


Abstract
Ribonucleotide reductase (RNR) is a critical enzyme of nucleotide metabolism, synthesizing precursors for DNA replication and repair. In prokaryotic genomes, RNR genes are commonly targeted by mobile genetic elements, including free standing and intron-encoded homing endonucleases and inteins. Here, we describe a unique molecular solution to assemble a functional product from the RNR large subunit gene, nrdA that has been fragmented into two smaller genes by the insertion of mobE, a mobile endonuclease. We show that unique sequences that originated during the mobE insertion and that are present as C- and N-terminal tails on the split NrdA-a and NrdA-b polypeptides, are absolutely essential for enzymatic activity. Our data are consistent with the tails functioning as protein interaction domains to assemble the tetrameric (NrdA-a/NrdA-b)2 large subunit necessary for a functional RNR holoenzyme. The tails represent a solution distinct from RNA and protein splicing or programmed DNA rearrangements to restore function from a fragmented coding region and may represent a general mechanism to neutralize fragmentation of essential genes by mobile genetic elements.
INTRODUCTION
In prokaryotic genomes, proteins are generally encoded by continuous open reading frames, reflecting evolutionary and functional constraints to maintain function in a single polypeptide. Fragmented coding regions can result from the insertion of introns or inteins, often into conserved sequences that correspond to functionally critical regions of the interrupted genes (1). Splicing pathways remove the intron or intein, restoring a continuous open reading frame and protein function (2). Fragmented coding regions also occur naturally, where a protein is encoded by a single gene in one species, but has been split into multiple, smaller genes in related species (3–8). In these instances, function is likely restored by the interactions of independently translated polypeptides that assemble to form a functional complex.
Here, we examine how function is restored when a ribonucleotide reductase (RNR) gene has been fragmented by the insertion of a mobile genetic element such that RNR active site residues are partitioned between two genes. In phage Aeh1 that infects Aeromonas hydrophila, the nrdA gene for the large subunit of aerobic RNR has been fragmented into two smaller genes, nrdA-a and nrdA-b, by the transposition of the homing endonuclease mobE (Figure 1A) (8). RNRs are functionally critical enzymes that catalyze the synthesis of deoxyribonucleotides used for DNA replication and repair (9) and are common targets of homing endonucleases (10–12). Class Ia RNRs require the presence of the NrdB component encoded by the nrdB gene and in bacteria and phage the active holoenzyme is generally a tetramer composed of a dimer of the large subunit NrdA protein (α2) and a dimer of the small subunit NrdB protein (β2; see Figure 1 for nomenclature). The mobE insertion splits the Aeh1 nrdA gene at a position that in the Escherichia coli NrdA structure lies between two adjacent β-strands in the RNR specific 10-stranded α/β-barrel that constitutes the active site (Figure 1B). Three active site residues, Cys-219 and Cys-431 in NrdA-a and Cys-31 in NrdA-b, are located in separate polypeptides, while the homologous residues in E. coli NrdA (Cys-225, Cys-439 and Cys-462) are present in the same polypeptide (13). An essential part of the reaction mechanism involves a transient disulfide bond formed between Cys-225 and Cys-462 in E. coli NrdA (9,14). The corresponding residues in Aeh1 are Cys-219 located in NrdA-a and Cys-31 located in NrdA-b. Moreover, in 1200 nrdA sequences from viruses as well as cellular organisms, none are split into multiple coding regions (15), suggesting strong evolutionary pressure to retain NrdA as a continuous polypeptide. Recent metagenomic data has, however, revealed the presence of nrdA genes fragmented by the insertion of inteins that presumably undergo trans-splicing to restore NrdA function (7). Remarkably, the Aeh1 holoenzyme is fully functional with specific activity equivalent to other characterized class Ia RNRs, but with an unusual (NrdA-a/NrdA-b)2NrdB2 or (αa/αb)2/β2, subunit composition (Figure 1C) (8). Thus, a composite active site and functional holoenzyme is assembled from residues on each polypeptide.
Figure 1.

The mobE insertion fragments the active site of the class I RNR of phage Aeh1. (A) Schematic of the Aeh1 nrdA-a, mobE and nrdA-b genes, with the GC content over a 100 nucleotide-sliding window plotted above. The average GC content over the operon (42%) is indicated by a dashed line. Note that the mobE and nrdA-b genes overlap. Shown below is an amino acid alignment of the Aeh1 NrdA-a and NrdA-b and related NrdA proteins, with the mobE insertion indicated by a right facing arrow (not to scale). Active site residues of E. coli NrdA are indicated by solid triangles and conserved or identical amino acids are shaded gray or black with white lettering, respectively. The Aeh1 NrdA-a and NrdA-b tails are highlighted by rectangles. (B) Left, the E. coli NrdA monomer, colored to indicate the split between the Aeh1 NrdA-a (yellow) and NrdA-b (green) proteins. Active site residues are depicted as red spheres. Right, model of the NrdA-a and NrdA-b fragments based on the E. coli NrdA structure, with active site residues indicated by red spheres. The NrdA-b fragment is rotated relative to its position in the monomer to highlight the active site residues. (C) Subunit composition of the large subunit or holoenzyme of the class Ia RNR for the prototypical E. coli enzyme or the phage Aeh1 enzyme.
In this report, we examine the mechanism by which the fragmented RNR of phage Aeh1 assembles to form a functional holoenzyme. Strikingly, we find that unique C- and N-terminal tails of NrdA-a and NrdA-b that likely originated during the mobE insertion are absolutely essential for enzymatic activity. Our data are consistent with the tails functioning as protein interaction domains to promote assembly of the RNR large subunit tetramer, rendering the insertion of mobE phenotypically neutral with respect to RNR function.
MATERIALS AND METHODS
Construction and purification of tail mutant proteins
The NrdA-aΔT mutant was constructed by the introduction of a stop codon at position 1324 of nrdA-a gene by site-directed mutagenesis of the wild-type nrdA-a gene in pACYCDuet-1. The NrdA-bΔT mutant was constructed by polymerase chain reaction (PCR) using primers to amplify an nrdA-b fragment lacking the first 54 nt and replacing an ACG codon (Thr18) with an AUG (Met) codon. The subsequent NrdA-bΔT construct was cloned from pCRBlunt into pACYC-Duet vector carrying the NrdA-aΔT mutant. Both mutant proteins were expressed from the same pACYC-Duet vector. Wild-type and mutant versions of the phage Aeh1 NrdA-a, NrdA-b and NrdB proteins were purified from E. coli BL21(DE3) cells overexpressing cloned versions of the genes as described (8).
Biophysical characterization
All ultracentrifugation studies were performed using a Beckman Optima XLA Analytical Ultracentrifuge in the Biomolecular Interaction and Conformation Facility in the Biochemistry Department at The University of Western Ontario. Equilibrium ultracentrifugation analyses were performed in triplicate using 0.27 mg/ml NrdA-a/NrdA-b heterodimer or 0.22 mg/ml NrdA-aΔT/NrdA-bΔT heterodimer. The reference channels were loaded with buffer consisting of 50 mM Tris–HCl pH 7.0, 1 mM dithiothreitol (DTT), 100 mM NaCl. Samples were centrifuged at 10, 15 and 20 k r.p.m. at 5°C for 16 h. One scan was performed after 16 h and another scan at 20 h. Data points were obtained by scanning in 0.002 cm step size increments, taking 10 replicate scans for each point. Data were analyzed using GraphPad Prism software, using the single ideal species model to solve for the molecular weight, M, of the sample according to the following equation
where Cr is the solute concentration at radius r, CF is the solute concentration at reference distance F, ω is the angular velocity, R is the gas constant and T is the temperature in Kelvin.
Velocity centrifugation studies were used to characterize the shape of the NrdA-a/NrdA-b and NrdA-aΔT/ NrdA-bΔT heterodimers using concentrations of 0.3 mg/ml and 0.68 mg/ml, respectively. Velocity runs for each sample were performed at 8°C at 40 k r.p.m., with one scan taken every 10 min for a total of 30 scans. The sedimentation co-efficient, s, was determined using the Lamm equation:
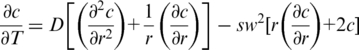 |
where c is the solute concentration, t is time, r is radius, D is the solute diffusion constant and ω is the angular velocity.
RNR enzymatic assays
Enzymatic activity measurements were performed as described previously (8). The NrdA-a/NrdA-b protein concentration was 0.11–0.22 µM and the NrdB concentration 1.9 µM. The mutant NrdA proteins were assayed at 2.4–4.7 µM together with 19 µM NrdB.
GEMMA analyses
The general procedure and instrumental setup was as described (16). In samples containing the NrdA proteins, a running buffer consisting of 40 mM ammonium acetate buffer, 0.005% Tween-20 and 1 mM DTT was used, while the NrdB protein in the absence of NrdA was analyzed in 20 mM ammonium acetate. The NrdA wild-type protein concentration was 0.01 mg/ml (0.11 µM), while the mutant proteins had a concentration of 0.04 mg/ml (0.47 µM). The NrdB protein was analyzed at a concentration of 0.005–0.02 mg/ml (0.12–0.46 µM). Capillary pressures of 1.4–2.0 p.s.i. were used and each data shown is the sum of 2–10 scans. Using GEMMA, the particle mass can usually be determined within ±5.6% (17).
Surface plasmon resonance studies
The interaction of wild-type NrdA protein with NrdB was analyzed by surface plasmon resonance (SPR) as described (18). The interactions between the mutant NrdA and the NrdB protein were analyzed using the same protocol but with 0.1–3.5 µM NrdA in the absence of dATP and 0.25–6 µM NrdA in the presence of 1 mM dATP (GE Healthcare).
Mass spectrometry
In-gel digestions with AspN1 and identification of peptide sequences were performed at the Functional Proteomics Facility and Biological Mass Spectrometry Facility at The University of Western Ontario. The NrdA-a and NrdA-b tail sequences were identified by Q-TOF MS/MS after separation of peptides by liquid chromatography.
RESULTS
The tail sequences of NrdA-a and NrdA-b are essential for enzymatic activity
In considering a mechanism by which the holoenzyme would be assembled, we noted the presence of unique C- and N-terminal extensions, or tails, on NrdA-a and NrdA-b, respectively (Figure 1B). The sequences encoding these unique tails are AT-rich compared to the surrounding Aeh1 genes (Figure 1A) and likely originated from foreign DNA that was fused in-frame to the 3′- and 5′-ends of nrdA-a and nrdA-b during the mobE transposition event. Database searches with the tail sequences failed to detect any significant matches. We rationalized that the tail sequences would not have been retained in the Aeh1 genome unless they were essential for NrdA function. To gain insight into potential function(s) of the tails, we first determined if the tails are present in the mature forms of NrdA-a and NrdA-b. We used dATP-sepharose chromatography to purify NrdA-a, NrdA-b and NrdB from Aeh1-infected A. hydrophila extracts (8). Previous mass spectrometry analyses of peptides after in-gel digestion with trypsin positively identified the proteins as NrdA-a, NrdA-b and NrdB (8), but failed to identify peptides from the tail regions. We thus repeated the in-gel digestion with the AspN1 protease and identified peptides from the C-terminal tail of NrdA-a (DQYKSLRY) and the N-terminal tail of NrdA-b (MIEHERIYEVYE), indicating that the tails are not post-translationally processed (Supplementary Figure S1). We next deleted the tails from cloned versions of the nrdA-a and nrdA-b genes to create the tail mutant (ΔT) proteins NrdA-aΔT and NrdA-bΔT (Supplementary Figure S2). We expressed combinations of wild-type and ΔT versions of NrdA-a and NrdA-b and found that the NrdA-a/NrdA-b and NrdA-aΔT/NrdA-bΔT combinations were soluble. Other combinations of wild-type and mutant NrdAs were not studied further. The purified NrdA-a/NrdA-b and NrdA-aΔT/NrdA-bΔT proteins were independently mixed with Aeh1 NrdB and assayed for enzymatic activity by measuring the reduction of CDP to dCDP. Crucially, the specific activity of the NrdA-aΔT/NrdA-bΔT proteins was ∼2000-fold reduced relative to the NrdA-a/NrdA-b proteins, almost abolishing enzymatic activity (Table 1). We conclude that the unique tails, present in the mature forms of NrdA-a and NrdA-b, are essential for enzymatic activity.
Table 1.
Biochemical properties of wild-type and mutant versions of NrdA-a and NrdA-b
| NrdA-a/NrdA-b heterodimera | Molecular weight (kDa) |
Sedimentation coefficient (S)d | Specific activity (U/mg)e | ||
|---|---|---|---|---|---|
| Predicted | AUCb | GEMMAc | |||
| αa/αb | 90.8 | 93.9 ± 0.7 | 91 ± 5.1 | 6.1 | 946 ± 12 |
| αaΔT/αbΔT | 84.6 | 91.7 ± 2.5 | 77 ± 5.7 | 4.5 | 0.4 ± 0.06 |
aα refers to the large subunit, while subscript a or b refers to the split NrdA component (i.e. αa refers to NrdA-a and αb refers to NrdA-b). The subscript ΔT refers to the tail mutant of that protein. See Figure 1 for nomenclature.
bAnalytical ultracentrifugation analyses with NrdA-a/NrdA-b and NrdA-aΔT/NrdA-bΔTproteins at ∼0.2 mg/ml and no added nucleotide (see also Supplementary Figure S2).
cGEMMA analyses with 0.01–0.04 mg/ml protein and no added nucleotide; mean of 2–3 experiments.
dS20,w values were calculated from velocity centrifugation analyses using 0.25 mg/ml NrdA-a/NrdA-b and 0.68 mg/ml NrdA-aΔT/NrdA-bΔTproteins and corrected for water at 20°C (See also Supplementary Figure S2).
eAssayed in the presence of NrdB with DTT as the reductant. One unit corresponds to 1 nmol of dCDP formed per min and specific activity is units/mg of protein.
The tails are not required for NrdA-a/NrdA-b heterodimer formation
We hypothesized that one mechanism by which the tails are required for RNR activity would be to promote or stabilize interactions between NrdA-a and NrdA-b. To test this hypothesis, we examined the oligomeric status of the split wild-type and mutant ΔT NrdA proteins by gas-phase electrophoretic mobility macromolecular analysis (GEMMA) (17), a technique that measures the diameter of protein complexes in the gas phase at low protein concentrations. In the absence of exogenously added nucleotides, the sizes of the major species observed are consistent with heterodimers of the wild type (αa/αb) and tail mutant proteins (αaΔT/αbΔT), respectively (Figure 2A and B; Table 1). Furthermore, we analyzed the wild-type and ΔT NrdA proteins by analytical ultracentrifugation at higher protein concentrations (∼0.2–0.6 mg/ml) in the absence of nucleotides and found that the sizes were consistent with the proteins existing as heterodimeric αa/αb and αaΔT/αbΔT species (Table 1 and Supplementary Figure S2). Velocity ultracentrifugation, which in contrast to equilibrium ultracentrifugation can detect changes in the shape of proteins, showed that the heterodimeric wild-type and ΔT complexes possessed different sedimentation coefficients (6.1S versus 4.5S) that are too large to be accounted for by differences in the predicted molecular weights of the heterodimers (Supplementary Figure S2). These results indicated that deletion of the tails affects the shape of the αaΔT/αbΔT complex relative to the αa/αb complex. Potential differences in shape were also evident upon gel-filtration chromatography, as the elution profiles of the αa/αb and αaΔT/αbΔT heterodimers were shifted relative to each other (Supplementary Figure S2). Collectively, these data indicate that the tails are not required for interaction of αa with αb to form a heterodimer and that deletion of the tails affects the shape of the heterodimer.
Figure 2.

The mutant αaΔT/αbΔT heterodimer is defective in dATP-mediated dimerization. Shown are representative GEMMA analyses of the αa/αb or αaΔT/αbΔT heterodimers with no added nucleotide (traces A and B), or with 50 mM dATP (traces C and D). The concentration of the αa/αb heterodimer was 0.01 mg/ml (0.11 µM) and the concentration of the αaΔT/αbΔT heterodimer was 0.04 mg/ml (0.47 µM). For each condition, the composition and predicted sizes (in kDa) of the species are indicated. The baseline has been shifted by 1000 intensity counts for each trace.
The tail mutants exhibit a dimerization defect
Class Ia RNRs are allosterically regulated by nucleoside triphosphates binding at two different types of sites on NrdA. Binding of ATP, dATP, dTTP or dGTP to one type of site regulates the substrate specificity of the enzyme and binding of ATP or dATP to another site regulates the overall activity, with ATP usually activating and dATP inhibiting enzyme activity (9,16). In general, the allosteric effector nucleotides promote formation of dimers or higher oligomers of NrdA and also strengthen the interaction between NrdA and NrdB. We therefore investigated whether addition of allosteric nucleoside triphosphates regulated the oligomeric status of the split Aeh1 wild-type and ΔT NrdA proteins. We used GEMMA to assay various nucleotide effector conditions and found that dimerization of the αa/αb heterodimer was promoted by 50 µM dATP, as a species with a predicted molecular mass of 173 kDa was observed (compare Figure 2A and C), consistent with a (αa/αb)2 dimer of heterodimers. The only nucleotide that could promote dimer formation was dATP, as addition of other effector nucleotides and/or substrate had no effect on dimerization at the tested concentrations (Supplementary Figure S3). Strikingly, we did not observe formation of an (αaΔT/αbΔT)2 dimer of heterodimers with the tail mutant proteins in the presence of 50 µM dATP (compare Figure 2B and D). Other nucleotides also had no effect on dimerization of the αaΔT/αbΔT heterodimer at the concentrations tested (Supplementary Figure S3).
Assembly of the holoenzyme is compromised with the tail mutants
We next examined the interactions of the wild-type and mutant split NrdA proteins with the NrdB protein (Figure 3). Studies with other class Ia RNRs have shown that NrdB, which together with NrdA forms the functional holoenzyme, is essential for enzyme activity (9). GEMMA analysis of Aeh1 NrdB alone revealed a peak consistent with β-dimers (73 kDa) and a much lower abundance species that is consistent with a β-tetramer (156 kDa) (Figure 3A). When we analyzed interactions between the Aeh1 wild-type NrdA-a/NrdA-b heterodimer and NrdB by GEMMA, two major species were observed, one consistent with an αa/αb heterodimer (91 kDa) and the other consistent with a (αa/αb)2β2 holoenzyme (240 kDa) (Figure 3B). Two peaks with higher predicted molecular weights (331 and 510 kDa) but of lower abundance could represent higher-order oligomers of the split NrdA proteins and NrdB (Figure 3B), as has been observed with the E. coli RNR that forms an α4β4 oligomer in the presence of dATP (16). Addition of dATP to the Aeh1 RNR resulted in an increased amount of (αa/αb)2β2 holoenzyme formed (Figure 3D), likely due to the formation of a dATP-stimulated (αa/αb)2 complex that is subsequently available to interact with NrdB. In contrast, addition of other nucleotides or substrate did not stimulate further formation of the holoenzyme (Supplementary Figure S3).
Figure 3.

The mutant αaΔT/αbΔT heterodimer cannot form a holoenzyme in the presence of NrdB. Shown are GEMMA analyses of the NrdB alone (trace A), the αa/αb or αaΔT/αbΔT heterodimers with NrdB (traces B and C) and the αa/αb or αaΔT/αbΔT heterodimers with NrdB and dATP (traces D and E). The concentrations of the large subunit proteins were as in Figure 2 and the concentration of the NrdB protein was 0.005–0.02 mg/ml (0.12–0.46 µM). For each condition analyzed, the composition and predicted sizes (in kDa) of the species are indicated. For trace B, the species labeled with an asterisk has a size of 331 kDa, with a predicted (αa/αb)2β4 composition. For traces B and D, the species labeled hash has a size of 510 kDa and a predicted (αa/αb)4β4 composition. The baseline has been shifted by 1000 intensity counts for each trace.
Intriguingly, the mutant ΔT NrdA proteins had a strongly reduced ability to interact with NrdB and form the active holoenzyme. When the interaction between the NrdA-aΔT, NrdA-bΔT and NrdB proteins was analyzed by GEMMA, a predominant species was observed consistent with a mixture of αaΔT/αbΔT and β2 complexes (Figure 3C). A higher molecular weight complex of 156 kDa was also observed, but in a reduced abundance relative to the 81 kDa complex and likely represents a β-tetramer. Significantly, no species characteristic of an assembled holoenzyme complex with a (αaΔT/αbΔT)2β2 composition was observed at the tested protein concentrations. Addition of 50 µM dATP or other nucleotides had no effect on promoting a (αaΔT/αbΔT)2β2 complex (Figure 3E and Supplementary Figure S3).
To test if higher concentrations of dATP would promote complex formation, we turned to surface plasmon resonance experiments that are more tolerant of higher nucleotide concentrations than GEMMA. In these experiments, Aeh1 NrdB was immobilized to the biosensor chip (18). With the split wild-type NrdA proteins, the KD for interaction with NrdB was estimated to be 0.18 µM in absence of added nucleotide effectors (Table 2; Supplementary Figure S4). Notably, addition of increasing amounts of dATP greatly stimulated the interaction, with the addition of 1 mM dATP resulting in a KD value of 0.007 µM. In contrast, the affinity constant for the interaction between the mutant ΔT NrdA proteins and NrdB was 1.2 µM and did not change significantly in the presence of 1 mM dATP (Table 2). Thus, two independent experimental approaches show that holoenzyme assembly is compromised with the ΔT mutant proteins, providing an explanation for the 2000-fold lower enzymatic activity relative to the wild-type NrdA proteins.
Table 2.
Effect of dATP on interaction of wild-type and ΔT versions of NrdA-a and NrdA-b with NrdB as measured by surface plasmon resonance (See also Supplementary Figure S4)
| dATP (mM) |
KD (µM) |
|
|---|---|---|
| NrdA-a/NrdA-b | NrdA-aΔT/NrdA-bΔT | |
| 0 | 0.18 ± 0.04 | 1.2 ± 0.3 |
| 1 | 0.007 ± 0.0003 | 1.4 ± 0.03 |
DISCUSSION
Molecular lego: assembly of a fragmented active site by protein interaction domains
Collectively, our data indicate that the unique tail sequences fused to the C- and N-termini of NrdA-a and NrdA-b are essential for enzymatic activity. We show that the tails are not required for interaction of the NrdA-a and NrdA-b fragments, as the ΔT mutants do not affect assembly of the αa/αb heterodimer. Rather, our data are consistent with a model whereby the tails function as interaction domains to promote assembly of the large subunit (αa/αb)2 dimer of heterodimers (Figures 4 and 5). Modeling of the split Aeh1 NrdA-a and NrdA-b proteins using the E. coli NrdA structure reveals that the tail sequences could lie in proximity to two α-helices verified by mutational studies of the E. coli protein as the major dimerization determinants between NrdA monomers (Figure 4) (13,14,19). The requirement for the tail sequences to assemble an (αa/αb)2 dimer of heterodimers clearly implies that the Aeh1 NrdA dimer interface was compromised by the mobE insertion. Although the molecular details of how the tails function remains to be elucidated, we note that the C-terminal tail of NrdA-a is positively charged (pI = 10.4), while the N-terminal tail of NrdA-b is negatively charged (pI = 3.8), suggesting that assembly of the (αa/αb)2 large subunit could be driven by direct charge–charge interactions between the tails, by interactions between the tails and opposing NrdA subunits or by a combination of both types of interactions. The proximity of the tails to active site residues on NrdA-a and NrdA-b is compelling and we cannot discount the possibility that the tails also function to promote assembly of the composite active site.
Figure 4.

The dimer interface between two E. coli NrdA monomers, colored to indicate the Aeh1 αa (yellow) and αb (green) polypeptides (modified from PDB file 4R1R). Atoms corresponding to Cys431 and Glu433 in NrdA-a and Cys31 in NrdA-b are shown as red spheres and the position of the mobE insertion is indicated in the model. Also shown are the substrate GDP (blue) and the allosteric effector dTTP (pink).
Figure 5.

Summary of the genetic organization, expression and assembly of the fragmented class Ia RNR from phage Aeh1. The map of the Aeh1 RNR operon (gDNA) shows an early phage promoter (right facing arrow) that drives expression of the operon (32). The NrdA-a, NrdA-b and NrdB polypeptides are independently translated from this message (mRNA) (8), while expression of MobE is inhibited by an RNA secondary structure (33). Our current data indicate that the NrdA-a and NrdA-b polypeptides self assemble to form the αa/αb heterodimer (active site residues depicted as spheres), while dimerization to form the (αa/αb)2 large subunit is stimulated by dATP. The ΔT mutants are defective in dimerization of the αa/αb heterodimer. Holoenzyme formation is stimulated by dATP and includes the small subunit dimer, β2. The structures of the individual polypeptides were modified from the E. coli NrdA (PBD file 4R1R) and NrdB (PDB file 1AV8) structures. In the holoenzyme model [based on Ref. (34)], the (αa/αb)2 large subunit is rotated 90° vertically relative to the free large subunit and the β2 subunit is rotated 90° horizontally relative to the free dimer.
Regardless of the mechanism by which the tails function, it is clear that they render the insertion of mobE phenotypically neutral with respect to NrdA function. Assembly of the fragmented RNR active site described here is distinct from other examples of composite active sites assembled from residues on separate polypeptides (20,21), because the mobE insertion has, in essence, provided the molecular antidote to fragmentation in the form of the tails that reassemble the split large subunit. Precedent for short peptides functioning as protein interaction domains stems from observations showing that protein fragments can be assembled by short unrelated peptides present as N- and C-terminal extensions (22,23).
What is the evolutionary origin of the mobE insertion and tail sequences? The observation that mobE is located in the intergenic region separating the nrdA and nrdB genes of a number of T-even like phages, except for Aeh1, strongly suggests that a transposition-like event created the split nrdA-a/nrdA-b genes of phage Aeh1 (8,24). Moreover, a number of fragmented genes are associated with homing endonucleases (5,7,25,26), lending support to the notion that genome rearrangements caused by homing endonucleases may occur more frequently than previously appreciated. Such transposition events could also explain the origin of the tail sequences, which would have been inherited with the endonuclease gene during the initial recombination event. We previously argued that natural selection would select for phage variants in which a mechanism arose to restore a functional RNR, as those phage would exhibit a significant replicative advantage (8). Acquisition of the foreign DNA associated with the mobE insertion, followed by fusion to the surrounding nrdA-a and nrdA-b gene fragments and selection for oppositely charged peptide sequences that promoted assembly of the polypeptides is one possible evolutionary scenario that could explain the origin and persistence of the tail sequences. Strikingly, similar observations regarding the charge distribution of peptides that promoted the interaction of split proteins were found from experiments that fused short random DNA fragments to fragmented genes to restore function (27–29), suggesting that acquisition of novel function by randomly acquired DNA is not an evolutionary bottleneck. Alternatively, we note that RNR genes in bacterial and phage genomes are often interrupted by self-splicing introns and inteins (10,11,30). Some of these elements also encode HNH family homing endonucleases similar to mobE, suggesting that the tail sequences and mobE could be remnants of a degenerate self-splicing intein analogous to the split inteins associated with fragmented genes recently identified from metagenomic data (7). Intriguingly, the protein–protein interactions necessary for splicing of split inteins were proposed to occur through electrostatic interactions between the N- and C-intein fragments (7,31), paralleling our hypothesis for the function of the NrdA-a and NrdA-b tails.
Tail-like sequences similar to those present on NrdA-a and NrdA-b may be associated with other fragmented genes, but their significance overlooked. Only detailed biochemical analyses such as those described here could distinguish the tails as functionally critical from variable length N- and C-termini with no presumed function. We would also anticipate that tail-like sequences are optimized to reassemble different protein fragments and would thus vary in sequence, making the tails difficult to detect by similarity-based searches. One notable example is the mobA insertion that splits the well-characterized type II topoisomerase of phage T4 into two smaller genes (Supplementary Figure S5). Intriguingly, the two split gene products, gp39 and gp60, possess tail-like extensions. While the gp39 and gp60 tails show little sequence similarity to the NrdA-a and NrdA-b tails, they exhibit a similar charge distribution and it is tempting to speculate that the gp39 and gp60 tails function analogously to assemble the T4 topoisomerase. The occurrence of similar tail-like sequences in different biological systems may represent a convergent evolutionary solution to promote the assembly of protein fragments within the context of a cellular environment.
In summary, our results highlight the involvement of mobile elements in promoting recombination events that influence gene structure and function and also describe a novel molecular solution for assembly of a fragmented coding region that does not involve RNA or protein splicing, or programmed DNA rearrangements. Although the tail sequences we describe here are associated with a particular class of mobile element, tail-like sequences could potentially reassemble coding regions fragmented by a variety of mobile elements, or by recombination events not involving mobile elements.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research (MOP 97780 to D.R.E.); the Swedish Research Council (to B.-M.S.); Carl Trygger’s Foundation (to A.H.). Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Greg Gloor and David Haniford for reading of the manuscript and Lee-Ann Briere for assistance in analyzing ultracentrifugation data.
REFERENCES
- 1.Swithers KS, Senejani AG, Fournier GP, Gogarten JP. Conservation of intron and intein insertion sites: implications for life histories of parasitic genetic elements. BMC Evol. Biol. 2009;9:303. doi: 10.1186/1471-2148-9-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belfort M, Derbyshire V, Cousineau B, Lambowitz A. Mobile introns: pathways and proteins. In: Craig N, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. NY: ASM Press; 2002. pp. 761–783. [Google Scholar]
- 3.Waters E, Hohn MJ, Ahel I, Graham DE, Adams MD, Barnstead M, Beeson KY, Bibbs L, Bolanos R, Keller M, et al. The genome of Nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc. Natl Acad. Sci. USA. 2003;100:12984–12988. doi: 10.1073/pnas.1735403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelman Z, Pietrokovski S, Hurwitz J. Isolation and characterization of a split B-type DNA polymerase from the archaeon Methanobacterium thermoautotrophicum ΔH. J. Biol. Chem. 1999;274:28751–28761. doi: 10.1074/jbc.274.40.28751. [DOI] [PubMed] [Google Scholar]
- 5.Petrov VM, Ratnayaka S, Karam JD. Genetic insertions and diversification of the PolB-type DNA polymerase (gp43) of T4-related phages. J. Mol. Biol. 2010;395:457–474. doi: 10.1016/j.jmb.2009.10.054. [DOI] [PubMed] [Google Scholar]
- 6.Gorbalenya AE. Non-canonical inteins. Nucleic Acids Res. 1998;26:1741–1748. doi: 10.1093/nar/26.7.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dassa B, London N, Stoddard BL, Schueler-Furman O, Pietrokovski S. Fractured genes: a novel genomic arrangement involving new split inteins and a new homing endonuclease family. Nucleic Acids Res. 2009;37:2560–2573. doi: 10.1093/nar/gkp095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich NC, Torrents E, Gibb EA, Sahlin M, Sjöberg B-M, Edgell DR. Insertion of a homing endonuclease creates a genes-in-pieces ribonucleotide reductase that retains function. Proc. Natl Acad. Sci. USA. 2007;104:6176–6181. doi: 10.1073/pnas.0609915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nordlund P, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 10.Landthaler M, Begley U, Lau NC, Shub DA. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res. 2002;30:1935–1943. doi: 10.1093/nar/30.9.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nord D, Sjöberg B-M. Unconventional GIY-YIG homing endonuclease encoded in group I introns in closely related strains of the Bacillus cereus group. Nucleic Acids Res. 2008;36:300–310. doi: 10.1093/nar/gkm1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarevic V, Soldo B, Dusterhoft A, Hilbert H, Mauel C, Karamata D. Introns and intein coding sequence in the ribonucleotide reductase genes of Bacillus subtilis temperate bacteriophage SPβ. Proc. Natl Acad. Sci. USA. 1998;95:1692–1697. doi: 10.1073/pnas.95.4.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uhlin U, Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 14.Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg B-M, Eklund H. Binding of allosteric effectors to ribonucleotide reductase protein R1: reduction of active-site cysteines promotes substrate binding. Structure. 1997;5:1077–1092. doi: 10.1016/s0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 15.Lundin D, Torrents E, Poole AM, Sjöberg B-M. RNRdb, a curated database of the universal enzyme family ribonucleotide reductase, reveals a high level of misannotation in sequences deposited to Genbank. BMC Genomics. 2009;10:589. doi: 10.1186/1471-2164-10-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rofougaran R, Crona M, Vodnala M, Sjöberg B-M, Hofer A. Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 2008;283:35310–35318. doi: 10.1074/jbc.M806738200. [DOI] [PubMed] [Google Scholar]
- 17.Bacher G, Szymanski WW, Kaufman SL, Zollner P, Blaas D, Allmaier G. Charge-reduced nano electrospray ionization combined with differential mobility analysis of peptides, proteins, glycoproteins, noncovalent protein complexes and viruses. J. Mass Spectrom. 2001;36:1038–1052. doi: 10.1002/jms.208. [DOI] [PubMed] [Google Scholar]
- 18.Crona M, Furrer E, Torrents E, Edgell DR, Sjöberg B-M. Subunit and small-molecule interaction of ribonucleotide reductases via surface plasmon resonance biosensor analyses. Protein Eng. Des. Sel. 2010;23:633–641. doi: 10.1093/protein/gzq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birgander PL, Bug S, Kasrayan A, Dahlroth SL, Westman M, Gordon E, Sjöberg B-M. Nucleotide-dependent formation of catalytically competent dimers from engineered monomeric ribonucleotide reductase protein R1. J. Biol. Chem. 2005;280:14997–15003. doi: 10.1074/jbc.M500565200. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature. 2006;442:831–835. doi: 10.1038/nature04975. [DOI] [PubMed] [Google Scholar]
- 21.Trotta CR, Paushkin SV, Patel M, Li H, Peltz SW. Cleavage of pre-tRNAs by the splicing endonuclease requires a composite active site. Nature. 2006;441:375–377. doi: 10.1038/nature04741. [DOI] [PubMed] [Google Scholar]
- 22.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl Acad. Sci. USA. 1998;95:12141–12146. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat. Struct. Mol. Biol. 2005;12:403–407. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- 24.Sandegren L, Nord D, Sjöberg B-M. SegH and Hef: two novel homing endonucleases whose genes replace the mobC and mobE genes in several T4-related phages. Nucleic Acids Res. 2005;33:6203–6213. doi: 10.1093/nar/gki932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paquin B, Laforest MJ, Lang BF. Interspecific transfer of mitochondrial genes in fungi and creation of a homologous hybrid gene. Proc. Natl Acad. Sci. USA. 1994;91:11807–11810. doi: 10.1073/pnas.91.25.11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sethuraman J, Majer A, Friedrich NC, Edgell DR, Hausner G. Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol. Biol. Evol. 2009;26:2299–2315. doi: 10.1093/molbev/msp145. [DOI] [PubMed] [Google Scholar]
- 27.Kaiser CA, Preuss D, Grisafi P, Botstein D. Many random sequences functionally replace the secretion signal sequence of yeast invertase. Science. 1987;235:312–317. doi: 10.1126/science.3541205. [DOI] [PubMed] [Google Scholar]
- 28.Ruden DM, Ma J, Li Y, Wood K, Ptashne M. Generating yeast transcriptional activators containing no yeast protein sequences. Nature. 1991;350:250–252. doi: 10.1038/350250a0. [DOI] [PubMed] [Google Scholar]
- 29.Ma J, Ptashne M. A new class of yeast transcriptional activators. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- 30.Perler FB. InBase: the Intein Database. Nucleic Acids Res. 2002;30:383–384. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dassa B, Amitai G, Caspi J, Schueler-Furman O, Pietrokovski S. Trans protein splicing of cyanobacterial split inteins in endogenous and exogenous combinations. Biochemistry. 2007;46:322–330. doi: 10.1021/bi0611762. [DOI] [PubMed] [Google Scholar]
- 32.Gibb EA, Edgell DR. Multiple Controls Regulate the Expression of mobE, an HNH Homing Endonuclease Gene Embedded within a Ribonucleotide Reductase Gene of Phage Aeh1. J. Bacteriol. 2007;189:4648–4661. doi: 10.1128/JB.00321-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibb EA, Edgell DR. An RNA Hairpin Sequesters the Ribosome Binding Site of Homing Endonuclease mobE Gene. J. Bacteriol. 2009;191:2409–2413. doi: 10.1128/JB.01751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.