study.
Another interesting finding of our study was that the in vivo CDA activity, as assessed by the MR, proved to be the strongest determinant of GEM CL (Figure 1A) and may be a reliable parameter to predict the total inactivation capacity in individual patients. Plasma CDA activity measured in vitro represented a negligible fraction of total CL and was devoid of any predictive value (Figure 1D, P= 0.97). An extremely weak correlation between CDA activity and GEM AUC(0,∞) (r2=−0.08) has been reported by Sugiyama et al. [10] in a larger study. Overall, the usefulness of measuring plasma CDA activity to identify ‘slow’ metabolizers seems, at best, to be of limited value.
Unlike CDA, the contribution of dCK to GEM CL cannot be determined, because the related metabolic product (dFdCMP) is not measurable in plasma, but remains trapped inside the cells. Nevertheless, an estimate of dCK involvement may be deduced indirectly by observing that the line correlating MR with CL (Figure 1A) has a positive intercept of 27.0 l h−1 m−2. This value corresponds to a null metabolic ratio and should therefore reflect the residual CL due to the sum of dCK metabolism and renal excretion. As the renal CL of unchanged gemcitabine is about 5% of the total CL (5.3 l h−1 m−2, on average, in our series), the metabolic CL due to dCK should be around 22 l h−1 m−2, which represents about 20% of total CL. In other words, it seems that the activity of the enzyme at a standard infusion rate of 30 min can transform one-fifth of the drug administered into active compound. To our knowledge, this is the first report inferring the percentage of the active compound produced in vivo by metabolism of the drug.
Lastly, our data demonstrate that age is an independent predictor of reduced GEM CL (P= 0.048). According to the linear correlation shown in Figure 1B, GEM CL decreases on average by 27% from the age of 51 to 82 years (126 l h−1 m−2vs. 92 l h−1 m−2). On theoretical grounds this effect may be ascribed to a decrease in CDA and dCK activity or to an age-related decline in renal function. Our finding of a lower MR in patients more than 70 years old suggests that CDA activity may decrease with ageing. At present, no information is available on the influence of age on dCK activity. The last hypothesis of a reduced renal excretion, though compatible with the physiology of ageing, seems unlikely given the minor contribution of the kidney to GEM elimination and in view of the results of two studies which reported no correlation between the clearance of creatinine and GEM [10, 28]. Gender was not a determinant of GEM CL in our series but Vz was significantly lower in females than in males. On the whole, our results on the influence of age and gender on GEM pharmacokinetics are in full agreement with those reported by Sugiyama et al. [10] in a larger population.
Our results can also be analyzed in the perspective of clinical applications. High interpatient variability requires methods able to predict GEM pharmacokinetics in individual patients, in order to tailor doses. It does not seem practical to use the multiple regression equation including the GEM MR, hENT genotype and age in order to predict individual GEM CL. In addition, this approach only accounts for 67% of the interindividual variability of CL (adjusted r2= 0.67). Instead, determination of GEMend to predict AUC according to the empirical equation AUC = 0.09 + 0.50 × GEMend is simpler and has a higher predictive value (r2= 0.77). Gan et al. [29] reported a similar good correlation (r2= 0.75) in a series of 21 patients treated with increasing GEM doses (750–1500 mg m−2 in 30 min). Their results were further confirmed by Grimison et al. [30] after both 30 min and 100 min infusions. From the clinical point of view, a single sample determination at the end of infusion may therefore be convenient, to identify patients at risk of excessive or insufficient drug exposure.
In conclusion, this study provides new insights into GEM pharmacology by quantifying the relative influence on GEM CL of hENT1 genotype, CDA and dCK activities and age and proposes a limited sampling method to predict GEM AUC(0,∞). Further studies are needed to evaluate whether these pharmacogenetic and pharmacokinetic variables can also predict the clinical outcome.
Competing interests
There are no competing interests to declare.
This research was supported by grants from the CARIPARO Foundation.
REFERENCES
- 1.Wong A, Soo RA, Yong WP, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41:77–88. doi: 10.1080/03602530902741828. [DOI] [PubMed] [Google Scholar]
- 2.Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN, Plunkett W. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9:491–8. doi: 10.1200/JCO.1991.9.3.491. [DOI] [PubMed] [Google Scholar]
- 3.Damaraju VJ, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22:7524–36. doi: 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- 4.Myers SN, Goyal RK, Roy JD, Fairfull LD, Wilson JW, Ferrel ER. Functional single nucleotide polymorphism haplotypes in the human equilibrative nucleoside transporter 1. Pharmacogenet Genomics. 2006;16:315–20. doi: 10.1097/01.fpc.0000189804.41962.15. [DOI] [PubMed] [Google Scholar]
- 5.Gray JH, Mangravite LM, Owen RP, Urban TJ, Chan W, Carlson EJ, Huang CC, Kawamoto M, Johns SJ, Stryke D, Ferrin TE, Giacomini KM. Functional and genetic diversity in the concentrative nucleoside transporter, CNT1, in human populations. Mol Pharmacol. 2004;65:512–9. doi: 10.1124/mol.65.3.512. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz Van Haperen R, Veerman G, Braakuis BJ, Vermorken JB, Boven E, Leyva A, Peters GJ. Deoxycytidine kinase and deoxycytidine deaminase activities in human tumour xenografts. Eur J Cancer. 1993;29A:2132–7. doi: 10.1016/0959-8049(93)90048-k. [DOI] [PubMed] [Google Scholar]
- 7.Skeith KJ, Wefuran J, Oswald R, Davis P. Serum cytidine deaminase as a measure of disease activity in rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol. 1993;20:1309–15. [PubMed] [Google Scholar]
- 8.Watanabe SC, Uchida T. Expression of cytidine deaminase in human solid tumors and its regulation by 1α,25-dihydroxyvitamin D3. Biochim Biophys Acta. 1996;1312:99–104. doi: 10.1016/0167-4889(96)00024-9. [DOI] [PubMed] [Google Scholar]
- 9.Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34:1274–81. doi: 10.1016/s0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 10.Sugiyama E, Kaniwa N, Kim SR, Kikura-Hanajiri R, Hasegawa R, Maekawa K, Saito Y, Ozawa S, Sawada J, Kamatani N, Furuse J, Ishii H, Yoshida T, Ueno H, Okusaka T, Saijo N. Pharmacokinetics of gemcitabine in Japanese cancer patients: the impact of cytidine deaminase polymorphism. J Clin Oncol. 2007;25:32–42. doi: 10.1200/JCO.2006.06.7405. [DOI] [PubMed] [Google Scholar]
- 11.Yonemori K, Ueno H, Okusaka T, Yamamoto N, Ikeda M, Saijo N, Yoshida T, Ishii H, Furuse J, Sugiyama E, Kim SR, Kikura-Hanajiri R, Hasegawa R, Saito Y, Ozawa S, Kaniwa N, Sawada J. Severe drug toxicity associated with a single-nucleotide polymorphism of the cytidine deaminase gene in a Japanese cancer patient treated with gemcitabine plus cisplatin. Clin Cancer Res. 2005;11:2620–4. doi: 10.1158/1078-0432.CCR-04-1497. [DOI] [PubMed] [Google Scholar]
- 12.Paira S, Roverano S, Rillo O, Barrionuevo A, Mahieu S, Millen N. Cytidine deaminase in polymyalgia rheumatica and elderly onset rheumatoid arthritis. Clin Rheumatol. 2005;24:460–3. doi: 10.1007/s10067-004-1058-5. [DOI] [PubMed] [Google Scholar]
- 13.Veltkampt SA, Beijnen JH, Schellens JHM. Prolonged versus standard gemcitabine infusion: translation of molecular pharmacology to new treatment strategy. Oncologist. 2008;13:261–76. doi: 10.1634/theoncologist.2007-0215. [DOI] [PubMed] [Google Scholar]
- 14.Fogli S, Danesi R, De Braud F, De Pas T, Curigliano G, Giovannetti G, Del Tacca M. Drug distribution and pharmacokinetic/pharmacodynamic relationship of paclitaxel and gemcitabine in patients with non-small-cell lung cancer. Ann Oncol. 2001;12:1553–9. doi: 10.1023/a:1013133415945. [DOI] [PubMed] [Google Scholar]
- 15.Lanz C, Früh M, Thormann W, Cerny T, Lauter BH. Rapid determination of gemcitabine in plasma and serum using reversed-phase HPLC. J Sep Sci. 2007;30:1811–20. doi: 10.1002/jssc.200600534. [DOI] [PubMed] [Google Scholar]
- 16.Grunewald R, Kantarijan H, Du M, Faucher K, Tarassoff P, Plunkett W. Gemcitabine in leukaemia: a phase I clinical trial, plasma, and cellular pharmacology study. J Clin Oncol. 1992;10:406–13. doi: 10.1200/JCO.1992.10.3.406. [DOI] [PubMed] [Google Scholar]
- 17.Bouffard DY, Laliberté J, Momparler RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45:1857–61. doi: 10.1016/0006-2952(93)90444-2. [DOI] [PubMed] [Google Scholar]
- 18.Mangravite LM, Badagnani I, Giacomini KM. Nucleoside transporters in the disposition and targeting of nucleoside analogs in the kidney. Eur J Pharmacol. 2003;479:269–81. doi: 10.1016/j.ejphar.2003.08.076. [DOI] [PubMed] [Google Scholar]
- 19.Govindarajan R, Bakken AH, Hudkins KL, Lai Y, Casado FJ, Pastor-Anglada M, Tse CM, Hayashi J, Unadkat JD. In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1809–R1822. doi: 10.1152/ajpregu.00293.2007. [DOI] [PubMed] [Google Scholar]
- 20.Pennycooke M, Chaudary N, Shuralyova I, Zhang Y, Coe IR. Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem Biophys Res Commun. 2001;280:951–9. doi: 10.1006/bbrc.2000.4205. [DOI] [PubMed] [Google Scholar]
- 21.Achiwa H, Oguri T, Sato S, Maeda H, Niimi T, Ueda R. Determinants of sensitivity and resistance to gemcitabine: the roles of human equilibrative nucleoside transporter 1 and deoxycytidine kinase in non-small cell lung cancer. Cancer Sci. 2004;95:753–7. doi: 10.1111/j.1349-7006.2004.tb03257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mori R, Ishikawa T, Ichikawa Y, Taniguchi K, Matsuyama R, Ueda M, Fujii Y, Endo I, Togo S, Danenberg PV, Shimada H. Human equilibrative nucleoside transporter 1 is associated with the chemosensitivity of gemcitabine in human pancreatic adenocarcinoma and biliary tract carcinoma cells. Oncol Rep. 2007;17:1201–5. [PubMed] [Google Scholar]
- 23.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–57. [PubMed] [Google Scholar]
- 24.Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet C, Cass C, Lai R, Mackey JR. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res. 2004;10:6956–61. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 25.Maréchal R, Mackey JR, Lai R, Demetter P, Peeters M, Polus M, Cass CE, Young J, Salmon I, Devière J, Van Laethem JL. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin Cancer Res. 2009;15:2913–9. doi: 10.1158/1078-0432.CCR-08-2080. [DOI] [PubMed] [Google Scholar]
- 26.Farrel JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, Abrams R, Benson AB, Macdonald J, Cass CE, Dicker AP, Mackey JR. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187–95. doi: 10.1053/j.gastro.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 27.Giovanetti E, Del Tacca M, Mey V, Funel N, Nannizzi S, Ricci S, Orlandini C, Boggi U, Campani D, Del Chiaro M, Iannopollo M, Bevilacqua G, Mosca F, Danesi R. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res. 2006;66:3928–35. doi: 10.1158/0008-5472.CAN-05-4203. [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Galettis P, Links M, Mitchell PL, McLachlan AJ. Population pharmacokinetics of gemcitabine and its metabolite in patients with cancer: effect of oxaliplatin and infusion rate. Br J Clin Pharmacol. 2007;65:326–33. doi: 10.1111/j.1365-2125.2007.03040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gan HK, Mitchell PL, Galettis P, Davis ID, Cebon J, de Souza P, Links M. A phase 1 and pharmacokinetics study of gemcitabine and oxaliplatin in patients with solid tumors. Cancer Chemother Pharmacol. 2006;58:157–64. doi: 10.1007/s00280-005-0152-y. [DOI] [PubMed] [Google Scholar]
- 30.Grimison P, Galettis P, Manners S, Jelinek M, Metharom E, de Souza PL, Liauw W, Links MJ. Randomized crossover study evaluating the effect of gemcitabine infusion dose rate: evidence of auto-induction of gemcitabine accumulation. J Clin Oncol. 2007;25:5704–9. doi: 10.1200/JCO.2007.10.7078. [DOI] [PubMed] [Google Scholar]