

Abstract
This report examines the molecular mechanism by which high-fidelity DNA polymerases select nucleotides during the replication of an abasic site, a non-instructional DNA lesion. This was accomplished by synthesizing several unique 5-substituted indolyl 2′-deoxyribose triphosphates and defining their kinetic parameters for incorporation opposite an abasic site to interrogate the contributions of π-electron density and solvation energies. In general, the Kd, app values for hydrophobic non-natural nucleotides are ∼10-fold lower than those measured for isosteric hydrophilic analogs. In addition, kpol values for nucleotides that contain less π-electron densities are slower than isosteric analogs possessing higher degrees of π-electron density. The differences in kinetic parameters were used to quantify the energetic contributions of desolvation and π-electron density on nucleotide binding and polymerization rate constant. We demonstrate that analogs lacking hydrogen-bonding capabilities act as chain terminators of translesion DNA replication while analogs with hydrogen bonding functional groups are extended when paired opposite an abasic site. Collectively, the data indicate that the efficiency of nucleotide incorporation opposite an abasic site is controlled by energies associated with nucleobase desolvation and π-electron stacking interactions whereas elongation beyond the lesion is achieved through a combination of base-stacking and hydrogen-bonding interactions.
INTRODUCTION
The central dogma of DNA replication is that the efficiency and fidelity of this process is dependent upon the formation of correct hydrogen-bonding interactions between an incoming nucleotide and its templating partner (1). The mutual recognition of adenine (A) by thymine (T) and of guanine (G) by cytosine (C) involves the ability of the DNA polymerase to consummate the formation of precise hydrogen-bonding interactions between each partner. The specific pairing of these hydrogen bonding groups provide high-fidelity DNA polymerases with enough flexibility to accurately recognize four distinct pairing partners (A:T, G:C, T:A and C:G), yet remain stringent enough to maintain exquisite genomic fidelity.
Under normal conditions, replicative DNA polymerases use hydrogen-bonding interactions to generate low error frequencies of approximately one mistake every 106 opportunities (2–4). However, polymerization fidelity and efficiency can become altered when the chemical composition of the templating strand is modified by DNA damaging agents such as methylmethane sulfonate and nitrosurea (5). Since these agents alter hydrogen-bonding information, the risk of mutagenesis increases as the frequency of misincorporation events is enhanced. Mutagenesis, in some cases, can benefit an organism by generating genomic diversity required for evolution and/or adaptation (6). However, mutagenesis in humans is typically associated with disease development, most notably cancer (7,8). To better understand the underlying origins of disease development, it is important to define how genomic errors are generated by misreplicating damaged DNA.
One commonly formed DNA lesion is an abasic site that, due to the lack of coding information, is highly pro-mutagenic (9). Although abasic sites are non-instructional, high-fidelity DNA polymerases such as the bacteriophage T4 DNA polymerase preferentially utilize dATP (10). This phenomenon is commonly referred to as the ‘A-rule’ of translesion DNA synthesis (11–18). While the mutagenic consequences of the ‘A-rule’ are well documented (19–21), the mechanistic basis for the preferential utilization of dATP has not been firmly established. Our investigations of the ‘A-rule’ have focused on characterizing the kinetic behavior of the library of non-natural nucleotides illustrated in Figure 1A during insertion opposite this DNA lesion (22–28). While these analogs mimic the core structure of dATP, the introduction of various functional groups to the 5-position of the indole ring provides a way to systematically modulate biophysical features such as shape, size, hydrophobicity, dipole moment and π-electron surface area. The kinetic parameters for their utilization were used to develop a structure–activity relationship (SAR) defining key biophysical features required for optimal incorporation opposite an abasic site (Figure 1B) (23). The parabolic nature of this plot reveals that the catalytic efficiency (kpol/Kd, app) for nucleotide incorporation is dependent upon an optimal π-electron surface area that most closely mimics that of a natural Watson–Crick base pair.
Figure 1.

(A) Library of non-natural nucleotides used to interrogate the mechanism accounting for the preferential utilization of dATP during the replication of an abasic site. For clarity, only the nucleobases of the 5-substituted indolyl nucleotides are shown. (B) SAR highlighting the importance of π-electron surface area of the incoming nucleotide for efficient incorporation opposite an abasic site. See text for details.
This article further examines this model by quantifying the energetic contributions required for nucleotide binding and insertion of isosteric analogs of two unique non-natural nucleotides, 5-NITP (22) and 5-PhITP (27), that are themselves incorporated opposite an abasic site with high catalytic efficiencies >106 M−1s−1. The strategy employed here is to modulate the degree of π-electron density and hydrophobicity without compromising the overall shape, size or conformation of the parental nucleotide. Kinetic analyses indicate that the binding affinity of the incoming nucleotide, designated as Kd, app, is linked with the overall shape and hydrophobicity of the incoming nucleotide whereas the maximal rate constant for incorporation or kpol value depends heavily on the presence of π-electron density. These results are used to propose an enzymatic mechanism accounting for the roles of desolvation, steric fit and π-electron density during translesion DNA synthesis. In addition, elongation beyond the abasic site is dependent upon a combination of base-stacking properties and hydrogen-bonding potential of the nucleotide that is paired opposite the DNA lesion. This final aspect has important implications toward the rational design of novel nucleotides that can be used as chemical probes for DNA synthesis, for expanding the genetic code and/or as potential therapeutic agents.
MATERIALS AND METHODS
Reagents and general methods: anhydrous sodium methoxide, phosphoryl oxychloride, tetrakis triphenylphosphine palladium [Pd(PPh3)4] and 1,8-bis(dimethylamino)naphthalene were purchased from ACROS. Sodium hydride (60% dispersion in mineral oil), tributylammonium pyrophosphate and dimethoxyethane (DME) were purchased from Sigma-Aldrich. Trimethylphosphate and tributylamine were purified via fractional distillation under reduced pressure. Acetonitrile was purified by refluxing with CaH2 for 2 h followed by simple distillation. DMF was purified by distillation with ninhydrin. All anhydrous solvents were dried over 4 Å molecular sieves and stored under argon. All other solvents and reagents were purchased at the highest purity. Water-sensitive reactions were performed in oven-dried glassware under argon. The intermediates and products were characterized by nuclear magnetic resonance (NMR), UV–vis absorbance and mass spectrometry. 1H, 31P and 19F NMR spectra were recorded in a Varian-400 FT NMR spectrometer. All chemical shifts are reported in ppm and the coupling constants are in Hertz (Hz). 1H NMR spectra were taken in deuterated chloroform (CDCl3), or dimethylsulfoxide (DMSO-d6) using tetramethylsilane (TMS) as the external reference. 19F-NMR spectra were taken in CDCl3 or D2O and trichlorofluoromethane was used as the external standard. 31P-NMR spectra were taken in D2O with 50 mM Tris–HCl and 2 mM EDTA and phosphoric acid (85%) was used as the external standard. High-resolution electrospray ionization mass spectrometry (Hi-Res ESI-MS) was performed on an IonSpec HiRes ESI-FTICRMS at the University of Cincinnati Mass Spectrometry facility. The configuration of the nucleoside was confirmed by 1H NMR NOE difference spectroscopy on a Varian Inova 600 NMR instrument. Thin layer chromatography was carried out on a Whatman Silica Gel UV254 plates. Column chromatography was performed on a Fisher Scientific Silica Gel, sizes 32–63. HPLC analysis and purification were carried out using the JASCO Model PU-2089 instrument with the reverse phase C-18 Vydac column. UV absorption of the nucleosides used for their quantification was performed on a Cary 50 spectrophotometer.
Method A: synthesis of 5-substituted indolyl 2′-deoxynucleosides
To a solution of 5-substituted indole nucleobase (1 eq) dissolved in dry acetonitrile (50 ml) was added NaH (60% dispersion in mineral oil) (5 eq). The reaction mixture was stirred at room temperature for an hour under argon prior to the addition of the Hoffer’s chlorosugar (1.5 eq) prepared using established protocols (29,30). The resulting mixture was further stirred at room temperature under argon for 16 h and immediately rota-evaporated to dryness. The remaining solid residue was re-suspended in dry methanol followed by the addition of anhydrous sodium methoxide until the pH of the reaction mixture reaches 12. The reaction mixture was again stirred for 16 h at room temperature under argon before being evaporated to dryness under reduced pressure. The crude product was purified by silica flash column chromatography.
Synthesis of 1-(2-deoxy-β-d-erythropentafuranosyl)-5-methylcarboxylate-1H-indole 1
This compound was prepared using method A with 5-methylcarboxylate indole (300 mg, 1.7 mmol, 1 eq) as the starting material. The crude product was purified by silica flash column chromatography (10–25% methanol in chloroform). The isolated product was a light brown foam and the yield was 330 mg. 1H NMR (DMSO, 400 MHz, 298 K, TMS) δ: 2.23–2.28 (m, 1H, 2′-H), 2.47–2.54 (m, 1H, 2′-H), 3.48–3.56 (m, 2H, 5′-H), 3.83–3.85 (m, 1-H, 4′-H), 3.85 (s, 3H, –OCH3), 4.35–4.36 (m, 1H, 3′-H), 4.93 (t, J = 5.93 Hz, 1H, 5′-OH), 5.32 (d, J = 4.1 Hz, 1H, 3′-OH), 6.43 (tapp, J = 6.8 Hz, 1H, 1′-H), 6.67 (d, J = 3.13 Hz, 1H, Ar), 7.69 (d, J = 8.9 Hz, 1H, Ar), 7.73 (d, J = 3.5 Hz, 1H, Ar), 7.77 (dd, J = 8.7 Hz, 1.6 Hz, 1H, Ar), 8.25 (d, J = 1.1 Hz, 1H, Ar). UV (MeOH) λmax (nm) 240 (ε = 104 591 cm−1 M−1). HiRes ESI-MS (+): calculated mass spectrum formula C15H17NO5 for M+Na: 314.1004; experimental mass spectrum: 314.1017
Synthesis of 1-(2-deoxy-β-d-erythropentafuranosyl)-5-carboxylate-1H-indole 2
This compound was prepared starting from 1 (204 mg, 0.7 mmol) dissolved in methanol (10 ml). To this reaction mixture was added NaOH (220 mg, 5.5 mmol) solution dissolved in 1:1 H2O:MeOH solvent (10 ml). The reaction mixture was stirred vigorously at room temperature and the reaction was monitored by TLC using 3:1 Hexane:EtOAc as the solvent system. Conversion to the carboxylic acid derivative was visualized via bromocresol green TLC staining (31). After 72 h, 0.5 N HCl was added to the reaction mixture until pH <6, followed by extraction with 20 ml of ethyl acetate (3×). The organic layers were combined and dried with Na2SO4 and evaporated to dryness under reduced pressure. The resulting product was a brownish crystalline solid and the yield was 200 mg. 1H NMR (DMSO, 400 MHz, 298 K, TMS) δ: 2.18–2.24 (m, 1H, 2′-H), 2.45–2.48 (m, 1H, 2′-H), 3.44–3.51 (m, 2H, 5′-H), 3.78–3.81 (m, 1-H, 4′-H), 4.30–4.33 (m, 1H, 3′-H), 4.88 (br s, 1H, 5′-OH), 5.73 (br s, 1H, 3′-OH), 6.37–6.40 (tapp, J = 7.1 Hz, 1H, 1′-H), 6.62 (d, J = 3.8 Hz, 1H, Ar), 7.62 (d, J = 8.8 Hz, 1H, Ar), 7.67 (d, J = 3.5 Hz, 1H, Ar), 7.72 (dd, J = 8.7 Hz, 1.6 Hz, 1H, Ar) 8.18 (d, J = 1.4 Hz, 1H, Ar), 12.5 (br s, 1H, –COOH). UV (MeOH) λmax (nm) 235 (ε = 36 074 cm−1 M−1). HiRes ESI-MS (+): calculated mass spectrum formula C14H15NO5 for M+H: 278.1028; experimental mass spectrum: 278.1027.
Synthesis of 5-perfluorophenylindole 5
To the mixture of 1-bromo-2,3,4,5,6-pentafluorobenzene (1.25 g, 5 mmol), Pd(PPh3)4 (0.17 g, 0.15 mmol) in DME (20 ml) was added 5-boronic acid indole (0.89 g, 5.5 mmol) followed by sodium bicarbonate (1.26 g, 15 mmol) in water (15 ml). The reaction mixture was refluxed for 3 h and cooled to room temperature to quench the reaction. DME was immediately evaporated under reduced pressure followed by the addition of ether (50 ml) to the two-layer mixture for extraction. The aqueous phase was extracted with ether (3×) and the organic phases were pooled, dried over MgSO4, filtered and evaporated to dryness. The crude product was purified by flash column chromatography (30–50% ethyl acetate in hexane). 5-perfluorophenylindole was a yellow solid (1.06 g, 74% yield). 1H NMR (CDCl3, 400 MHz, 298 K, TMS) δ: 6.62–6.63 (m, 1H, 3-H), 7.22 (dd, J = 4.0, 1.2 Hz, 1H, 7-H), 7.29 (tapp, J = 2.8 Hz, 1H, 2-H), 7.50 (d, J = 8.4 Hz, 1H, 6-H), 7.71 (s, 1H, 4-H), 8.30 (br s, 1H, 1-H). 19F-NMR (CDCl3, 376 MHz, 293 K, CCl3F) δ: −163.4–−163.2 (m, 2F), −157.6 (t, 1F, J = 21.1 Hz), −143.8 (dd, 2F, J = 31.7, 7.9 Hz). HiRes ESI-MS (+): calculated mass spectrum formula C14H6F5N for M+H: 284.0499; experimental mass spectrum: 284.0493.
Synthesis of 1-(2-deoxy-β-d-erythropentafuranosyl)-5-perfluorophenyl-1H-indole 6
This compound was prepared as described in method A using 5 (300 mg, 1.7 mmol, 1 eq) as the starting material. The crude product was purified by silica flash column chromatography using hexane:ethyl acetate (1:2) as eluent. The isolated product was a light brown solid and the yield was 445 mg. 1H NMR (DMSO, 400 MHz, 298 K, TMS) δ: 2.15–2.20 (m, 1H, 2′-H), 2.42–2.48 (m, 1H, 2′-H), 3.41–3.49 (m, 2H, 5′-H), 3.74–3.77 (m, 1-H, 4′-H), 4.29–4.33 (m, 1H, 3′-H), 4.83 (br s, 1H, 5′-OH), 5.23 (br s, 1H, 3′-OH), 6.35 (tapp, J = 6.47 Hz, 1H, 1′-H), 6.52–6.53 (d, J = 3.28 Hz, 1H, 3-H), 7.15 (d, J = 8.80 Hz, 1H, Ar), 7.58–7.62 (m, 3H, Ar). 19F-NMR (CDCl3, 376 MHz, 293 K, CCl3F) δ: −163.2–−163.1 (m, 2F), −157.3 (t, 1F, J = 21.1 Hz), −143.8 (dd, 2F, J = 23.7, 8.7 Hz). UV (MeOH) λmax (nm) 248 (ε = 18 437 cm−1 M−1). HiRes ESI-MS (+): calculated mass spectrum formula C19H14F5NO3 for M+H: 400.0972; experimental mass spectrum: 400.0977.
Synthesis of 1-(2-deoxy-β-d-erythropentafuranosyl)-5-(2,3,5,6-tetrafluoro-4-methoxyphenyl)-1H-indole 7
This compound was prepared based on a slightly modified protocol (32) starting from 6 (300 mg, 0.8 mmol) dissolved in ether (5 ml). To this reaction mixture was added methanol (45 ml) and sodium methoxide until the pH of the solution is >12. The reaction mixture was stirred for 36 h at room temperature under argon and the product formation was monitored by 19F-NMR. This process afforded 70% conversion of the starting material to the desired product which was a yellowish brown solid and the yield was 243 mg. 1H NMR (DMSO, 400 MHz, 298 K, TMS) δ: 2.14–2.20 (m, 1H, 2′-H), 2.41–2.43 (m, 1H, 2′-H), 3.40–3.49 (m, 2H, 5′-H), 3.74–3.77 (m, 1-H, 4′-H), 4.01 (s, 3H, –OCH3), 4.26–4.29 (m, 1H, 3′-H), 4.83 (t, J = 8.0 Hz, 1H, 5′-OH), 5.22 (d, J = 4.0 Hz, 1H, 3′-OH), 6.34 (tapp, J = 7.4 Hz, 1H, 1′-H), 6.51 (d, J = 4.0 Hz, 1H, 3-H), 7.13 (dd, J = 8.7 Hz, 1.4 Hz, 1H, Ar), 7.58–7.64 (m, 4H, Ar). 19F-NMR (CDCl3, 376 MHz, 293 K, CCl3F) δ: −159.0 (dd, 2F, J = 24.1, 8.6 Hz), −145.6 (dd, 2F, J = 23.7, 10.2 Hz). UV (MeOH) λmax (nm) 250 (ε = 30 342 cm−1 M −1). HiRes ESI-MS (+): calculated mass spectrum formula C20H17F4NO4 for M+Na: 434.0992; experimental mass spectrum: 434.0982
Method B: synthesis of non-natural deoxynucleoside triphosphates
The triphosphates were prepared as previously described (24,25,28,33) starting from the corresponding nucleosides. Triphosphorylation was initiated by forming the 5′-monophosphorodichloridated intermediate by the dropwise addition of POCl3 in the reaction mixture containing the 2′deoxyribonucleoside (0.07 mmol) and 1,8-bis(dimethylamino)naphthalene (0.11 mmol) dissolved in 0.37 ml of trimethylphosphate pre-chilled at 0°C. This mixture was stirred for 3 h under argon and was then spontaneously treated with 0.5 M DMF solution of tributylammonium pyrophosphate (0.37 mmol) and tributylamine (0.37 mmol) and further stirred for 15 min at room temperature. Tributylammoniumbicarbonate (1 M TEAB, pH 7.5) was added to quench the reaction and stirred at room temperature for 2 h to yield the nucleotide. The course of the reaction was monitored by TLC using the solvent system of 1-propanol:ammonium hydroxide:water (6:3:1). The crude product was concentrated by rota-evaporation under reduced pressure prior to purification by preparative reverse phase HPLC using 0.1 M TEAB as mobile phase A and 50% ACN in 0.1 M TEAB as mobile phase B. The desirable nucleotides were lyophilized to dryness and characterized by mass spectrometry and NMR. The concentration of the triphosphate was determined using the extinction coefficient at the UV λmax for the corresponding nucleoside (34).
1-(2-Deoxy-β-d-erythropentafuranosyl)-5-methylcarboxylate-1H-indole triphosphate 3
Using 1 as the starting material, 3 was prepared and purified as described in method B. 31P-NMR (D2O, 162 MHz, H3PO4) δ: −5.4 (d, γ-P), −10.1 (d, α-P), −21.2 (t, β-P). HiRes ESI-MS (−): calculated mass spectrum formula C15H20NO14P3 for M-H: 530.0024; experimental mass spectrum: 530.0036.
1-(2-Deoxy-β-d-erythropentafuranosyl)-5-carboxylate-1H-indole triphosphate 4
Compound 3 was converted to 4 by reacting with 4 N NaOH in methanol/water (2:1) overnight at room temperature in the presence of 15-crown-5 ether. The course of the reaction was monitored by TLC using 1-propanol:ammonium hydroxide:water (6:3:1) as the solvent system for elution and the conversion of the methyl carboxylate to the carboxylate moiety was visualized via bromocresol green staining which detects carboxylic acid (31). Reverse-phase HPLC was also used to determine the progression of the deprotection process. 5-MeCITP was eluted at 46% B (13 min retention time) using a C-18 analytical column from Vydac; buffer A: 0.1 M TEAB; buffer B: 50%ACN in 0.1 M TEAB, linear gradient of 20–50% within 15 min at a flow rate of 1 ml min−1 and monitored at a wavelength of 235 nm. Using the same conditions, 5-CITP was eluted at 27% B (3.5 min retention time). 31P-NMR (D2O, 162 MHz, H3PO4) δ: −4.9 (d, γ-P), −9.9 (d, α-P), −20.8 (t, β-P). HiRes ESI-MS (−): calculated mass spectrum formula C14H16NO14P3Na for M-H: 537.96868; experimental mass spectrum: 537.96875.
1-(2-Deoxy-β-d-erythropentafuranosyl)-5-perfluorophenyl-1H-indole triphosphate 8
Using 6 as the starting material, this compound was prepared and purified as described in method B. 19F-NMR (D2O, 376 MHz, 293 K, CCl3F) δ: −164.31–−164.16 (m, 2F); −158.15 (t, J = 21.6 Hz, 1F), −145.2 (dd, J = 24.4 Hz, 8.6 Hz, 2F). 31P-NMR (D2O, 162 MHz, 293 K, H3PO4) δ: −5.8 (d, γ-P), −10.2 (d, α-P), −21.4 (t, β-P). HiRes ESI-MS (−): calculated mass spectrum for formula C19H16F5NO12P3 for M-H: 637.9811; experimental mass spectrum: 637.9809
1-(2-Deoxy-β-d-erythropentafuranosyl)-5-(2,3,5,6-tetrafluoro-4-methoxyphenyl)-1H-indole triphosphate 9
Using 7 as the starting material, this compound was prepared and purified as described in method B. 19F-NMR (D2O, 376 MHz, 293 K, CCl3F) δ: −159.50 (dd, J = 24.3 Hz, 8.9 Hz, 2F), −146.72 (dd, J = 23.5 Hz, 9.1 Hz, 2F). 31P-NMR (D2O, 162 MHz, 293 K, H3PO4) δ: −5.6 (d, γ-P), −10.1 (d, α-P), −21.2 (t, β-P). HiRes ESI-MS (−): calculated mass spectrum for formula C20H20F4NO13P3 for M-H: 650.0011; experimental mass spectrum: 650.0001.
Enzymatic assays
Materials
Magnesium acetate and Trizma base were from Sigma. [γ-32P]-ATP was purchased from Perkin Elmer Life and Analytical Sciences (Boston, MA, USA). Boric acid, urea, acrylamide and bis-acrylamide were from National Diagnostics (Rochester, NY, USA). Oligonucleotides, including those containing a tetrahydrofuran moiety mimicking an abasic site, were synthesized by Operon Technologies (Almeda, CA, USA). All other materials were obtained from commercial sources and were of the highest quality available. The exonuclease-deficient mutant of gp43 (Asp-219 to Ala mutation) was purified and quantified as previously described (35,36). Single-stranded and duplex DNA substrates used in this study were purified and quantified as previously described (35) and the 5′-ends of the DNA substrates were 32P-radiolabeled using a conventional reaction protocol with γ-32P-ATP and T4 polynucleotide kinase (GibcoBRL).
Enzyme assays
The assay buffer used in all kinetic studies with the exonuclease-deficient bacteriophage T4 DNA polymerase consisted of 25 mM Tris–OAc (pH 7.5), 150 mM KOAc and 10 mM 2-mercaptoethanol. All enzymatic assays were performed at 25°C and polymerization reactions were monitored by analysis of the product in 20% sequencing gels as previously described (37). Gel images were obtained with a PhosphorImager instrument using the Optiquant software supplied by the manufacturer. Product formation was quantified by calculating the ratio of extended (14-mer) and non-extended (13-mer) 32P-radiolabeled primer which was corrected for substrate in the absence of polymerase (zero point). The total product was derived by multiplying the corrected ratios with the total concentration of primer/template used in each assay. All concentrations are listed as final solution concentrations.
Nucleotide incorporation assays
A rapid quench instrument (KinTek Corporation, Clarence, PA) was employed to monitor the time course in nucleotide incorporation by gp43 exo− opposite a single- and double abasic sites. kpol and Kd, app values were determined using single-turnover reaction conditions in which excess gp43 exo− (500 nM) was preincubated with DNA (250 nM) in an assay buffer and mixed with variable concentrations of the nucleotide analogue (5–500 µM) and 10 mM Mg2+. The reactions were quenched with 350 mM EDTA at variable times (0.001–10 s) and analyzed as previously described. The time course in product formation was fitted using the following equation:
| (1) |
y is the amount of product, A is the amplitude, kobs is the rate constant, t is time and C is a defined constant. Kd, app and kpol were determined by fits of the data points to the Michaelis–Menten equation:
| (2) |
where kobs is the apparent first-order rate constant, kpol is the maximal polymerization rate constant, Kd, app is the apparent dissociation constant for dXTP and [dXTP] is the concentration of nucleotide substrate.
Extension enzymatic assays
Rates of elongating beyond the incorporated nucleotide were measured under single-turnover conditions. In these experiments, gp43 exo− (500 nM) was incubated with 250 nM DNA in assay buffer containing 10 mM Mg2+ and mixed with 150 µM nucleotide. After four half-lives, an aliquot of the reaction was quenched with 350 mM EDTA to validate nucleotide insertion opposite the lesion. At this time, dGTP (500 µM) was then added. Aliquots were quenched with 350 mM EDTA at variable time points and analyzed as described above.
RESULTS
Synthesis of isosteric non-natural nucleotides
Two non-natural nucleotides designated as 5-CITP and 5-MeCITP (Figure 2A) were synthesized to further interrogate the role of hydrophobicity, π-electron interactions and steric constraints during translesion DNA synthesis. In this case, 5-MeCITP and 5-NITP are similar with respect to hydrophobicity and π-electron density yet are significantly different with respect to overall shape and size. In contrast, 5-CITP is nearly identical to 5-NITP with respect to shape, size and π-electron surface area yet differ with respect to hydrophobicity and solvation energies. These differences provide an excellent way to probe their roles during the misreplication of an abasic site. Both non-natural nucleotides were synthesized as outlined in Scheme 1. 5-MeCITP was prepared using the protocol established by Girgis et al. (34). Briefly, formation of the N-glycosidic bond between the 5-methylcarboxylate indole and Hoffer’s chlorosugar was achieved by employing sodium hydride as the base to form the sodium salt of the indole nucleobase in situ which then reacts with the electrophilic center of the anomeric carbon (C-1′) of the deoxyribose sugar. Deprotection of the 3′- and 5′-hydroxyl moieties of the sugar was accomplished by employing the Zemplén conditions to yield mainly the β-anomeric isomer as confirmed by nuclear-Overhauser effect (NOE) difference experiments (NOEs of H-2, H-7, H-2′α and H-2′β upon irradiation of H-1′) as well as the presence of an apparent triplet (tapp) peak for the H-1′ resonance with a coupling constant of ∼7.0 Hz which are characteristics for the β-anomer of N-nucleosides (38,39). The one-pot synthesis procedure of Smith et al. (33) was used to convert the nucleoside to the corresponding triphosphate by first reacting the substituted indolyl deoxyribonucleoside with POCl3 to form the 5′-monophosphorodichloridated nucleoside intermediate which was then treated with pyrophosphosphate to form the nucleoside 5′-triphosphate. 5-CITP was prepared from 5-MeCITP in which the methyl ester protecting group was cleaved using sodium hydroxide in the presence of 15-crown-5 ether to chelate the sodium ions that act as a Lewis acid and jeopardize the integrity of the triphosphate group. In fact, attempts to cleave the methyl ester protecting group in the absence of 15-crown-5 ether produced low amounts of the desired product (data not shown). The conversion of 5-MeCITP to 5-CITP was monitored via TLC stained with bromocresol green dye (31) and analytical HPLC as further described in the Experimental Section.
Figure 2.

Structures of (A) 5-NITP, 5-CITP and 5-MeCITP; (B) 5-PhITP, 5-F5-PhITP and 5-F4OMe-PhITP. ‘For clarity, only the nucleobases of the isosteric 5-substituted indolyl nucleotide analogues are shown’. (C) Nucleotide sequence of the DNA substrate used in kinetic studies. (D) Dependency of the apparent burst rate constant on the concentration of 5-CITP as measured under single-turnover conditions. Assays were performed using 500 nM gp43 exo−, 250 nM 13/20SP-mer, 10 mM Mg(OAc)2 and 5-CITP in variable concentrations of 10 (open circle), 25 (closed circle), 50 (open square), 100 (closed square), 200 (open diamond) and 500 µM (closed diamond). (E) Observed rate constants for incorporation (closed circle) were plotted against 5-CITP concentration. A fit of the data to the Michaelis–Menten equation yields a kpol of 67 ± 5 s−1 and a Kd, app of 172 ± 31 µM.
Scheme 1.

Synthetic scheme for the preparation of 5-NITP analogs, 5-MeCITP and 5-CITP. Reagents and conditios were as follow: (a) NaH, anhydrous ACN, RT, 16 h; (b) NaOMe, MeOH, pH >12, RT, 16 h; (c) (i) POCl3, Proton Sponge®, trimethylphosphate, 0°C; (ii) Tributylammonium pyrophosphate, DMF, tributylamine, RT, 15 min and (iii) 1 M TEAB, RT, 2 h; (d) NaOH, 3:2 MeOH:H2O, RT, 72 h and (e) NaOH, 2:1 MeOH:H2O, 15-crown-5 ether, RT.
Analogs of 5-PhITP were synthesized in which the π-electron density around the phenyl substituent group of 5-PhITP was reduced by substituting highly electronegative fluorines for all of the hydrogen atoms. As illustrated in Figure 2B, this substitution reduces the overall π-electron density without significantly perturbing the overall size of the phenyl ring (Supplementary Data S1). Scheme 2 outlines the synthesis of the fluorophenyl-substituted indolyl nucleotides. The 5-perfluorophenylindole nucleobase 5 was first prepared using the Suzuki–Miyaura coupling method (40) and attached to the deoxyribose sugar using the protocol established by Girgis et al. (34) as previously described above. The one-pot synthesis procedure of Smith et al. (33) was used to obtain the desired product, 5-F5-PhITP, which was verified by nuclear magnetic resonance spectroscopy and high-resolution mass spectrometry analyses (Supplementary Data S2A and S2B).
Scheme 2.

Synthetic scheme for the preparation of fluorinated analogs. Reagents and conditions were as follows: (a) Pd(PPh3)4, NaHCO3 in H2O, DME, reflux, 2 h; (b) NaH, anhydrous ACN, RT, 16 h; (c) NaOMe, MeOH, pH ≥12, RT, 16 h; (d) (i) POCl3, Proton Sponge®, trimethylphosphate, 0°C; (ii) Tributylammonium pyrophosphate, DMF, tributylamine, RT, 15 min and (iii) 1 M TEAB, RT, 2 h and (e) NaOMe, 1:10 (ether:MeOH), pH >12, RT, 36 h.
5-F4OMe-PhITP, a slightly larger structural analog of 5-F5-PhITP (Figure 2B), was synthesized to further interrogate the influence of nucleobase size on the kinetic parameters of nucleotide incorporation. 5-F4OMe-PhIdR, 7, was obtained using a slightly modified procedure (32) starting from 5-F5-PhIdR, 6. In this procedure, 6 was dissolved in 10% ether in anhydrous methanol followed by the addition of sodium methoxide until pH >12 and vigorously stirred for 36 h. 19F-NMR and high-resolution mass spectrometry indicated that the fluorine atom at the para position of the perfluorophenyl ring was substituted by the methoxy moiety under these conditions. Phosphorylation of 7 was performed as previously described (33) and characterized by NMR and mass spectrometry after purification (Supplementary Data S2C and S2D).
Nucleobase hydrophobicity and π-electron density influences translesion DNA synthesis
The kinetic parameters for the utilization of 5-CITP opposite an abasic site were measured using a defined DNA substrate illustrated in Figure 2C. Time courses in product formation were generated using single-turnover conditions in which 500 nM of the exonuclease-deficient bacteriophage T4 DNA (gp43 exo−) was incubated with 250 nM DNA containing an abasic site (designated as13/20SP-mer) and mixed with variable concentrations of non-natural nucleotide (5–1000 µM) and 10 mM Mg2+. Representative time courses provided in Figure 2D were fit to an equation describing a single exponential process (Equation 1) to define kobs, the rate constant in nucleotide incorporation. The plot of kobs versus 5-CITP concentration is hyperbolic (Figure 2E) and a fit of the data to the Michaelis–Menten equation (Equation 2) yields a kpol value of 67 ± 5 s−1, a Kd, app of 172 ± 31 µM and a kpol/Kd, app of 3.9×105 M−1s−1 for 5-CITP. The fast kpol of ∼70 s−1 is consistent with a role for π-electron density in modulating rate of the conformational change that precedes phosphoryl transfer. However, the Kd, app of 170 µM for 5-CITP is ∼10-fold higher than the hydrophobic analog, 5-NITP. This difference suggests that the binding of a non-natural nucleotide is governed largely by its hydrophobic nature (Table 2).
Table 2.
Summary of biophysical features for natural and non-natural nucleotides used during correct and translesion DNA synthesisa
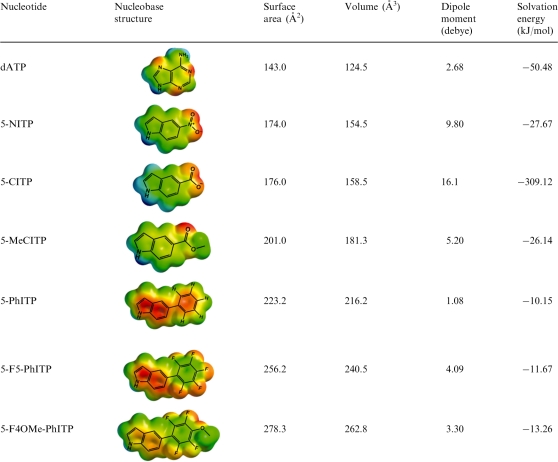 |
aSurface areas and volumes (used as an indicator of the relative size of the nucleobase), dipole moments and solvation energies (in the aqueous phase) for each nucleobase were calculated using Spartan ’08 software. The semi-empirical method of calculation with AM1 was used for geometry optimization and the Density Functional Theory level of calculation with B3LYP 6-31G** basis sets were used for energy minimization.
The kinetic parameters for 5-MeCITP were measured to further validate this mechanism. As reported in Table 1, the Kd, app of 13 µM for 5-MeCITP is ∼13-fold lower than that for 5-CITP and the increase in binding affinity is directly linked with the increase in hydrophobicity caused by the addition of the methyl group (Table 2). Moreover, 5-MeCITP displays a fast kpol value of 79 s−1 that is nearly identical to that of 5-CITP. The fast kpol value with both analogs is consistent with the presence of π-electron density generated by the carbonyl groups. Collectively, these results are consistent with a model in which nucleotide binding is directly linked with its hydrophobic nature while the rate constant for incorporation is influenced by the amount of π-electron density present in the incoming nucleotide.
Table 1.
Summary of kinetic parameters for incorporation of non-natural nucleotides opposite non-instructional DNA lesions and thymine catalyzed by the exonuclease-deficient bacteriophage T4 DNA polymerasea
| DNA | Analog | Kd, app (µM) | kpol (s−1) | kpol/Kd, app (M−1 s−1) |
|---|---|---|---|---|
| Single abasic | dATPb | 35 ± 5 | 0.15 ± 0.01 | 4300 |
| Single abasic | 5-PhITPc | 14 ± 3 | 53 ± 4 | 3 800 000 |
| Single abasic | 5-F5-PhITP | 2.3 ± 0.7 | 15 ± 1 | 6 500 000 |
| Single abasic | 5-F4OMe-PhITP | 4 ± 1 | 18 ± 1 | 4 500 000 |
| Single abasic | 5-NITPd | 18 ± 3 | 126 ± 7 | 7 000 000 |
| Single abasic | 5-CITP | 172 ± 31 | 67 ± 5 | 390 000 |
| Single abasic | 5-MeCITP | 13.3 ± 0.9 | 79.8 ± 1.2 | 6 000 000 |
| Double abasic | dATPe | 157 ± 28 | 0.55 ± 0.05 | 3500 |
| Double abasic | 5-PhITPe | 12 ± 2 | 45 ± 3 | 3 750 000 |
| Double abasic | 5-F5-PhITP | 17 ± 4 | 10.6 ± 0.8 | 624 000 |
| Double abasic | 5-F4OMe-PhITP | 14 ± 3 | 30 ± 3 | 2 143 000 |
| Double abasic | 5-NITPe | 18 ± 5 | 70 ± 6 | 3 890 000 |
| Double abasic | 5-CITP | 172 ± 20 | 70 ± 4 | 407 000 |
| Double abasic | 5-MeCITP | 4.4 ± 0.5 | 82 ± 2 | 18 636 000 |
| Blunt end | dATPe | NDg | ND | ND |
| Blunt end | 5-PhITPe | 45 ± 8 | 0.007 ± 0.004 | 160 |
| Blunt end | 5-F5-PhITP | ND | ND | ND |
| Blunt end | 5-F4OMe-PhITP | ND | ND | ND |
| Blunt end | 5-NITPe | 21 ± 8 | 0.015 ± 0.004 | 710 |
| Blunt end | 5-CITP | ND | ND | ND |
| Blunt end | 5-MeCITP | 36.5 ± 9.9 | 0.047 ± 0.004 | 1290 |
| Thymine | dATPf | 10 ± 2 | 100 ± 10 | 10 000 000 |
| Thymine | 5-PhITPc | 25 ± 7 | 0.16 ± 0.01 | 6400 |
| Thymine | 5-F5-PhITP | 10 ± 2 | 0.072 ± 0.002 | 7000 |
| Thymine | 5-F4OMe-PhITP | 3.2 ± 0.6 | 0.062 ± 0.002 | 19 000 |
| Thymine | 5-NITPd | 9 ± 1 | 0.9 ± 0.1 | 100 000 |
| Thymine | 5-CITP | 818 ± 136 | 1.4 ± 0.1 | 1700 |
| Thymine | 5-MeCITP | 69 ± 26 | 7.0 ± 0.8 | 100 000 |
aAssays were performed using 500 nM gp43 exo−, 250 nM DNA substrates and variable concentrations of non-natural nucleotide in the presence of 10 mM Mg2+.
Kinetic values were taken from the following references: b(57), c(27), d(22), e(42) and f(35) for comparison.
gND: not determined as incorporation was too inefficient to measure accurately.
The role of π-electron density during translesion DNA synthesis was further investigated by measuring the kinetic parameters for the π-electron-deficient analogs, 5-F5-PhITP and 5-F4OMe-PhITP, opposite an abasic site (Table 2). Representative time courses for the incorporation of 5-F5-PhITP and the plot of kobs versus 5-F5-PhITP concentration are provided as Supplementary Data S3A and S3B. A fit of the data to the Michaelis–Menten equation yields a kpol of 15 ± 1 s−1, a Kd, app value of 2.3 ± 0.7 µM and a kpol/Kd, app of 6.5×106 M−1 s−1 (Table 1). It is surprising that the overall catalytic efficiency of 5-F5-PhITP is ∼2-fold higher than 5-PhITP since introducing fluorine groups should have reduced the amount of π-electron density within the aromatic phenyl ring and thus ‘lowered’ the efficiency of incorporation. However, close inspection of the kinetic parameters indicates that reducing π-electron stacking potential indeed lowers the polymerization rate constant by 3-fold (15 s−1 versus 53 s−1 using 5-F5-PhITP and 5-PhITP, respectively). As such, the higher catalytic efficiency for the fluorinated analog results from a 6-fold reduction in the Kd, app value (compare a Kd, app of 2.3 µM for 5-F5-PhITP with 14 µM for 5-PhITP). This effect coincides with the slightly larger size of 5-F5-PhITP compared with 5-PhITP and could reflect better shape complementarity with the DNA lesion. An alternative explanation is that the fluorinated nucleobase is more hydrophobic as it has a lower solvation energy (41).
These mechanisms were evaluated by monitoring the incorporation of 5-F4OMe-PhITP opposite the abasic site (Supplementary Data S4A and S4B). Kinetic analyses yield a kpol of 18 ± 1 s−1, a Kd, app of 4 ± 1 µM and a kpol/Kd, app of 4.5×106 M−1s−1 for this π-electron-deficient analog (Table 1). The kpol value for 5-F4OMe-PhITP is 3-fold lower than 5-PhITP, again indicating that reductions in π-electron stacking potential slow the rate constant for this kinetic step. In addition, the Kd, app of 4 µM for 5-F4OMe-PhITP is lower than that of 14 µM measured with 5-PhITP. This ∼4-fold increase in binding affinity coincides with the slightly larger size and lower solvation energy associated with the fluorinated nucleobase. While this suggests that shape complementarity is an important determinant in substrate binding, it should be noted that the Kd, app of 4 µM for 5-F4OMe-PhITP is ∼2-fold higher than that for 5-F5-PhITP (Kd, app of 2.3 µM). This reduction in binding affinity argues that optimizing the size of the nucleobase to fit within the void of the abasic site has a minimal effect on binding affinity.
Incorporation opposite other non-instructional DNA lesions
The results presented above are consistent with a mechanism in which nucleotide binding is controlled by the hydrophobic nature of the incoming nucleotide while the polymerization rate constant is influenced by π-electron density. To test whether this mechanism applies to other non-instructional DNA lesions, we measured the kinetic parameters for these non-natural nucleotides using DNA containing two adjacent abasic sites. As summarized in Table 1, the Kd, app and kpol values for most non-natural nucleotides are similar regardless of whether the DNA contains a single abasic site or two adjacent DNA lesions. The best example for this behavior is 5-CITP as it displays identical Kd, app values of 170 µM and identical kpol values of 70 s−1 using either DNA substrate.
However, there are some important exceptions that highlight nuances in the replication of non-instructional DNA lesions. For instance, the Kd, app values for both fluorinated non-natural nucleotides are ∼5-fold higher at the double abasic site compared with a single abasic site. This difference could reflect weaker binding interactions caused by the larger void of adjacent abasic sites compared with a single-DNA lesion. In addition, differential effects are observed on the kpol value for both fluorinated nucleotides. The kpol for 5-F5-PhITP is slightly slower measured opposite a double abasic site compared with a single abasic site. Surprisingly, an opposite effect is observed with 5-F4OMe-PhITP as the kpol value of 30 s−1 measured opposite the double abasic site is 2-fold faster than that of 15 s−1 measured with a single-DNA lesion. In this case, the overall catalytic efficiency (kpol/Kd, app) for 5-F4OMe-PhITP is nearly identical between the two types of DNA lesion. This occurs as larger Kd, app values are offset by faster kpol values. Similar effects are observed with the natural nucleotide, dATP, as its binding affinity is weakened 5-fold while the rate constant for incorporation is increased 4-fold as a consequence of expanding the size of the ‘void’ in DNA.
More significant differences are observed during blunt-end extension, another form of non-instructional DNA lesion (42). As indicated in Table 1, only 5-MeCITP is utilized during blunt-end DNA synthesis, albeit with a low catalytic efficiency of 1290 M−1s−1. This low efficiency reflects a large diminuation in the rate constant for incorporation (kpol = 0.047 s−1) rather than a negative influence on binding affinity (Kd, app = 37 µM). In fact, the kinetic parameters for 5-MeCITP are very similar to those measured using 5-NITP (22), another hydrophobic non-natural nucleotide that possesses significant π-electron density. Indeed, both features appear to be essential for blunt-end incorporation as alterations in one parameter or the other adversely affects this activity. For example, 5-PhITP is utilized far more effectively during blunt-end extension compared with the fluorinated analogs, 5-F5-PhITP and 5-F4OMe-PhITP. The reduced capacity of the fluorinated analogs to act as efficient substrates is likely due to deficiencies in π-electron density. Although 5-CITP possesses π-electron density, it is not utilized during blunt-end DNA synthesis since its extremely hydrophilic nature adversely affects its binding affinity. Collectively, these data indicate that efficient nucleotide incorporation opposite any non-instructional DNA lesion depends upon a combination of low solvation energy coupled with significant π-electron density.
Incorporation opposite templating nucleobases
The incorporation of these analogs opposite a templating thymine were also measured (Table 1). Although all of the non-natural nucleotides reported here are inserted opposite T, their overall catalytic efficiencies are ∼100-fold lower compared with the non-instructional abasic site. Their reduced efficiencies are caused by lower kpol values rather than through perturbations in their Kd, app values and this result is similar to that obtained using the other non-natural nucleotides displayed in Figure 1A. Despite this difference, it is clear that insertion of a non-natural nucleotide opposite a templating nucleobase is also influenced by the combination of nucleobase hydrophobicity and π-electron density. For example, fluorinated analogs of 5-PhITP have higher catalytic efficiencies compared with 5-PhITP (Table 1). In both cases, the increase in catalytic efficiency for the fluorinated analogs is caused by significant decreases in their Kd, app values that compensate for slower kpol values.
Extension beyond templating DNA and non-instructional lesions
We previously demonstrated that the bacteriophage T4 DNA polymerase extends beyond dATP or dGTP when paired opposite an abasic site (10,26). However, this high-fidelity DNA polymerase cannot extend beyond any of the 5-substituted indolyl nucleotides illustrated in Figure 1A. The lack of extension could be caused by alterations in the conformation of the primer template (43) or by a lack of hydrogen-bonding functional groups that serve as recognition elements for elongation (44). To differentiate between these models, we used the experimental protocol outlined in Figure 3A to quantify extension beyond the non-natural nucleotides developed in this study. These experiments employed single-turnover conditions in which DNA polymerase (500 nM) and DNA substrate (250 nM) were mixed with 150 µM non-natural nucleotide and 10 mM Mg2+ to allow for incorporation opposite the abasic site. After four half-lives (time required to obtain 95% mispair formation), 500 µM dGTP was added to initiate elongation beyond the abasic site. Aliquots were quenched with EDTA at variable times and analyzed via denaturing gel electrophoresis to visualize product formation. Representative data provided in Figure 3B shows that the T4 DNA polymerase does not extend beyond hydrophobic nucleotides such as 5-NITP, 5-F5-PhITP, 5-F4OMe-PhITP and 5-PhITP since only 14-mer product is observed (Figure 4B, lanes 2, 5, 6 and 7, respectively). In contrast, the T4 DNA polymerase extends beyond 5-CITP (lane 3) and 5-MeCITP (lane 4) almost as efficiently as dATP (lane 8). In fact, quantifying the amount of primer extension reveals that the efficiency for extending beyond 5-CITP and 5-MeCITP is only 20% lower than that for dATP (Figure 3C). These data support a mechanism in which the presence of functional groups that can participate in hydrogen-bonding interactions is an important determinant for elongation.
Figure 3.

Measuring extension beyond an abasic site. (A) Experiments were performed under single-turnover conditions in which 500 nM bacteriophage T4 DNA polymerase was incubated with 250 nM 13/20SP-mer in assay buffer containing 10 mM Mg2+ and mixed with 150 µM dXTP for 5 min. To initiate extension beyond the lesion, 500 µM dGTP, the correct nucleotide for the next three insertion positions, was added and the reactions were quenched with 350 mM EDTA at 180 s. (B) Denaturing gel electrophoresis data for the incorporation and extension of nucleotides opposite an abasic site catalyzed by the bacteriophage T4 DNA polymerase. Only nucleotides containing hydrogen-bonding functional groups (dATP, 5-CITP and 5-MeCITP) showed extension beyond the abasic site. (C) Quantification of gel electrophoresis data provided in panel B. Blue bars represent percentage of 14-mer product (incorporation of nucleotide opposite abasic site) while red bars represent percentage of 14-mer product that is elongated after addition of dGTP.
Figure 4.

Simplified model illustrating the kinetic steps involved in desolvation and π–π stacking interactions during the replication of an abasic site. See text for complete details. The model was generated using the Spartan ’08, ChemBioOffice 2010 and Microsoft Office 2010 Powerpoint software packages.
DISCUSSION
Although hydrogen bonding interactions are widely associated with efficient and faithful DNA replication, other biophysical features such as π–π stacking, desolvation and geometrical constraints also contribute to this process (45). Unfortunately, it is difficult to accurately define the individual contributions of one feature versus another since they are interrelated. This is best exemplified by examining the consequences of exposing nucleic acid to DNA damaging agents. For example, alkylation of the O6 position of guanine creates a miscoding DNA lesion, O6-methylguanine, that has altered hydrogen bonding potential compared with the unmodified nucleobase. However, alkylation also influences other biophysical features including nucleobase hydrophobicity, tautomeric form and shape/size of the nucleobase. These global changes make it difficult to unambiguously assign the energetic contributions of hydrogen bonding, π–π stacking interactions, desolvation energies and geometrical constraints during correct and pro-mutagenic DNA synthesis.
To circumvent these problems, we studied the replication of an abasic site, a non-instructional DNA lesion that is devoid of a templating base and thus alleviates complications arising from hydrogen bonding interactions. In addition, we utilized a series of isosteric 5-substituted indolyl deoxynucleotides to probe the mechanism of nucleotide incorporation opposite this lesion. Since these nucleotide analogs contain modified functional groups attached to a common indole scaffold, the degree of π-electron density, desolvation energies, dipole moments and shape/size can be varied (Table 2) and quantified to evaluate their influence on the two most important kinetic steps associated with polymerization. These include nucleotide binding and the conformation change step preceding phosphoryl transfer. In this study, the use of single-turnover reaction conditions allows the first round of enzyme turnover to be accurately measured. This feature provides constraints on the derived kinetic parameters such that Kd, app values represent close approximations to true binding affinities for the non-natural nucleotides. In addition, we have demonstrated through elemental effects on nucleotide incorporation (10,46), pulse chase (47,48) and fluorescence analyses (49) that kpol values reflect the conformational change step that precedes phosphoryl transfer. As a result, measurable differences in these kinetic parameters as a function of non-natural nucleotide can be used to quantify the importance of various biophysical features.
The utility of this approach is apparent by comparing the kinetic parameters for utilizing 5-NITP and 5-CITP during translesion DNA synthesis. These particular nucleotides are essentially identical with respect to shape, size, π-electron density and dipole moment. However, their solvation energies are significantly different so that any variation in their catalytic efficiencies can be attributed to the energetic requirements for nucleobase desolvation. In fact, the differences in kpol/Kd, app values of 7×106 M−1s−1 and 3.9×105 M−1s−1 for 5-NITP and 5-CITP, respectively, indicate that desolvation contributes ∼1.71 kcal/mol (The overall change in Gibb's free energies was calculated using the equation, ΔΔGo = RT ln K, where R = 1.9872 cal mol−1 K−1, T = 298 K and K is the ratio of respective kinetic parameters for the incorporation of these non-natural nucleotides opposite the lesion.) to the overall efficiency for nucleotide incorporation opposite the abasic site. The influence of desolvation toward nucleotide binding (Kd, app value) and the conformational change step (kpol value) can be further broken down using an identical approach. The energetic contribution of desolvation is 1.34 kcal/mol for nucleotide binding, while that for the conformational change step is 0.37 kcal/mol. As such, this analysis indicates that nucleobase desolvation plays a more significant role during the binding of non-natural nucleotides compared with the conformational change step preceding phosphoryl transfer.
Although desolvation may occur during the conformational change step, we envisioned that this kinetic step is more sensitive to the presence of π-electron density. This conclusion is based upon differences in the measured kpol values for 5-PhITP and the fluorinated analogs, 5-F5-PhITP and 5-F4OMe-PhITP. Since these analogs possess similar shapes, sizes and desolvation energies, the quantifiable disparities in their kpol values should primarily be attributed to energetic differences associated with π-electron density. Using the kpol values of 53 s−1 versus 15 s−1 for 5-PhITP and 5-F5-PhITP, respectively, a ΔΔGo value of 0.75 kcal/mol is calculated for the conformational change step. This energetic difference may not be as large as expected for π–π stacking interactions that must occur inside the interior of duplex DNA. However, reducing the π-electron density within the phenyl ring of 5-PhITP via substitution of hydrogen with electronegative fluorine (5-F5-PhITP) also introduces a partial positive charge at the center of the perfluorinated analog. As a result, favorable π–π stacking interactions could be replaced by electrostatic quadrupolar interactions that are similar to π-cation interactions. Indeed, studies have shown that fluorinated aryl compounds have the ability to base stack with electron-rich aromatic rings through quadrupolar electrostatic interactions (50–52). This argument is consistent with the ∼2-fold higher kpol value of 5-NITP (126 s−1) compared with 5-CITP (67 s−1). While both molecules possess similar π-electron densities, the positive charge on the nitrogen of 5-NITP may slightly enhance its base-stacking abilities (53). However, we note that the dipole moments and/or van der Waals contacts could also contribute to the mechanism of nucleotide incorporation opposite an abasic site. Further experimentation is being performed to address these features.
These data were used to propose the model outlined in Figure 4 that highlights the roles of nucleobase desolvation and π–π stacking interactions during the replication of an abasic site. In this model, nucleobase desolvation primarily occurs during the initial binding step. This is based upon the fact that most hydrophobic analogs bind with relatively high affinity (∼20 µM) regardless of their overall shape and size. In addition, the Kd, app values for hydrophobic 5-substituted indolyl nucleotides such 5-NITP and 5-MeCITP are 10-fold lower than that for the hydrophilic 5-CITP analog. Following nucleotide binding, the polymerase:DNA:dXTP complex undergoes a conformational change that aligns the α-phosphate of the incoming nucleotide with the 3′-OH of the primer for phosphoryl transfer. This step is controlled predominantly by the π-electron density present on the incoming nucleotide and allows for proper π–π stacking interactions within an intrahelical conformation to commence phosphoryl transfer. With the bacteriophage T4 DNA polymerase, this kinetic step is also influenced by base-stacking interactions with the downstream templating nucleobase since polymerization using blunt-end DNA or DNA containing a double abasic site occurs with reduced kpol values without a negative effect on Kd, app.
This model can also be applied to describe the roles of desolvation, π–π stacking, steric fit and hydrogen-bonding interactions during normal DNA synthesis. In the presence of a templating nucleobase, it is clear that desolvation must also occur during the initial binding step as water molecules must be effectively stripped away from functional groups that eventually participate in hydrogen-bonding interactions. In this case, energetic penalties caused by the removal of water molecules are adequately offset by formation of favorable enthalpic contributions by hydrogen bonding interactions between correctly paired nucleobases. Subsequent formation of proper π–π stacking interactions occur during the conformational change step then allows for phosphoryl transfer and primer elongation to occur. As such, there are two major energetic differences between correct versus translesion DNA synthesis. The first is with respect to desolvation and enthalpic compensation during the binding of a nucleotide. During correct DNA synthesis, energetic penalties associated with nucleobase desolvation are offset by the formation of correct and complementary hydrogen bonding interactions. Enthalpic compensation via hydrogen-bonding interactions does not occur at non-instructional DNA lesions such as an abasic site and this deficiency adversely affects the mechanism of nucleotide binding. The other major difference is the involvement of nucleobase desolvation during the conformational change step. During correct DNA synthesis, there is no need for nucleobase desolvation during this kinetic step as functional groups on the templating and incoming nucleobase are already locked into proper hydrogen bonding interactions. During the replication of a DNA lesion, however, desolvation likely occurs during the conformational change step and this energetic demand also reduces the overall efficiency of nucleotide incorporation. A provocative implication is that the primary determinant for efficient and faithful DNA synthesis is the ability of the DNA polymerase to effectively perform nucleobase desolvation.
Primer extension depends upon hydrogen-bonding interactions and shape complementarity
While π-electron density and hydrophobicity are important for efficient nucleotide incorporation opposite an abasic site, these features are not required for primer elongation. In fact, nucleotide extension depends heavily upon the presence of functional groups that provide hydrogen-bonding interactions. This conclusion is based upon the observation that the bacteriophage T4 polymerase does not extend beyond 5-PhITP or 5-NITP but can extend beyond 5-CITP and 5-MeCITP when paired opposite an abasic site. Elongation beyond 5-CITP and 5-MeCITP likely occurs since both analogs contain a carbonyl group that can hydrogen bond with active site amino acids necessary for elongation. This observation is similar to results indicating that most high-fidelity DNA polymerases are incapable of extending non-natural nucleotides unless the formed base pair contains at least one functional group that participate as a hydrogen bond acceptor or donor (44).
The presence of hydrogen-bonding groups does not always guarantee that a non-natural nucleotide will be elongated. This is evident as the bacteriophage T4 DNA polymerase cannot extend beyond 5-CITP or 5-MeCITP when they are paired opposite thymine (data not shown), the predicted pairing partner for these non-natural nucleotides. The lack of extension argues that the conformation of the primer-template also plays an important role in defining if a base pair will be efficiently extended or not. This interpretation is consistent with work published by Romesberg indicating that extension beyond a base-pair containing a non-natural nucleotide depends upon the proper geometry of the terminal base pair (43,54). Our data extend these findings and indicate that efficient elongation depends upon the presence of hydrogen bonding groups that must be in a precise geometrical orientation. If either condition is not met, primer elongation cannot occur. This model may also explain why most mispairs formed between natural nucleotide and DNA lesions are poorly elongated by high-fidelity DNA polymerases (55,56).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Institutes of Health (CA118408 to A.J.B.); National Cancer Institute Training Programs in Cancer Pharmacology (CA148052 to E.A.M.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Cheng YK, Pettitt BM. Stabilities of double- and triple-strand helical nucleic acids. Prog. Biophys. Mol. Biol. 1992;58:225–257. doi: 10.1016/0079-6107(92)90007-s. [DOI] [PubMed] [Google Scholar]
- 2.Goodman MF, Fygenson KD. DNA polymerase fidelity: from genetics toward a biochemical understanding. Genetics. 1998;148:1475–1482. doi: 10.1093/genetics/148.4.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunkel TA, Bebenek K. DNA replication fidelity. Annu. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 4.Mitra R, Pettitt BM, Rame GL, Blake RD. The relationship between mutation rates for the (C-G)–>(T-A) transition and features of T-G mispair structures in different neighbor environments, determined by free energy molecular mechanics. Nucleic Acids Res. 1993;21:6028–6037. doi: 10.1093/nar/21.25.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishina Y, Duguid EM, He C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006;106:215–232. doi: 10.1021/cr0404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sniegowski PD, Gerrish PJ, Johnson T, Shaver A. The evolution of mutation rates: separating causes from consequences. Bioessays. 2000;22:1057–1066. doi: 10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 9.Lhomme J, Constant JF, Demeunynck M. Abasic DNA structure, reactivity, and recognition. Biopolymers. 1999;52:65–83. doi: 10.1002/1097-0282(1999)52:2<65::AID-BIP1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 10.Berdis AJ. Dynamics of translesion DNA synthesis catalyzed by the bacteriophage T4 exonuclease-deficient DNA polymerase. Biochemistry. 2001;40:7180–7191. doi: 10.1021/bi0101594. [DOI] [PubMed] [Google Scholar]
- 11.Avkin S, Adar S, Blander G, Livneh Z. Quantitative measurement of translesion replication in human cells: evidence for bypass of abasic sites by a replicative DNA polymerase. Proc. Natl Acad. Sci. USA. 2002;99:3764–3769. doi: 10.1073/pnas.062038699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boiteux S, Laval J. Coding properties of poly(deoxycytidylic acid) templates containing uracil or apyrimidinic sites: in vitro modulation of mutagenesis by deoxyribonucleic acid repair enzymes. Biochemistry. 1982;21:6746–6751. doi: 10.1021/bi00269a020. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence CW, Borden A, Banerjee SK, LeClerc JE. Mutation frequency and spectrum resulting from a single abasic site in a single-stranded vector. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 15.Mozzherin DJ, Shibutani S, Tan CK, Downey KM, Fisher PA. Proliferating cell nuclear antigen promotes DNA synthesis past template lesions by mammalian DNA polymerase delta. Proc. Natl Acad. Sci. USA. 1997;94:6126–6131. doi: 10.1073/pnas.94.12.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sagher D, Strauss B. Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: uniqueness of adenine nucleotides. Biochemistry. 1983;22:4518–4526. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 17.Shibutani S, Takeshita M, Grollman AP. Translesional synthesis on DNA templates containing a single abasic site. A mechanistic study of the “A rule”. J. Biol. Chem. 1997;272:13916–13922. doi: 10.1074/jbc.272.21.13916. [DOI] [PubMed] [Google Scholar]
- 18.Strauss BS. The “A” rule revisited: polymerases as determinants of mutational specificity. DNA Repair. 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 19.Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 2002;16:1872–1883. doi: 10.1101/gad.1009802. [DOI] [PubMed] [Google Scholar]
- 20.Otterlei M, Kavli B, Standal R, Skjelbred C, Bharati S, Krokan HE. Repair of chromosomal abasic sites in vivo involves at least three different repair pathways. EMBO J. 2000;19:5542–5551. doi: 10.1093/emboj/19.20.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loeb LA. Apurinic sites as mutagenic intermediates. Cell. 1985;40:483–484. doi: 10.1016/0092-8674(85)90191-6. [DOI] [PubMed] [Google Scholar]
- 22.Reineks EZ, Berdis AJ. Evaluating the contribution of base stacking during translesion DNA replication. Biochemistry. 2004;43:393–404. doi: 10.1021/bi034948s. [DOI] [PubMed] [Google Scholar]
- 23.Vineyard D, Zhang X, Donnelly A, Lee I, Berdis AJ. Optimization of non-natural nucleotides for selective incorporation opposite damaged DNA. Org. Biomol. Chem. 2007;5:3623–3630. doi: 10.1039/b712480e. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Donnelly A, Lee I, Berdis AJ. Rational attempts to optimize non-natural nucleotides for selective incorporation opposite an abasic site. Biochemistry. 2006;45:13293–13303. doi: 10.1021/bi060418v. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Lee I, Berdis AJ. Evaluating the contributions of desolvation and base-stacking during translesion DNA synthesis. Org. Biomol. Chem. 2004;2:1703–1711. doi: 10.1039/b401732c. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Lee I, Berdis AJ. A potential chemotherapeutic strategy for the selective inhibition of promutagenic DNA synthesis by nonnatural nucleotides. Biochemistry. 2005;44:13111–13121. doi: 10.1021/bi050584n. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Lee I, Berdis AJ. The use of nonnatural nucleotides to probe the contributions of shape complementarity and pi-electron surface area during DNA polymerization. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Lee I, Zhou X, Berdis AJ. Hydrophobicity, shape, and pi-electron contributions during translesion DNA synthesis. J. Am. Chem. Soc. 2006;128:143–149. doi: 10.1021/ja0546830. [DOI] [PubMed] [Google Scholar]
- 29.Max H. alpha-Thymidin. Chemische Berichte. 1960;93:2777–2781. [Google Scholar]
- 30.Rolland V, Kotera M, Lhomme J. Convenient preparation of 2-deoxy-3,5-di-o-p-toluoyl-α-D-erythro-pentofuranosyl chloride. Synthetic Comm. 1997;27:3505–3511. [Google Scholar]
- 31.Ramette R. The dissociation quotient of bromcresol green: A class study of ionic strength effects. J. Chem. Educ. 1963;40:252. [Google Scholar]
- 32.Filler R, Ayyangar NR, Gustowski W, Kang HH. New reactions of polyfluoroaromatic compounds. Pentafluorophenylalanine and tetrafluorotyrosine. J. Org. Chem. 1969;34:534–538. [Google Scholar]
- 33.Smith CL, Simmonds AC, Felix IR, Hamilton AL, Kumar S, Nampali S, Loakes D, Brown DM. DNA polymerase incorporation of universal base triphosphates. Nucleos. Nucleot. Nucleic Acids. 1998;17:541–554. [Google Scholar]
- 34.Girgis NS, Cottam HB, Robins RK. Synthesis of 2′-deoxyribofuranosyl indole nucleosides related to the antibiotics SF-2140 and neosidomycin. J. Heterocycl. Chem. 1988;25:361–365. [Google Scholar]
- 35.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 36.Rush J, Konigsberg WH. Rapid purification of overexpressed T4 DNA polymerase. Prep. Biochem. 1989;19:329–340. doi: 10.1080/10826068908544921. [DOI] [PubMed] [Google Scholar]
- 37.Mizrahi V, Benkovic P, Benkovic SJ. Mechanism of DNA polymerase I: exonuclease/polymerase activity switch and DNA sequence dependence of pyrophosphorolysis and misincorporation reactions. Proc. Natl Acad. Sci. USA. 1986;83:5769–5773. doi: 10.1073/pnas.83.16.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schweitzer BA, Kool ET. Aromatic nonpolar nucleosides as hydrophobic isosteres of pyrimidines and purine nucleosides. J. Org. Chem. 1994;59:7238–7242. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren RXF, Chaudhuri NC, Paris PL, Rumney, Kool ET. Naphthalene, phenanthrene, and pyrene as DNA base analogues: Synthesis, structure, and fluorescence in DNA. J. Am. Chem. Soc. 1996;118:7671–7678. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995;95:2457–2483. [Google Scholar]
- 41.Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. Chembiochem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 42.Berdis AJ, McCutcheon D. The use of non-natural nucleotides to probe template-independent DNA synthesis. Chembiochem. 2007;8:1399–1408. doi: 10.1002/cbic.200700096. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda S, Leconte AM, Romesberg FE. Minor groove hydrogen bonds and the replication of unnatural base pairs. J. Am. Chem. Soc. 2007;129:5551–5557. doi: 10.1021/ja068282b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morales JC, Kool ET. Functional hydrogen-bonding map of the minor groove binding tracks of six DNA polymerases. Biochemistry. 2000;39:12979–12988. doi: 10.1021/bi001578o. [DOI] [PubMed] [Google Scholar]
- 45.Kool ET. Hydrogen bonding, base stacking, and steric effects in dna replication. Annu. Rev. Biophys. Biomol. Struct. 2001;30:1–22. doi: 10.1146/annurev.biophys.30.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Hays H, Berdis AJ. Manganese substantially alters the dynamics of translesion DNA synthesis. Biochemistry. 2002;41:4771–4778. doi: 10.1021/bi0120648. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Motea E, Lee I, Berdis AJ. Replication of a universal nucleobase provides unique insight into the role of entropy during DNA polymerization and pyrophosphorolysis. Biochemistry. 2010;49:3009–3023. doi: 10.1021/bi901523y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devadoss B, Lee I, Berdis AJ. Is a thymine dimer replicated via a transient abasic site intermediate? A comparative study using non-natural nucleotides. Biochemistry. 2007;46:4486–4498. doi: 10.1021/bi602438t. [DOI] [PubMed] [Google Scholar]
- 49.Lee I, Berdis A. Fluorescent analysis of translesion DNA synthesis by using a novel, non-natural nucleotide analogue. Chembiochem. 2006;7:1990–1997. doi: 10.1002/cbic.200600128. [DOI] [PubMed] [Google Scholar]
- 50.Zivkovic A, Engels JW. Florobenzene as artificial nucleobases-base pairing and stacking interactions. Nucleos. Nucleot. Nucleic Acids. 2007;26:559–562. doi: 10.1080/15257770701490019. [DOI] [PubMed] [Google Scholar]
- 51.Mathis G, Schutz R, Hunziker J. Towards a DNA-like duplex without hydrogen bonds. Nucleos. Nucleot. Nucleic Acids. 2003;22:1183–1185. doi: 10.1081/NCN-120022831. [DOI] [PubMed] [Google Scholar]
- 52.Lai JS, Qu J, Kool ET. Fluorinated DNA bases as probes of electrostatic effects in DNA base stacking. Angew. Chem. Int. Ed. Engl. 2003;42:5973–5977. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]
- 53.Zahn KE, Belrhali H, Wallace SS, Doublie S. Caught bending the A-rule: crystal structures of translesion DNA synthesis with a non-natural nucleotide. Biochemistry. 2007;46:10551–10561. doi: 10.1021/bi7008807. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda S, Romesberg FE. Optimization of interstrand hydrophobic packing interactions within unnatural DNA base pairs. J. Am. Chem. Soc. 2004;126:14419–14427. doi: 10.1021/ja047291m. [DOI] [PubMed] [Google Scholar]
- 55.Reha-Krantz LJ. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochim. Biophys. Acta. 2010;1804:1049–1063. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 56.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 57.Newbold RF, Warren W, Medcalf AS, Amos J. Mutagenicity of carcinogenic methylating agents is associated with a specific DNA modification. Nature. 1980;283:596–599. doi: 10.1038/283596a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.