

Abstract
A potent class of anticancer, human farnesyltransferase (hFTase) inhibitors has been identified by “piggy-backing” on potent, antimalarial inhibitors of Plasmodium falciparum farnesyltransferase (PfFTase). On the basis of a 4-fold substituted ethylenediamine scaffold, the inhibitors are structurally simple and readily derivatized, facilitating the extensive structure–activity relationship (SAR) study reported herein. Our most potent inhibitor is compound 1f, which exhibited an in vitro hFTase IC50 value of 25 nM and a whole cell H-Ras processing IC50 value of 90 nM. Moreover, it is noteworthy that several of our inhibitors proved highly selective for hFTase (up to 333-fold) over the related prenyltransferase enzyme geranylgeranyltransferase-I (GGTase-I). A crystal structure of inhibitor 1a co-crystallized with farnesyl pyrophosphate (FPP) in the active site of rat FTase illustrates that the para-benzonitrile moiety of 1a is stabilized by a π–π stacking interaction with the Y361β residue, suggesting a structural explanation for the observed importance of this component of our inhibitors.
Introduction
As the successful treatment of cancer remains a challenging goal, research into novel, selective, and less toxic chemotherapeutic agents is gathering pace.1-3 Indeed, increasing understanding of the cellular processes that lead to cancer has identified additional targets for the design of such chemotherapeutics. Ras, the protein product of the ras oncogene, is a small GTPase that is important in signal transduction, cell growth, and cell proliferation.4 Mutations in Ras, which cause the protein to persistently bind GTP and thus become constitutively active, can lead to unregulated cell division; such Ras mutants are found in approximately 30% of human tumors.5,6 In the 1980s, it was reported that Ras required farnesylation to enhance its hydrophobicity and thereby facilitate its anchorage to the plasma membrane, a process necessary for its signaling function.7,8 Accordingly, it was envisioned that inhibition of the enzyme that performs this post-translational modification, protein farnesyltransferase (FTasea), would offer an indirect method of blocking the function of Ras oncoproteins. Indeed, in addition to inhibiting FTase in vitro,9-12 farnesyltransferase inhibitors (FTIs) have demonstrated anti-tumor activity in several animal models.2,9 Clinically, however, the results are mixed. For example, a lack of activity was reported when Tipifarnib13 (R115777) was used against advanced colorectal and pancreatic cancers.14,15 In contrast, extremely encouraging results were observed when Tipifarnib was used against breast cancer in combination with cytotoxic agents.16,17 In recent years, it has become clear that aberrant Ras activity is not the only target for FTIs, and it is likely that other FTase substrates, such as Rheb, are also involved in oncogenesis.18-21 Nonetheless, despite the now-apparent complexity of this system and the unclear molecular mechanisms by which FTIs operate, the past decade has seen many FTIs established as antiproliferative agents of high efficacy and low toxicity, validating the continued research into more drug-like FTIs as alternative chemotherapeutics for cancer.1-3
The prenyltransferases are a family of zinc metalloenzymes that catalyze the prenylation (addition of a prenyl group through a thioether linkage) of a particular set of proteins, many of which are crucial to signal transduction pathways, causing their localization to the plasma membrane and other cellular compartments and so rendering them biologically active.22 There are three members of the prenyltransferase family: farnesyltransferase (FTase), geranygeranyltransferaseI (GGTase-I), and geranygeranyltransferase-II (GGTase-II). FTase catalyzes the transfer of a farnesyl (C15 isoprenoid) group from the cosubstrate farnesyl pyrophosphate (FPP) to the cysteine residue within the C-terminus Ca1a2X tetra-peptide sequence of the target protein (including Ras and Rheb), where C = cysteine, a = an aliphatic amino acid, and X = methionine (M), serine (S), alanine (A), or glutamine (Q).23 Likewise, GGTase-I catalyzes the corresponding S-geranylgeranylation by accelerating the transfer of the geranylgeranyl group (C20 isoprenoid) from geranylgeranyl pyrophosphate (GGPP) to the cysteine within the C-terminus Ca1a2X sequence of the substrate protein (including Rho, Rap, and Ral),24 where this time X is usually leucine (L), isoleucine (I), or phenylalanine (F).23 It is the identity of the X residue that dictates if a target protein is farnesylated or geranylgeranylated, and is so-called the specificity residue. Finally, in a similar fashion, GGTase-II transfers two geranylgeranyl groups to protein trafficking Rab proteins that contain Cys-CysorCys-Ala-Cyssequencesatthe C-terminus.23
Previous research within our laboratories has focused on the design of peptidomimetic inhibitors of FTase based on the Ca1a2X tetrapeptide substrate.25-28 Herein we describe a novel series of ethylenediamine-based, mammalian FTase inhibitors as anticancer compounds that were discovered by a “piggy-back” approach after the success of the core scaffold in a series of antimalarial plasmodial FTase inhibitors.29 We present an extensive structure–activity relationship (SAR) study of these inhibitors with both in vitro and whole cell data, including relative activities against FTase and GGTase-I. Additionally, we discuss our efforts to improve inhibitor potency against mammalian FTase, and we present crystallographic data that reveals the actual binding mode of our inhibitors within the active site of the enzyme.
Results and Discussion
Design
We have previously reported on the design and synthesis of inhibitors of Plasmodium falciparum farnesyltransferase (PfFTase).29,30 An homology model of the active site of PfFTase suggests the presence of four sub-pockets.31 By employing the computational modeling program GOLD,32 we identified that an ethylenediamine scaffold with both nitrogens doubly substituted in order to gain simultaneous access to these four subpockets might furnish inhibitors of PfFTase. Indeed, compounds based on this scaffold, including 1a (Figure 1), in which the choice of the four substituents (para-benzonitrile, imidazolylmethyl, arylmethyl, and heterocycle-substituted sulfonyl) was influenced by the BMS series of tetrahydrobenzodiazepine-based33 and tetrahydroquinoline (THQ)-based34 FTase inhibitors, proved particularly potent inhibitors of PfFTase, both in vitro and in infected erythrocytes.29,30 Although PfFTase is significantly larger than rat FTase (rFTase) in both the α- (472 vs 379 residues) and β-subunits (923 vs 437 residues), the differences are mainly due to insertions in the PfFTase protein sequence, and overall there is minimal difference in the residues that form the active site.29,31 Of the sequence segments in the model of PfFTase, there is 23% identity (53% similarity) for the α-subunit between PfFTase and rFTase isoforms and 37% (56%) for the β-subunit. Therefore, we anticipated that our PfFTase inhibitors may also inhibit mammalian FTase, binding in the same sub-domains but, in certain instances, to differing active site residues (i.e., PfFTase31 → rFTase (PDB ID: 1JCR35): S150 → N165α, F151 → Y166α, N317 → H201α), providing a route to inhibitor selectivity for the mammalian isoform of the enzyme.
Figure 1.

One high scoring active site conformation of inhibitor 1a (green) as identified by flexible ligand GOLD32 docking experiments (A) using a Connoly analytical surface graphical representation developed in PyMOL,37 red hydrophobic to blue hydrophilic, and (B) using a “cartoon” graphical representation developed in PyMOL37 and overlaid with the peptide inhibitor CVFM (orange) from the rFTase crystal structure. Binding surface of rat FTase (PDB ID: 1JCR35), values in parentheses refer to corresponding residues in PfFTase;31 small molecules colored by atom type: FPP colored red (farnesyl) and blue (pyrophosphate); blue spheres = water molecules; purple sphere = zinc ion.
Specifically, as with an almost identical analogue of 1a in the homology model of the active site of PfFTase (Figure 2 in ref 29b), we hypothesized that the more basic of the two imidazoles, the 1-methyl-1H-imidazol-5-ylmethyl substituent of 1a, would bind the zinc ion (itself held in place by D297β (D659), C661β (C299), and H362β (H838), where the labels in parentheses represent the corresponding residues in PfFTase31). In addition, we envisioned that the para-benzonitrile moiety would bind in the mostly hydrophobic pocket constructed from the tetramethylene portion of the side chain of K164α (K149) and Y166α (F151) and whose deepest point forms a hydrophilic domain (H201α (N317) and N165α (S150)). We considered that the benzyl moiety would bind in the hydrophobic pocket comprising W102β (W452), W106β (W456), and Y361β (Y837), and finally that the sulfonylimidazole would bind in the hydrophilic domain formed by R202β (R564) and three water molecules participating in a hydrogen-bonded network between S99β (S449) and Q167α (Q152).
Figure 2.

(A) Co-crystal structure of inhibitor 1a (yellow, and colored by atom type) and FPP bound to rFTase (PDB ID: 3E32),38 and (B) co-crystal structure of FPP and inhibitor 1a overlaid with the tetrapeptide inhibitor CVFM (orange, and colored by atom type) from PDB ID: 1JCR.35
To maintain consistency with the GOLD docking experiments of our ethylenediamine-based inhibitors in the homology model of the active site of PfFTase,29,30 the only constraint imposed in the flexible ligand GOLD docking with compound 1a in the active site of rFTase (PDB ID: 1JCR) was that the more basic, non-sulfonylated imidazole should again bind the Zn2+ ion. Due to the flexible nature of the ethylenediamine scaffold in 1a, we did not want to bias the docking results any further and used no additional constraints. Moreover, despite the similarities of the pendant groups in 1a and the former clinical candidate (R)-7-cyano-2,3,4,5-tetrahydro-1-(1H-imidazol-4-ylmethyl)-3-(phenylmethyl)-4-(2-thienylsulfonyl)-1H-1,4-benzodiazepine (BMS-214662, 233), we elected not to use the knowledge of the crystal structure of 236 given its more rigid tetrahydrobenzodiazepine scaffold. As was anticipated owing to the similarities in the active sites of PfFTase and rFTase, several low energy GOLD docked poses of compound 1a in rFTase demonstrated an almost identical binding mode to that observed with a very close analogue of 1a in PfFTase, with the biggest difference being that the benzyl group, rather than binding in the W102β, W106β, Y361β sub-pocket, is engaged in a π–π stacking interaction with Y361β (compare Figure 2 in ref 29b with Figure 1a above). Figure 1A illustrates one such high scoring (low energy) docked pose of compound 1a in green and colored by atom type, using the graphical representation (Connolly analytical surface, PyMOL37) and orientation employed in previous publications.29,30 The binding surface of rFTase shown incorporates the cosubstrate farnesyl pyrophosphate (FPP: farnesyl, red; pyrophosphate, blue). This binding mode of 1a overlays well with the tetrapeptide inhibitor CVFM from the rFTase crystal structure as shown in Figure 1B, in which we have used an alternative graphical representation (“cartoon”, PyMOL37) and orientation that have also been presented by us recently.38 For simplicity, the latter graphical representation shall be used throughout the remainder of this manuscript. Given the highly flexible nature of the ligand, coupled with the fact that the other high scoring poses from our studies (data not shown) were generally those in which the scaffold projected functionalities to positions similar to those seen in Figure 1, we feel that it is likely that the molecule, in solution, would occupy pockets as previously predicted as part of an ensemble of binding motifs.
Initially, we selected a focused set of our PfFTase inhibitors and analyzed their abilities to inhibit human FTase (hFTase), as well as GGTase-I, both in vitro and in whole cells (Table 1). The amino acid sequences of rat and human FTase are 95% identical with complete sequence and structural conservation around the active site,35 so it is reasonable to analyze inhibition of hFTase with respect to docking studies in rFTase. Briefly, in vitro inhibition assays for hFTase and GGTase-I were carried out by measuring the incorporation of [3H]FPP and [3H]GGPP into recombinant H-Ras-CVLS and H-Ras-CVLL, respectively, as described previously.39 Whole cell inhibition of farnesylation and geranylgeranylation were determined based on the level of inhibition by synthetic compounds of H-Ras and Rap1A processing, respectively.40 In all cases, IC50 data represent the average of three independent assays (n = 3), unless otherwise stated, and errors are given as standard deviations. The importance of both the para-substitution of the aniline component and Nπ-methylation (R1 = Me) of the imidazole that we observed in the inhibition of PfFTase29 was reflected in the mammalian isoform of the enzyme. Unsubstituted aniline 3 exhibited little activity toward the inhibition of hFTase. Incorporation of bromine into the para position of the aniline ring, however, led to an order of magnitude increase in potency with in vitro IC50s for hFTase improving from 6300 ± 360 nM for 3 to 730 ± 20 nM for 4. Additionally, H-Ras processing IC50s of the FTIs were enhanced from > 10 μM to 5.7 ± 1.2 μM. A further order of magnitude increase in FTase inhibition in vitro was achieved upon Nπ-methylation of the imidazole, with 5 exhibiting an IC50 of 79 ± 30 nM, and an associated increase in whole cell activity (H-Ras processing IC50 = 1.6 ± 1.3 μM) was also observed. Even greater hFTase inhibitory activity was achieved by the replacement of bromine with cyano in the para position of the aniline ring (1a:IC50 =56 ± 29 nM). It is interesting to note that a considerable improvement in selectivity for hFTase over GGTase-I was also observed; 1a was approximately 7-fold more selective for hFTase than was 5. In addition, the trends observed here in the inhibition of the mammalian isoform of FTase are mirrored by those observed in the disruption of the plasmodial isoform.29 Given these very encouraging data, we embarked on an SAR study of our ethylenediamine-based inhibitors in order to optimize the inhibitory activity of 1a against hFTase.
Table 1.
Enzyme Inhibition and Whole Cell Data of a Focused Set of Ethylenediamine-Based PfFTase Inhibitors
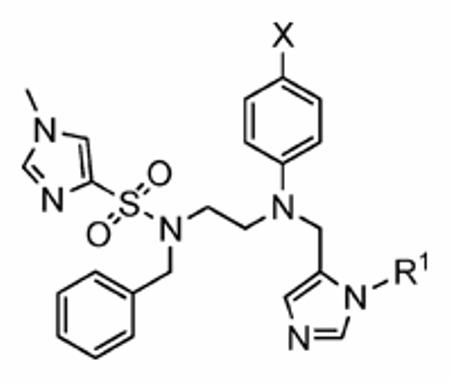 | |||||||
|---|---|---|---|---|---|---|---|
| compd |
IC50 (nM)a |
processing
IC50 (μM)c |
|||||
| no. | X | R1 | hFTase | GGTase-I | selectivityb | H-Ras | Rap1A |
| 3 (FTI-2581) | H | H | 6300 ± 360 | > 10000 | 1.6 | > 10 | > 10 |
| 4 (FTI-2584) | Br | H | 730 ± 20 | 4400 | 6.0 | 5.7 ± 1.2 | > 10 |
| 5 (FTI-2586) | Br | Me | 79 ± 30 | 530 ± 170 | 6.7 | 1.6 ± 1.3 | > 10 |
| 1a (FTI-2585) | CN | Me | 56 ± 29 | 2700 ± 2200 | 48 | 1.9 ± 1.2 | > 10 |
IC50 = inhibitor concentration required to achieve 50% inhibition of h FTase or GGTase-I in vitro.
Ratio of GGTase-I to FTase IC50s.
Processing IC50 = inhibitor concentration required to achieve 50% inhibition of farnesylation of H-Ras or geranylgeranylation of Rap1A in whole cells. In all cases, IC50 data represents the average of three independent assays (n = 3), unless otherwise stated, and errors are given as standard deviations.
Crystal Structure
We recently reported the crystal structure of the 1a:FPP:rFTase ternary complex (Figure 2A; PDB ID: 3E32).38 As a comparison, in Figure 2B we have superimposed the crystal structure of 1a with that of the tetrapep-tide substrate CVFM (PDB ID: 1JCR). Interestingly, the crystal structure shows that the ethylenediamine-based FTIs actually adopt a different binding mode to that predicted (Figure 1B). Figure 2A illustrates that the para-benzonitrile substituent of 1a is oriented toward the product exit groove and is partially stabilized by a stacking interaction with Y361β. The rings are approximately 30° offset from parallel, with the distance between them ranging from 3.8 to 4.9 Å. The binding pocket also consists of F360β (not shown in Figure), Y93β, L96β, and W106β, creating a generally hydrophobic environment for this moiety. Inhibitor 1a possesses two N-methylimidazole groups: one coordinates the catalytic zinc ion (2.0 Å distance) through its nonmethylated τ-nitrogen in place of the Ca1a2X box cysteine residue, while the second (at the sulfonamide position) is stacked between the first N-methylimidazole (3.7 Å distance) and the first isoprene of FPP (4.1 Å distance). The non-methylated τ-nitrogen of the 1-methyl-1H-imidazole-4-sulfonyl group also makes a single, weak polar contact (3.7 Å distance) to the side chain hydroxyl of Y361β. The phenyl substituent of 1a binds in a pocket formed by W106β, W102β, and L96β, essentially the a2 residue binding site of the Ca1a2X substrate, and occupying almost exactly the same space as the phenylalanine side chain of CVFM (Figure 2B).
Several similarities exist between the chemical substituents and binding mode of the ethylenediamine inhibitor 1a with tetrahydrobenzodiazepine 2; their crystal structures are overlaid in Figure 3. The binding modes of the para-benzonitrile group and the non-sulfonylated N-methylimidazole group of the ethylenediamine inhibitor 1a are nearly indistinguishable from the similar substituents in 2, with the imidazole of 2 coordinating the catalytic zinc ion like the non-sulfonylated N-methylimidazole in 1a. The scaffold nitrogen atoms bearing these substituents in both 1a and 2 occupy similar locations. The para-benzonitrile groups extend into the exit groove, with the positions of the cyano group nitrogen atoms from the two molecules differing by only ~0.4 Å, a value close to the estimated error in crystallo-graphic coordinates. Finally, the phenyl substituents of both compounds take advantage of the aromatic character of the Ca1a2Xa2 residue site, interacting with tryptophan residues 102β and 106β. The sulfonamide positions of both compounds exhibit the greatest differences in binding modes. As a consequence, the scaffold nitrogen bearing this group in the tetrahydrobenzodiazepine ring of 2 is not oriented in a spatially similar location to the nitrogen bearing the sulfonylated N-methylimidazole of 1a, which is sandwiched between the first isoprene of the lipid substrate and the zinc-coordinating N-methylimidazole moiety. By contrast, the thienyl group of 2 is largely solvent-exposed and binds in a manner resembling the a1 residue of the Ca1a2X motif. This ring is primarily stabilized by stacking on the tetrahydrobenzodiazepine ring scaffold itself, as opposed to interacting with the enzyme active site or lipid substrate.
Figure 3.
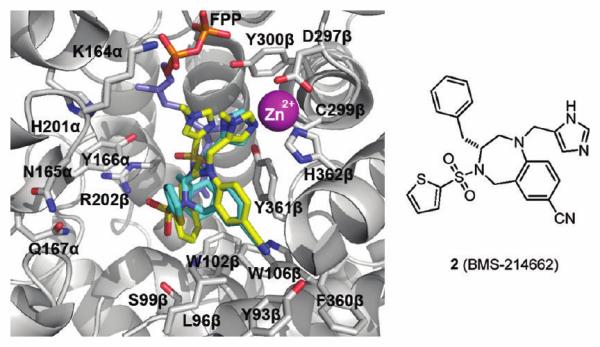
Overlay of the crystal structures of the ternary complexes of 1a38 (yellow, colored by atom type) and 236 (cyan, colored by atom type) with FPP in rFTase.
The crystal structure of the ternary complex of 1a:FPP: rFTase was solved toward the end of this research; the medicinal chemistry research program reported herein was actually driven by piggy-backing on our inhibitors of PfFTase and, to a lesser extent, the GOLD docking result presented in Figure 1. At the end of the SAR discussion, the FTase inhibition data will be evaluated in the context of both this crystal structure and the GOLD docking models.
Chemistry
“Piggy-back” mammalian FTase inhibitors (FTIs) were prepared as described previously,29 with new FTIs furnished by synthetic routes depicted in Schemes 1–3. Inhibitors incorporating the para-benzonitrile moiety were accessed by the route in Scheme 1. Briefly, mono-N-Bocethylenediamine (6) was arylated with para-fluorobenzonitrile at 120 °C for 48 h in DMSO, affording secondary aniline 7.29b Installation of the imidazole was achieved by double deprotonation of 7 with LDA at −78 °C and subsequent chemo-selective alkylation with 5-chloromethyl-1-methyl-1H-imidazole 3 HCl,29b affording 829b in a moderate yield. After Boc deprotection, completion of the syntheses of FTase inhibitors 1 was conducted in one of two ways: either sulfonylation of 9,29b followed by alkylation of the resultant sulfonamide, or reductive amination of 9, and then sulfonylation of the resultant secondary amine. Finally, purification of the FTIs was achieved by silica gel flash column chromatography, followed by rpHPLC. Full details can be found in the Experimental Section.
Scheme 1a.

a (a) para-Fluorobenzonitrile, DIPEA, DMSO, 120 °C, 48 h, 89%; (b) (1) LDA, THF, −78 °C, 30 min, (2) 5-chloromethyl-1-methyl-1H-imidazole · HCl,29 NaH, −78 °C, 1 h, 52% (98% brsm); (c) TFA-CH2-Cl2, 1:1, rt, 30min, 99%; (d) (1) R2CHO, AcOH, 4 Å MS, MeOH, rt, 30min, (2) NaCNBH3, rt, 16 h, 72–84%; (e) R2SO2Cl,DIPEA,CH3CN, 0 °C → rt, 16 h, 82–93%; ((f) R3Br, Cs2CO3, DMF, rt, 16 h, 79–82%.
Scheme 3a.

a (a) 1-Methyl-1H-imidazole-4-sulfonyl chloride, DIPEA, CH3CN, 0 °C → rt, 16 h, 95%; (b) 4-bromomethyl-N-(2-pyrimidinyl)-piperidine, Cs2CO3, DMF, rt, 4 d, 94%; (c) TFA-CH2Cl2-TIPS-H2O, 47.5:47.5:2.5:2.5, rt, 1 h, 100%; (d) R4F, DIPEA, DMSO, 120 °C, 24–48 h, 48–97%; (e) (1) NaH, 0 °C, 30 min, (2) 5-chloromethyl-1-methyl-1H-imidazole 3 HCl,29b 0 °C → rt, 2–3h, 63–96%.
Sulfonyl R2 SAR
With R1 constrained as methyl and R3 as benzyl, a series of eight R2 sulfonyl groups was surveyed (Table 2), all of which proved potent inhibitors of hFTase. The largest and most basic 5-dimethylamino-naphthalene-1-sulfonyl derivative 1h was the least active inhibitor (hFTase IC50 = 160 ± 110 nM) of this series, with the most potent being the pyridine-2-sulfonyl compound 1f (hFTase IC50 =25 ± 20 nM). Additionally, these inhibitors were more selective for hFTase over GGTase-I, with selectivities ranging from around 8-fold to 333-fold. Many of these compounds also demonstrated very good whole cell activity, with pyridine-2-sulfonyl derivative 1f proving one of the most effective inhibitors (H-Ras processing IC50 = 0.09 ± 0.06 μM). Despite its potency in vitro (hFTase: IC50 = 56 ± 29 nM), 1-methyl-1H-imidazole-4-sulfonyl derivative 1a had the poorest H-Ras processing IC50 (1.9 ± 1.2 μM). This might be a consequence of the basicity of imidazole, and its protonation could hinder cellular entry. Regardless of this, the potent in vitro activity of 1a against hFTase, coupled with the fact that we already had several inhibitors functionalized with the 1-methyl-1H-imidazole-4-sulfonyl group in-house from the related PfFTase project,29 led us to retain this particular sulfonyl group as a benchmark while varying other parts of the molecule. Selectivity of these FTIs for the hFTase isoform over the PfFTase isoform might then later be achieved by taking advantage of the previously mentioned three amino acid differences in the subpocket that is predicted to bind the para-benzonitrile ring.
Table 2.
Enzyme Inhibition and Whole Cell Data of Ethylenediamine-Based FTIs Exhibiting a Range of R2 Sulfonyl Groups
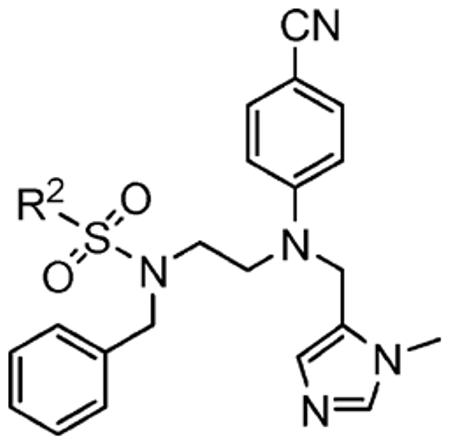 | ||||||
|---|---|---|---|---|---|---|
| Compound | IC50 (nM) | Selectivity | Processing IC50 (μM) | |||
| Number | R2 | hFTase | GGTase-I | H-Ras | Rap1A | |
|
1a (FTI-2585) |
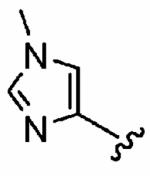
|
56 ± 29 | 2700 ± 2200 | 48 | 1.9 ± 1.2 | >10 |
|
1b (FTI-2640) |
|
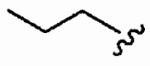
48 ± 35 | 4100 ± 1700 | 85 | 0.07 | >10 |
|
1c (FTI-2644) |
|
85 ± 0.7 | 5500 ± 2700 | 65 | NDa | ND |
|
1d (FTI-2592) |
|
41 ± 26 | 310 ± 50 | 7.6 | 0.3 ± 0.1 | >10 |
|
1e (FTI-2589) |
|
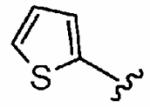
50 ± 13 | 10,000 | 200 | 0.5 ± 0.2 | >10 |
|
1f (FTI-2587) |
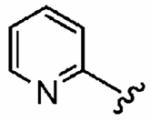
|
25 ± 20 | 820 ± 240 | 113 | 0.09 ± 0.06 | >10 |
|
1g (FTI-2590) |
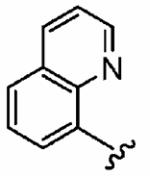
|
30 ± 14 | 10,000 | 333 | 0.6 ± 0.3 | >10 |
|
1h (FTI-2591) |
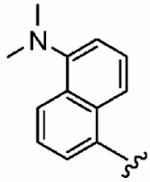
|
160 ± 110 | 10,000 | 63 | 1.5 ± 0.3 | >10 |
ND = not determined.
Sulfonamide R3 SAR
Thus, constraining R2 as 1-methy-1H-imidazole-4-sulfonyl, the R3 group was next investigated with a broad series of derivatives, which included previously reported29 and novel inhibitors (Table 3). Replacement of benzyl in the parent inhibitor 1a with a propargyl group (1aa) led to a reduction in both in vitro activity (IC50 = 720 ± 200 nM cf. 56 ± 29 nM) and whole cell activity, as well as in GGTase-I/FTase selectivity. However, the unsubstituted allyl derivative (1ab) was tolerated in vitro (IC50 =54 ± 30 nM), although not in whole cells (H-Ras processing IC50 >10 μM). Substitution of the allyl moiety with a methyl group at position-2 (1ac) led to a reduction in hFTase in vitro activity and, at the same time, afforded an improvement in GGTase-I, resulting in a reduced GGTase-I/FTase selectivity of about 5-fold. Again, whole cell activity was poor but this was recovered somewhat by replacement of the methyl in 1ac with a bromine atom, giving an H-Ras processing IC50 of 5.7 ± 1.5 μM for inhibitor 1ad. Extension of this sp2-hybridized series to the bulky tert-butylacetamido derivative 1ae was not tolerated, neither in vitro nor in whole cells. Incorporation of nitrogen into the para position of benzyl to give pyridine derivative 1af maintained activity in vitro (IC50 = 72 ± 20 nM), although it showed little activity in whole cells. Nonetheless, 1af exhibited substantially improved selectivity toward hFTase, with a GGTase-I/FTase selectivity of > 139-fold.
Table 3.
Enzyme Inhibition and Whole Cell Data of Ethylenediamine-Based FTIs Exhibiting a Range of R3 Sulfonamides
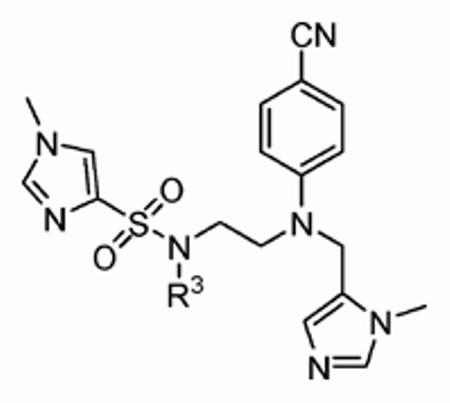 | ||||||
|---|---|---|---|---|---|---|
| Compound | IC50 (nM) | Selectivity | Processing IC50 (μM) | |||
| Number | R3 | hFTase | GGTase-I | H-Ras | Rap1A | |
|
1aa (FTI-2630) |
|
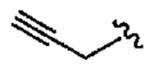
720 ± 200 | >10,000 | >14 | 6 (n = 2) | >10 |
|
1ab (FTI-2600) |
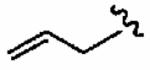
|
54 ± 30 | >10,000 | >14 | >10 | >10 |
|
1ac (FTI-2506) |
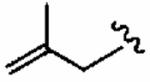
|
114 ± 80 | 600 ± 300 | 5.3 | >10 | >10 |
|
1ad (FTI-2611) |
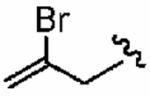
|
180 ± 100 | 5500 ± 1900 | 31 | 5.7 ± 1.5 | >10 |
|
1ae (FTI-2607) |
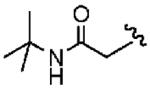
|
1400 ±900 | >10,000 | 7.1 | >10 | >10 |
|
1a (FTI-2585) |
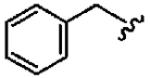
|
56 ± 29 | 2700 ± 2200 | 48 | 1.9 ± 1.2 | >10 |
|
1af (FTI-2601) |
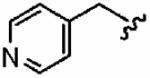
|
72 ± 20 | >10,000 | >139 | >10 | >10 |
|
1ag (FTI-2602) |
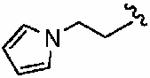
|
30 ± 33 | 510 ± 270 | 17 | 4.3 ± 1.5 | >10 |
|
1ah (FTI-2635) |
|
320 ± 190 | 6100 ± 1600 | 19 | 0.65 (n = 2) | >10 |
|
1ai (FTI-2536) |
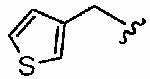
|
67 ± 38 | 940 ± 20 | 14 | 0.4 (n = 2) | >10 |
|
1aj (FTI-2538) |
|
870 ± 120 | 13,400 (n = 2) | 15 | 0.8 (n = 2) | >10 |
|
1ak (FTI-2639) |
|
2300 ± 1500 | 5900 ± 3000 | 2.6 | 0.4 (n = 2) | >10 |
|
1a (FTI-2533) |
|
510 ± 460 | 1360 ± 740 | 2.7 | >10 | >10 |
|
1am (FTI-2637) |
|
6200 ± 920 | >10,000 | >1.6 | >10 | >10 |
|
1an (FTI-2542) |
|
5300 ± 1700 | >10,000 | >1.9 | ND | ND |
|
1ao (FTI-2614) |
|
51 ± 26 | 780 ± 320 | 15 | 1 ± 0 | >10 |
|
1ap (FTI-2615) |
|
54 ± 30 | 630 ± 240 | 12 | 0.3 ± 0.2 | >10 |
|
1aq (FTI-2623) |
|
370 ± 210 | 580 ± 200 | 1.6 | 2 ± 1 | >10 |
|
1ar (FTI-2624) |
|
78 ± 12 | ND | - | 5 ± 3 | >10 |
|
1as (FTI-2625) |
|
370 ± 370 | 6500 ± 2900 | 18 | >10 | >10 |
|
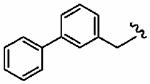
1at (FTI-2610) |
|
230 ± 150 | 550 ± 120 | 2.4 | 3 ± 0 | >10 |
|
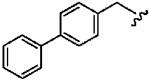
1au (FTI-2612) |
|
1700 ± 900 | 7300 ± 2200 | 4.3 | 5 ± 1 | >10 |
|
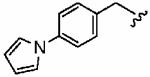
1av (FTI-2715) |
|
390 ± 100 | 5800 ± 3500 | 15 | 4 ± 1 | >10 |
|
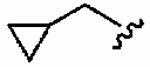
1aw (FTI-2631) |
|
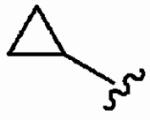
590 ± 300 | >10,000 | >17 | >10 | >10 |
|
1ax (FTI-2602) |
|
60 ± 10 | 530 ± 120 | 8.8 | 0.1 ± 0.07 | >10 |
|
1ay (FTI-2670) |
|
3700 ± 790 |
>10,000 | >2.7 | >2 | >2 |
|
1az (FTI-2722) |
|
510 ± 62 |
>10,000 | >20 | >10 | >10 |
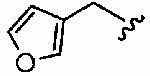
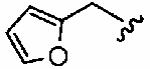
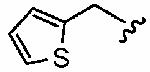
We next examined a focused set of five-membered heteroaromatics 1ag–1an. Pyrrole 1ag was around twice as active as the parent inhibitor 1a, disrupting hFTase with an IC50 value of 30 ± 33 nM. Conversely, the series of furans and thiophenes typically proved much poorer inhibitors, although interestingly FTase inhibitory activity was found to be sensitive to heteroatom position. Furan 1ah was more than twice as potent as isomeric 1aj, and thiophene 1ai was more than 34 times as potent as isomeric 1ak and approximately as active as 1a. Despite these moderate to poor in vitro data for hFTase (except for 1ai) all members of this series exhibited reasonable H-Ras processing whole cell data and in all cases the IC50 values were more potent than 1a. The remaining heteroaromatics (1al–1an) containing two heteroatoms were poor inhibitors of hFTase.
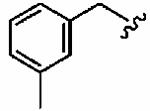
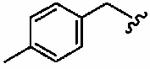
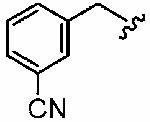
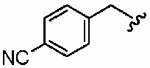
Introduction of a methyl group on the benzyl ring of 1a was tolerated in the ortho (1ao)and meta (1ap) positions (in vitro hFTase IC50sof51 ± 26 nM and 54 ± 30 nM, respectively) but not in the para (1aq) position (IC50 = 370 ± 210 nM). A similar trend was observed in whole cell activity, with meta-derivative 1ap the most potent (H-Ras processing IC50 =0.3 ± 0.2 μM). The members of this series demonstrated GGTase-I IC50 values between 580 and 780 nM, considerably reducing the GGTase-I/hFTase selectivities, relative to that of the parent benzyl inhibitor 1a. The greater potency of meta-substituted benzyl groups over their para counterparts was also observed for the cyano derivatives 1ar and 1as and for the phenyl derivatives 1at and 1au, a finding that was reflected in their corresponding H-Ras processing whole cell activities. Additionally, the same trend was noticed in the in vitro data of GGTase-I inhibition. The poor in vitro inhibition of hFTase by para-phenyl derivative 1au (IC50 = 1700 ± 900 nM) was improved by approximately 4-fold by replacement of the terminal phenyl group with the smaller pyrrole heterocycle (1av; IC50 = 390 ± 210 nM), which was also reflected in the whole cell assays (IC50 =5 ± 1 μM (1au) vs 4 ± 1 μM (1av)).
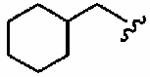
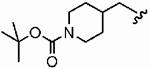
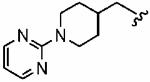
The small cyclopropylmethyl derivative 1aw was less active than the parent benzyl inhibitor 1a, but the larger cyclohexylmethyl derivative 1ax, a closer match to benzyl, proved a potent inhibitor in vitro (hFTase IC50 =60 ± 10 nM) and was one of our most potent inhibitors in whole cells (H-Ras processing IC50 = 0.1 ± 0.07 μM)). However, inhibitor 1ax was also quite active toward GGTase-I (IC50 = 530 ± 120 nM), giving a much-reduced hFTase/GGTase-I selectivity of about 9-fold. We envisaged probing deeper into the binding pocket that was predicted to bind the R3 group by modifying the cyclohexylmethyl substituent to a 4-piperidinylmethyl group; the piperidine nitrogen provided a handle from which to achieve further functionalization. Accordingly, N-Boc-piperidin-4-ylmethyl derivative 1ay and N-(2-pyrimidinyl)-4-ylmethyl derivative 1az were synthesized. Introduction of the N-Boc group at the 4 position of the cyclohexyl group (1ay) caused more than a 60-fold drop in hFTase inhibition relative to 1ax. Activity was recovered by more than 7-fold through replacement of the bulky tert-butoxycarbonyl group with the planar 2-pyrimidinyl group (1az:IC50 = 510 ± 62 nM; cf. 1ay: IC50 = 3700 ± 790 nM) although both piperidinylmethyl derivatives exhibited poor whole cell activities.
Aniline R4 SAR: The para-Position (X)
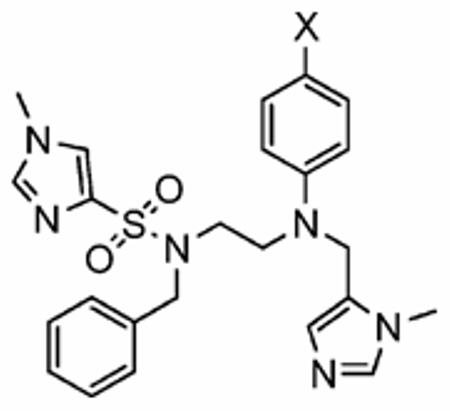
At the deepest point of the predominantly hydrophobic pocket of rFTase in which GOLD docking studies consistently predicted the para-benzonitrile moiety (the aniline “R4 group”) of our inhibitors would bind, there is a hydrophilic domain formed by N165α, Y166α, and H201α. To probe this site further, we replaced the cyano group (X) of the para-benzonitrile moiety with alternative polar groups, such as a carboxylic acid or a carboxamide, in the anticipation that such groups might be able to participate in additional hydrogen-bonding interactions and so furnish more potent hFTase inhibitors. We also investigated nonpolar substituents in the para position, such as Br, Ph and CO2t-Bu, which, provided the aniline group binds as predicted, would be expected to inhibit hFTase less well. Accordingly, para-(tert-butyl ester) derivative 11 was prepared in a similar manner to the synthetic route in Scheme 1 but employing tert-butyl para-fluorobenzoate in place of para-fluorobenzonitrile (see Experimental Section for full details). As shown in Scheme 2, treatment of 11 with TFA furnished the para-carboxylic acid 12, which was then converted to para-carboxamide 13 using HBTU and ammonium chloride. para-Phenyl derivative 14 was prepared from the previously reported intermediate 1-tert-butoxycarbonylamino-2-[biphenyl-4-yl-(3-methyl-3H-imidazol-4-ylmethyl)amino]-ethane (compound 9a in ref 29b; see Experimental Section for full details). As illustrated in Table 4, all replacements for the cyano group afforded compounds that were poorer inhibitors of hFTase in vitro. Interestingly, FTIs with bulky X groups (11: X = CO2t-Bu; 14: X = Ph) exhibited a reversed selectivity for GGTase-I over hFTase, suggesting the aniline-binding domain may be larger in GGTase-I than in hFTase.
Scheme 2a.

a (a) tert-Butyl para-fluorobenzoate, DMSO, 120 °C, 24 h, 96%; (b) 1-methyl-1H-imidazole-4-sulfonyl chloride, DIPEA, CH3CN, 0 °C → rt, 12 h, 90%; (c) BnBr, Cs2CO3, DMF, rt, 16 h, 92%; (d) (1) NaH, DMF, 0 °C, 30 min, (2) 5-chloromethyl-1-methyl-1H-imidazole · HCl,29b 0 °C → rt, 3 h, 76%; (e) TFA-CH2Cl2, 1:1, 3 h, rt, 96%; (f) NH4Cl, HBTU, DIPEA, DMF, rt, 16 h, 89%.
Table 4.
Enzyme Inhibition and Whole Cell Data of Ethylenediamine-Based FTIs Exhibiting a Range of para-Substituted Anilines
 | ||||||
|---|---|---|---|---|---|---|
| compd |
IC50
(nM) |
processing
IC50 (nM) |
||||
| no. | X | hFTase | GGTase-I | selectivity | H-Ras | Rap1A |
| 4 (FTI-2586) | Br | 79 ± 30 | 530 ± 170 | 6.7 | 1.6 ± 1.3 | >10 |
| 1a (FTI-2585) | CN | 56 ± 29 | 2700 ± 2200 | 48 | 1.9 ± 1.2 | >10 |
| 11 (FTI-2720) | CO2tBu | 5400 (n = 2) | 4900 (n = 2) | 0.9 | ND | ND |
| 12 (FTI-2721) | COOH | > 10000 | > 10000 | – | ND | ND |
| 13 (FTI-2728) | CONH2 | 5300 (n = 2) | > 10000 | > 2 | ND | ND |
| 14 (FTI-2727) | Ph | 6850 (n = 2) | 5000 ± 2600 | 0.7 | ND | ND |
Aniline R4 SAR: The Aromatic Ring
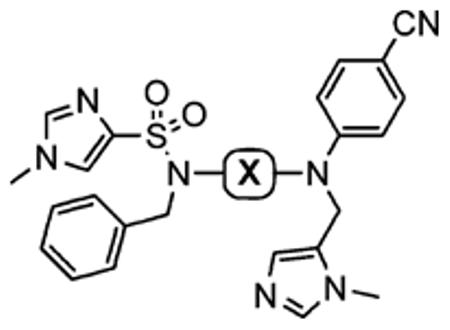
In addition to the para-substituent of the aniline affecting inhibitor activity, GOLD docking studies suggested that two of the polar amino acids within the para-benzonitrile-binding pocket might influence binding of the aniline ring itself. Specifically, Y166α could make contact with hydrogen bonding acceptor or donor groups in the ortho and meta positions of the aniline to improve inhibitor binding in mammalian FTase, and likewise, H201α might be exploited with an appropriate group in the meta position. Accordingly, a series of FTase inhibitors was prepared in which the para-benzonitrile portion was modified in an attempt to target these two amino acids; the synthetic route that was pursued is shown in Scheme 3. We selected compound 1az bearing the N-(2-pyrimidinyl)-piper-idin-4-ylmethyl R3 group as the benchmark, anticipating that its moderate activity would better highlight the effects of modifying the para-benzonitrile component. Again, with the only constraint being that the non-methylated τ-nitrogen of the 1-methyl-1H-imidazol-5-ylmethyl group should bind the active site Zn2+ ion, several GOLD docking experiments of compound 1az were peformed. While the R3 = N-(2-pyrimidinyl)-piperidin-4-ylmethyl and the 1-methyl-1H-imidazole-4-sulfonyl groups in 1az bound differently to the corresponding groups in 1a, the R4 = para-benzonitrile group under investigation bound in the same subpocket (tetramethylene side chain of K164α and side chain of Y166α) as the corresponding motif in 1a in four out of the five highest scoring (lowest energy) docking solutions (for example, Figure 4A). Furthermore, in most of those cases, the para-benzonitrile moieties of 1a and 1az overlaid excellently, as illustrated in Figure 4B. Thus, we were confident the SAR data that would be acquired by varying the R4 aniline component of 1az would be directly translatable to 1a and related inhibitors.
Figure 4.

One high scoring rFTase active site conformation of inhibitor 1az (purple, colored by atom type) as identified by flexible ligand GOLD32 docking experiments (A) in isolation and (B) overlaid with the high scoring active site conformation of inhibitor 1a (green, colored by atom type) that was presented in Figure 1B. The co-substrate FPP was included in the docking experiments as this forms part of the binding surface.
As depicted in Scheme 3, mono-N-Boc-ethylenediamine (6) was sulfonylated with 1-methyl-1H-imidazole-4-sulfonyl chloride to give 15 in 95% yield. Chemoselective alkylation of the more acidic and less hindered sulfonamide NH was then readily accomplished by treatment of 15 with N-(2-pyrimidinyl)-piperidin-4-ylmethyl bromide in the presence of cesium carbonate in DMF. After TFA-mediated Boc deprotection of 16, arylation of the resultant primary amine was achieved by heating 17 at 120 °C in DMSO with a series of aryl fluorides, giving the secondary anilines 18 in a range of yields from 48 to 97%. Finally, these secondary anilines were then smoothly alkylated in good to excellent yields by deprotonation with NaH in DMF, followed by reaction with 5-chloromethyl-1-methyl-1H-imidazole · HCl29b to furnish the FTase inhibitors 19a–19h (Table 5).
Table 5.
Enzyme Inhibition and Whole Cell Data of Ethylenediamine-Based FTIs Exhibiting a Range of R4 Anilines
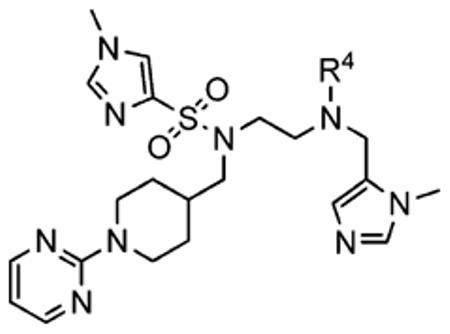 | ||||
|---|---|---|---|---|
| Compound | IC50 (nM) | Selectivity | ||
| Number | R4 | hFTase | GGTase-I | |
|
1az (FTI-2722) |
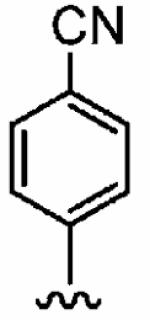
|
510 ± 62 | >10,000 | >20 |
|
19a (FTI-2718) |
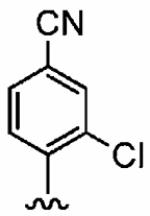
|
550 ± 110 | >10,000 | >18 |
|
19b (FTI-2733) |
|
520 ± 320 | >10,000 | >19 |
|
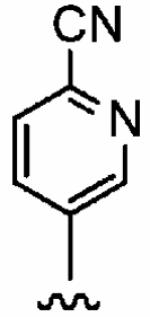
19c (FTI-2707) |
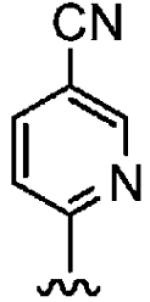
|
110 ± 26 | >10,000 | >91 |
|
19d (FTI-2709) |
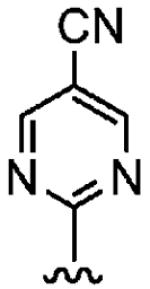
|
620 ± 100 | >10,000 | >16 |
|
19e (FTI-2712) |
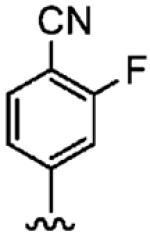
|
230 ± 110 | 9400 (n = 2) | 41 |
|
19f (FTI-2713) |
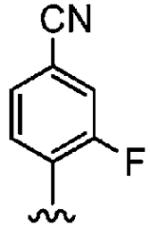
|
64 ± 8.6 | >10,000 | >156 |
|
I9g (FTI-2715) |
|
290 ± 180 | >10,000 | >34 |
|
19h (FTI-2719) |
|
490 ± 70 | 8750 ± 750 | 18 |
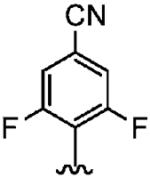
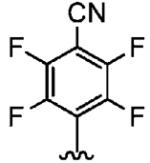
Positioning a chlorine (19a) at the ortho position had little effect relative to the parent compound 1az on the inhibition of hFTase. Conversely, replacement of an ortho-CH unit with nitrogen to give pyridine 19c conferred approximately 4-fold improvement in hFTase enzymatic inhibitory activity (19c 110 ± 26 nM vs 1az 510 ± 320 nM), while meta-substituted pyridine 19b offered no enhancement in activity, suggesting an important role for a hydrogen-bonding acceptor group in the ortho position. However, incorporation of nitrogens at both ortho positions, as in pyrimidine 19d, caused a reduction in the IC50 value back to approximately the same as that exhibited by the parent compound 1az. Inhibitor 19e with a fluorine at one of the meta positions was around twice as potent as 1az, while a fluorine at the ortho position led to an especially potent inhibitor (19f) with an hFTase IC50 of 64 ± 8.6 nM, approximately 8-fold as active as 1az and twice as potent as the ortho-pyridine 19c. Introduction of a second fluorine atom at the other ortho position (inhibitor 19g) caused a reduction in inhibition potency relative to the singly ortho-substituted inhibitor 19f, as was observed with the pyridine derivatives 19c and 19d (pyrimidine), respectively. Replacement of all four ring hydrogens in 19h offered no benefit to hFTase inhibition, relative to parent 1az. Finally, it is noteworthy that all derivatives based on the parent compound 1az generally exhibited very good to excellent selectivity for inhibition of hFTase over GGTase-I, particularly for ortho-pyridine 19c and ortho-fluoride 19f.
Scaffold Optimization
After optimizing the R1,R2,R3, R4, and X groups, we next investigated modifying the ethylenediamine core with a variety of alternative diamino-based scaffolds, whose structures are depicted in Table 6 and whose syntheses have been described elsewhere.30 1,3-Diaminopropane-based inhibitor 20a has increased conformational flexibility relative to the corresponding ethylenediamine-based inhibitor 1a. The data in Table 6 suggest that increasing the flexibility in this way caused more than a 7-fold drop in hFTase inhibitory activity. Likewise, reducing the conformational flexibility of the scaffold with the 1,2- and 1,3-diaminocyclopentyl derivatives 20b–20e also led to a decrease in hFTase inhibitor potency, with the cis-configurations exhibiting half the potencies of their trans-counterparts. Similar trends were observed in PfFTase.30 Of all the different scaffolds examined, the ethylenediamine unit was found to be optimal at delivering the four substituents into the proposed subpockets as well as furnishing the greatest GGTase-I/hFTase selectivity.
Table 6.
Enzyme Inhibition Data of FTIs Exhibiting a Range of Ethylenediamine-Analogue Scaffolds
 | ||||
|---|---|---|---|---|
| Compound | IC50 (nM) | Selectivity | ||
| Number | R4 | hFTase | GGTase-I | |
| 1a |
|
56 ± 29 | 2700 ± 2200 | 48 |
| 20a |
|
420 ± 210 | 4600 (n = 2) | 11 |
| 20b |
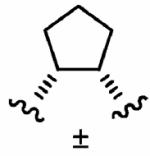
|
1000 ± 350 | >10,000 | >10 |
| 20c |
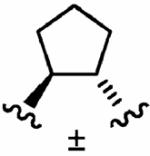
|
450 ± 70 | 9150 (n = 2) | 20 |
| 20d |
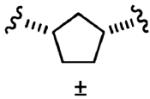
|
650 ± 220 | >10,000 | >15 |
| 20e |
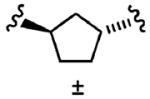
|
380 ± 100 | 7050 (n = 2) | 19 |
Optimization of FTIs
Finally, we designed three ethylenediamine-based inhibitors incorporating optimized R1, R2, R3, R4, and X substituents that were identified from our SAR studies, as illustrated by compounds 21a–21c in Table 7. Primary data considered in the selection of moieties that we hoped would furnish optimized inhibitors were in vitro FTase inhibition, whole cell H-Ras processing data, and to a lesser extent, GGTase-I/hFTase selectivity. The R1 group selected was methyl (Table 1), R2 was 2-pyridinesulfonyl (Table 2), R3 was cyclohexylmethyl (Table 3), R4 was para-benzonitrile (Table 4) substituted at the ortho position (Table 5), and the core scaffold incorporated was the simple ethylenediamine unit (Table 6), leading to inhibitors 21a, 21b, and 21c in Table 7. As a comparison, we have also included in Table 7 three of our most potent “unoptimized” FTIs: 1a, 1ax, and 1f. The trend observed in Table 5 upon varying the ortho-CH unit to CF and to N was reproduced with these optimized inhibitors, however none of compounds 21a–21c exhibited improved activity over the leads 1a, 1ax, and 1f, possibly due to the adoption of different binding geometries. Much reduced selectivities for hFTase over GGTase-I were also observed. Nonetheless, 21b was equipotent with 1ax in whole cells, whereby both inhibitors disrupted H-Ras processing with IC50 values of about 90 nM.
Table 7.
Enzyme Inhibition and Whole Cell Data of Optimized, Ethylenediamine-Based FTIs
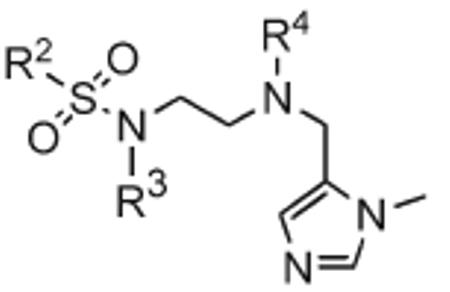 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | IC50 (nM) | Selectivity | Processing IC50 (μM) | |||||
| Number | R2 | R3 | R4 | hFTase | GGTase-I | H-Ras | Rap1A | |
|
1a (FTI-2585) |
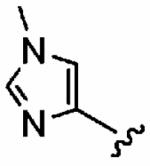
|
|
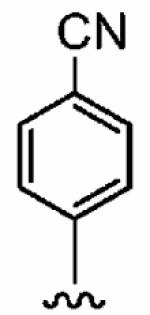
|
56 ± 29 | 2700 ± 2200 | 48 | 1.9 ± 1.2 | >10 |
|
1ax (FTI-2602) |
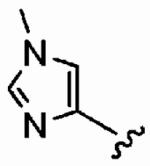
|
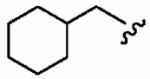
|
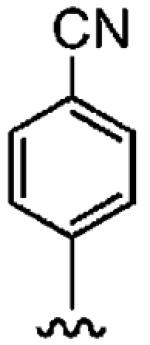
|
60 ± 10 | 530 ± 120 | 8.8 | 0.1 ±0.07 | >10 |
|
1f (FTI-2587) |
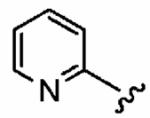
|
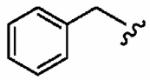
|
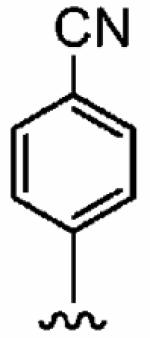
|
25 ± 20 | 820 ± 240 | 33 | 0.09 ± 0.06 | >10 |
|
21a (FTI-2736) |
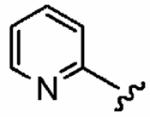
|
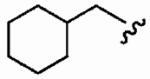
|
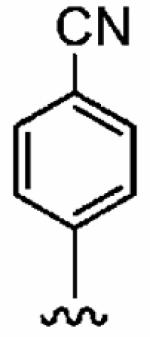
|
400 ± 160 | 610 ± 160 | 1.5 | 0.268 ± 0.212 | 6.2 ± 1.2 |
|
21b (FTI-2734) |
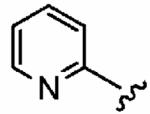
|
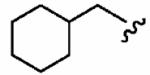
|
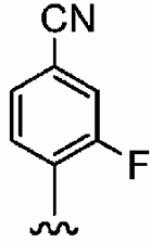
|
250 ± 190 | 520 ± 90 | 2.1 | 0.0887 ± 0.0258 | 2.7 ±1.5 |
|
21c (FTI-2735) |
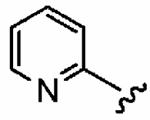
|
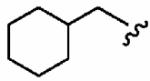
|
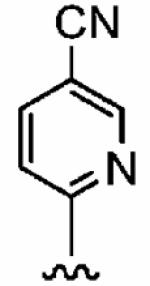
|
350 ± 170 | 690 ± 92 | 2 | 0.288 ± 0.251 | 19 |
Discussion of SAR Data in the Context of the Crystal Structure of 1a and the GOLD Docking of 1a
Figure 5 shows the crystal structure of 1a (yellow; taken from Figure 2A) overlaid with the flexible ligand GOLD docking of 1a (green; taken from Figure 1B). We correctly predicted that the more basic imidazole 1-methyl-1H-imidazol-5-ylmethyl would bind the zinc ion (which was the only constraint implemented in the GOLD docking experiments), and GOLD subsequently correctly placed the benzyl moiety of 1a in very close proximity to the sub-pocket formed by W102β, W106β, and Y361β. However, as Figure 5 illustrates, the para-benzonitrile and the sulfonylimidazole substituents were found to bind in different manners to those predicted. Specifically, the para-benzonitrile moiety was predicted to bind in the subpocket created by K164α, N165α, H201α, Y166α, but was actually found engaged in a weak π–π interaction with Y361β in the product exit groove, F360β, Y93β, L96β, W106β, while the sulfonylimidazole group was predicted to bind the Arg202β, yet was found stacked between the Zn-binding imidazole and the first isoprene of FPP.
Figure 5.
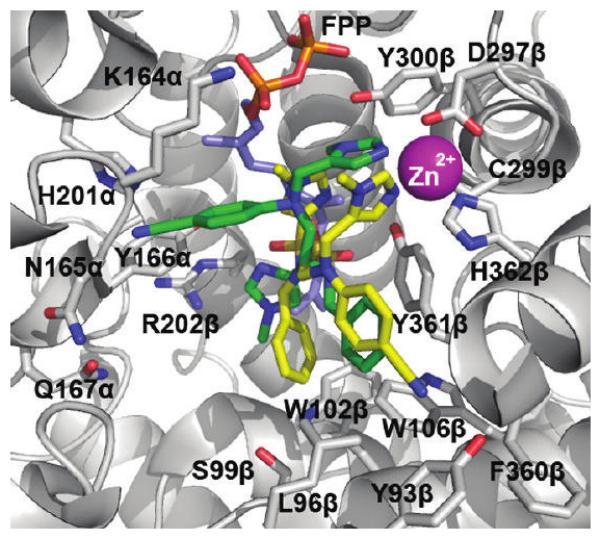
Overlay of the co-crystal structure of 1a (yellow, colored by atom type; PDB ID: 3E3238) and FPP with a high scoring (low energy) GOLD docked pose of 1a (green, colored by atom type) in the active site of rFTase.
Despite the GOLD prediction of a very different binding mode for the para-benzonitrile moiety, several potent inhibitors (e.g., 19f, Table 5) were serendipitously identified as a consequence of the para-benzonitrile making alternative but still significant contacts. The observed π–π stacking of the para-benzonitrile moiety against the Y361β residue suggests an explanation for the observed in vitro SAR data for the modifications made to the para-benzonitrile ring of 1a (Tables 1 and Table 4) and 1az (Table 5). π–π Stacking interactions are known to be energetically more favorable between an electron-rich arene, here Y361β, and an electron-poor arene, here the para-benzonitrile of inhibitor 1a. For the series of inhibitors in Table 4, substitution of the para position of the aniline with a strongly electron-withdrawing cyano group led to the most potent inhibitor (1a) while eliminating all substitution from the aniline ring of 1a and with R1 = H essentially abolished activity (compound 2, Table 1). Additionally, the product exit groove where the para-benzonitrile binds is of limited size and mostly hydro-phobic, suggesting that substituents larger than the linear cyano unit, such as the bulky tert-butyl ester in 11 or the phenyl ring in 14, and more polar than the cyano group, such as the acid in 12 and the carboxamide in 13, would be poorly tolerated. This was confirmed experimentally (Table 4). For the series of inhibitors in Table 5, replacement of the ortho (with respect to the aniline nitrogen) CH (1az) with either N (19c)or CF(19f) led to an approximate 5-fold or 8-fold improvement in inhibitor activity, respectively. This may be due to the introduced electron-withdrawing groups that render the aniline even more electron-poor, thereby enhancing its interaction with Y361β. On the other hand, replacement of the meta-CH (1az) with N (19b) resulted in no improvement in activity, while substitution of the meta-CH with a CF group (19e) led to only a 2-fold increase in inhibitor potency, as opposed to the 8-fold enhancement achieved by ortho-fluoride 19f. The crystal structure of 1a suggests that the incorporation of electronegative groups in the meta position might be poorly tolerated due to a clash with the anionic side chain of D359β, explaining the reduced enhancement in potency of inhibitors 19b and 19e (as a means of avoiding this unfavorable interaction, 180° rotation about the N–C aniline bond would direct the introduced N or CF group into a hydrophobic domain comprising L96β, W102β and W106β and would therefore also be unfavorable). Replacement of both ortho-CH groups with either two nitrogens (19d) or two CF groups (19g) was not tolerated relative to the mono-ortho-substituted inhibitors (19b and 19e, respectively). This observation may be a consequence of one of the electronegative groups being forced into the hydrophobic L96β, W102β and W106β region in order that the energetically favorable π–π stacking between the aniline ring of the inhibitor and Y361β is maintained.
Generally, there was little variation in activity among the R2 sulfonyl derivatives shown in Table 2, which may be a consequence of this moiety exhibiting insignificant interactions with the protein itself, being found stacked between the zinc-binding imidazole and FPP. However, there were clear trends in the hFTase inhibition data for the R3 series of sulfonamide analogues (Table 3). The crystal structure of 1a illustrates that the R3 phenyl ring can access the a2 residue binding site (L96β, W102β and W106β) of the Ca1a2X substrate. For the most part, small, hydrophobic R3 groups afforded potent hFTase inhibitors, likely due to the R3 group making van der Waals contacts with L96β, W102β and/or W106β. Within a particular series of R3 substituents, for example the ortho-, meta-, and para-tolyl derivatives 1ao, 1ap, and 1aq, respectively, ortho- and meta-functionalized compounds were tolerated, whereas para-functionalization was not. This may be a consequence of the para derivative being too large to access the a2 residue binding site. Indeed, the crystal structure of 1ay shows that the large N-Bocpiperidin-4-ylmethyl group traverses, rather than binds in, the a2 residue binding site and instead reaches into the X residue binding site.38 We have previously reported on the relative selectivities of our ethylenediamine-based inhibitors for the Plasmodium isoform of FTase over the mammalian isoform.29,30 With this crystal structure data in hand, along with several others,38 it should now be possible to tailor these compounds to become more selective for one enzyme isoform over the other.
Conclusions
In summary, we have developed a potent series of mammalian farnesyltransferase inhibitors (FTIs) based on an ethylenediamine scaffold. This class of compounds was identified by a “piggy-back” approach on our potent antimalarial inhibitors of plasmodium FTase. The simple and cost-effective ethylenediamine core allowed facile access to a diverse array of inhibitors, greatly facilitating lead inhibitor optimization. We have identified several inhibitors with double-digit nano-molar inhibition of hFTase in vitro, the most potent compound being 1f (IC50 = 25 nM). In most cases, potent inhibition of hFTase in vitro was accompanied by potent whole cell data (inhibition of H-Ras processing); for example, inhibitor 1f displayed a whole cell IC50 of 90 nM, one of the most active of the entire series. Moreover, in all but two cases inhibitors were selective for hFTase over GGTase-I, with the greatest selectivity (333-fold) exhibited by inhibitor 1g. Finally, we have solved the crystal structure of one of our ethylenediamine-based FTIs (1a) in the active site of rFTase, and this will assist in the future design of more potent and isoform-selective mammalian farnesyltransferase inhibitors.
Experimental Section
Ligand Docking Studies
Ligand energy minimization was peformed with the CVFF force field in InsightII on an SGI O2. Flexible ligand docking studies were subsequently performed with GOLD, version 3.0,32 on a Linux PC. Default GOLD parameters were utilized with the following exceptions: (i) maximal ligand flexibility was allowed; (ii) the affinity of nitrogen for zinc ion was increased in the GOLD parameters file to better reflect the ability of an imidazole ring to bind to the active site metal ion. Each ligand was used to seed the genetic algorithm 10 times.
Chemistry: General Methods
Solvents CH2Cl2,CH3CN, and DMF were dried on an Innovative Techonology SPS-400 dry solvent system. Anhydrous MeOH and DMSO were purchased from Sigma-Aldrich and used directly from their Sure-Seal bottles. Molecular sieves were activated by heating to 300 °C under vacuum overnight. All reactions were performed under an atmosphere of dry nitrogen in oven-dried glassware and were monitored for completeness by thin-layer chromatography (TLC) using silica gel (visualized by UV light, or developed by treatment with KMnO4 stain or Hanessian's stain). 1H and 13C NMR spectra were recorded on Bruker AM 400 MHz and Bruker AM 500 MHz spectrometers in either CDCl3, MeOH-d4, or DMSO-d6. Chemical shifts (δ) are reported in parts per million after calibration to residual isotopic solvent. Coupling constants (J) are reported in Hz. Mass spectrometry was performed using electrospray ionization on either a Varian MATCH-5 (HRMS) or Waters Micromass ZQ (LRMS). Before biological testing, all new target molecules (1b, 1c, 1ah–1an, 1aw, 11–14, 19a–19h, 21a–21c) were subjected to further purification by reversed-phase HPLC (rpHPLC). Analysis and purification by rpHPLC were performed using either a Phenonenex Luna 5 μm C18 (2) 250 mm × 21 mm column run at 15 mL/min (preparative) or a Microsorb-MV 300 A C18 250 mm × 4.6 mm column run at 1 mL/min (analytical), using gradient mixtures of (A) water with 0.1% TFA and (B) 10:1 acetonitrile/water with 0.1% TFA. Appropriate product fractions were pooled and lyophilized to dryness, affording the inhibitors as fluffy, white powders as their TFA salts. Inhibitor purity was confirmed by analytical rpHPLC using linear gradients from 100% A to 100% B, with changing solvent composition of either (I) 4.5% or (II) 1.5% per min after an initial 2 min of 100% A. For reporting HPLC data, percentage purity is given in parentheses after the retention time for each condition. FTase inhibitors 1a, 1d–1h, 1aa–1ag, 1ao–1av, 1ax–1az, 3–5, and 20a–20e have been previously reported in ref 29b.
General Procedure A (Reaction of Amines with Sulfonyl Chlorides)
The appropriate sulfonyl chloride (1.2 equiv) was added to a solution of the amine (1 equiv) and DIPEA (2 equiv) in anhydrous CH3CN (0.1 M) at 0°C. The reaction was warmed to room temperature and stirred for 16 h, at which time the solvent was evaporated. The residue was redissolved in CH2Cl2, washed with 5% NaHCO3, water, brine, dried (Na2SO4), filtered, and concentrated.
General Procedure B (Alkylation of Sulfonamides)
To a solution of the sulfonamide (1 equiv) and Cs2CO3 (2 equiv) in DMF (0.1 M) at 0 °C was added the appropriate alkyl bromide or chloride (1.1 equiv). The resulting mixture was stirred at room temperature overnight. The reaction was diluted with water, then extracted into EtOAc (×3). The combined EtOAc extractions were washed with 5% NaHCO3 (×3), brine, dried (Na2SO4), filtered, and concentrated.
General Procedure C (Reductive Amination of Primary Amines)
To a solution of 9 (1 equiv) in dry methanol (0.2 M) with 4 Å molecular sieves was added the appropriate aldehyde (1.1 equiv) and acetic acid (1.3 equiv). The reaction was stirred under nitrogen at room temperature for 30 min. Sodium cyanoborohydride (1.5 equiv) was added, and the resulting suspension was stirred at room temperature overnight. The reaction mixture was decanted, washing thoroughly with further MeOH, and then dry-loaded onto silica gel.
General Procedure D (Arylation of Primary Amines)
To a stirring solution of the primary amine (1 equiv) in DMSO (0.2 M) were added the aryl fluoride (1.2 equiv) and DIPEA (3 equiv). The reaction mixture was heated to 120 °C for 48 h. After allowing the reaction to cool, H2O was added, and the crude product was extracted with EtOAc (×3). The EtOAc extractions were combined, washed with water (×3), brine, dried (Na2SO4), filtered, and concentrated.
General Procedure E (Alkylation of Secondary Anilines)
The secondary aniline (1 equiv) was dissolved in DMF (0.07 M), and then the reaction was cooled to 0 °C. After 15 min, NaH (3 equiv) was added in one portion. After a further 15min, 5-chloromethyl-1-methyl-1H-imidazole·3 HCl31 (1.1 equiv) was added. The reaction was allowed to stir at 0 °C from 2–3 h, when TLC indicated the reaction was complete or had stalled. Upon quenching the reaction with brine (approximately 1 mL for 1 mmol scale), the reaction was diluted with water and extracted with EtOAc (×3). The EtOAc extractions were combined, and washed with 5% NaHCO3 (×3), brine, dried (Na2SO4), filtered, and concentrated.
General Procedure F (Cyanation of Aryl Bromides)
A stirring solution of the aryl bromide (1 equiv) in DMF (0.1 M) was degassed, and then Zn(CN)2 (0.6 equiv), Pd(PPh3)4 (0.1 equiv), Zn dust (0.05 equiv), and Zn(OAc)2 (0.05 equiv) were added. The mixture was heated to 120 °C for 2 h, after which time the reaction was allowed to cool, diluted with water, and extracted into EtOAc (×3). The EtOAc extractions were combined and washed with 5% NaHCO3 (×3), brine, dried (Na2SO4), filtered, and concentrated.
4-[(2-Aminoethyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile (9)
To a solution of 4-[(2-{N-tert-butoxycarbonyl}-aminoethyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile (compound 9c in ref 29b) (8.0 g, 30.6 mmol) in CH2Cl2 (75mL) cooled to 0 °C was added TFA (75mL). The reaction was warmed to room temperature and stirred for 30 min, after which time the solvent was concentrated in vacuo, and then the crude material was repeatedly re-dissolved in CHCl3 and re-evaporated until a constant weight was achieved, to afford the ditrifluoroacetic acid salt of 9 as a light-brown solid (14.8 g, 100%): δH (500 MHz, MeOH-d4)3.20(t, J =7.1Hz,2H,CH2CH2NH2), 3.81 (t, J = 7.1 Hz, 2H, CH2CH2NH2), 3.87 (s, 3H, CH3 (Im)), 4.82 (s, 2H, CH2Im), 6.96 (d, J = 9.0 Hz, 2H, 2 CH(Ar)), 7.19 (s, 1H, CH (Im)), 7.56 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 8.88 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 33.9, 37.1, 45.3, 48.2, 100.8, 114.0, 118.4, 120.4, 132.3, 134.6, 137.5, 151.0; HRMS (ESI) m/z calcd for [C14H17N5 + H] 256.1562, obsd 256.1559. Subsequently, the free base form of 9, which is the form used for all subsequent reactions, was furnished by passing the di-TFA salt through a short pad of silica gel (eluent CH2Cl2/MeOH/NH4OH, 92:7:1).
N-Benzyl, N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl} Propane-1-sulfonamide (1b)
Compound 9 was treated with propane-1-sulfonyl chloride, according to general procedure A, on a 0.15 mmol scale. The crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give N-{2-[(4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl} propane-1-sulfonamide as a colorless film (46 mg, 85%): δH (500 MHz, MeOH-d4)0.98 (t, J = 7.5 Hz, 3H, CH2CH2CH3), 1.71 (sextet, J = 7.5 Hz, 2H, CH2CH2CH3), 2.96 (m, 2H, NHCH2CH2N), 3.21–3.26 (m, 2H, CH2CH2CH3), 3.61 (t, J = 6.5 Hz, 2H, NHCH2CH2N), 3.84 (s, 3H, CH3 (Im)), 4.79 (s, 2H, CH2Im), 6.91 (d, J = 9.1 Hz, 2H, 2CH (Ar)), 7.20 (s, 1H, CH (Im)), 7.47 (d, J = 9.1 Hz, 2H, 2 CH (Ar)), 8.82 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 11.7, 16.9, 32.8, 39.7, 44.4, 50.6, 53.2, 98.9, 112.6, 117.7, 119.4, 131.5, 133.3, 136.2, 150.4; LRMS (ESI) m/z 384.2 [C17H23N5O2S+Na]. Subsequently, the sulfonamide (36 mg, 0.1 mmol) was treated with benzyl bromide according to general procedure B. After the usual workup and flash chromatography over silica gel (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), the title compound 1b was furnished as a glassy film (37 mg, 82%): δH (400 MHz, MeOH-d4)1.20(t, J = 7.4 Hz, 3H, CH2CH2CH3), 1.99 (m, 2H, CH2CH2CH3), 3.24 (t, J = 7.8 Hz, 2H, N(Bn)CH2CH2N), 3.40 (obscured, 4H, NHCH2CH2N, CH2CH2CH3), 3.94 (s, 3H, CH3 (Im)), 4.52 (s, 2H, CH2Ph), 4.65 (s, 2H, CH2Im), 6.75 (d, J = 8.9 Hz, 2H, 2 CH (Ar)), 7.25 (s, 1H, CH (Im)), 7.55 (m, 7H, 5 CH (Ph), 2 CH (Ar)), 9.00 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 13.7, 18.6, 34.6, 45.4, 46.8, 51.6, 53.4, 55.1, 100.3, 114.1, 119.6, 121.2, 129.8, 130.7, 133.1, 135.1, 138.2, 138.7, 151.9, 152.9; HRMS (ESI) m/z calcd for [C24H29N5O2S+H] 452.2120, obsd 452.2108; HPLC (I) tR = 13.45 min (100%), (II) tR = 19.72 min (100%).
N-Benzyl, N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl} Cyclopropanesulfonamide (1c)
First, amine 9 was reacted with cyclopropanesulfonyl chloride, according to general procedure A, on a 0.15 mmol scale. After workup and flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1), N-{2-[(4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl} cyclopropanesulfonamide was yielded as a colorless film (44 mg, 82%): δH (500 MHz, MeOH-d4) 0.98 (m, 4H, 2 CH2 (cyclopropyl)), 2.51 (m, 1H, CH (cyclopropyl)), 3.33 (t, J = 5.8 Hz, 2H, NHCH2CH2N), 3.67 (t, J = 5.8 Hz, 2H, NHCH2CH2N), 3.88 (s, 3H, CH3 (Im)), 4.76 (s, 2H, CH2Im), 6.95 (d, J = 8.83 Hz, 2H, 2 CH (Ar)), 7.25 (s, 1H, CH (Im)), 7.53 (d, J=8.84 Hz, 2H, 2CH (Ar)), 8.86 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 4.1, 29.0, 32.8, 39.9, 44.5, 50.5, 99.1, 112.7, 117.8, 119.4, 131.6, 133.4, 136.3, 150.4; LRMS (ESI) m/z 382.1 [C17H21N5O2S + Na]. Subsequently, the sulfonamide (36 mg, 0.1 mmol) was treated with benzyl bromide, according to general procedure B. The crude material was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 1c as a white foam (40 mg, 89%): δH (500 MHz, MeOH-d4) 0.95 (m, 4H, 2 CH2 (cyclopropyl)), 2.55 (m, 1H, CH (cyclopropyl)), 3.31 (m, 4H, NCH2CH2N), 3.71 (s, 3H, CH3 (Im)), 4.32 (s, 2H, CH2Ph), 4.43 (s, 2H, CH2Im), 6.51 (d, J = 8.89 Hz, 2H, 2 CH (Ar)), 7.02 (s, 1H, CH (Im)), 7.26–7.33 (m, 7H, 5 CH (Ph), 2 CH (Ar)), 8.76 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 5.3, 28.1, 34.3, 45.1, 46.3, 51.1, 54.9, 100.3, 113.7, 119.1, 120.8, 129.4, 129.9, 130.3, 132.7, 134.7, 137.8, 138.2, 151.5; HRMS (ESI) m/z calcd for [C24H27N5O2S + H] 450.1964, obsd 450.1985; HPLC (I) tR = 12.70 min (100%), (II) tR = 18.99 min (100%).
[N-(Furan-3-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1ah, 4.38)
Furan-3-carbaldehyde was reductively aminated with amine 9 according to general procedure C, on a 0.15 mmol scale. Purification by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) then gave 4-[{2-[(furan-3-ylmethyl)-amino]-ethyl}-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile as a film (39 mg, 78%): δH (400 MHz, MeOH-d4) 3.24 (obscured t, 2H, NHCH2CH2N), 3.78 (m, 5H, NHCH2CH2N, CH3 (Im)), 4.07 (s, 2H, CH2-furan), 4.75 (s, 2H, CH2Im), 6.50 (s, 1H, CH (furan)), 6.88 (d, J =9.0 Hz, 2H, 2 CH (benzonitrile)), 7.11 (s, 1H, CH (furan)), 7.42–7.53 (m, 3H, 2 CH (benzonitrile), CH (Im)), 7.62 (s, 1H, CH (furan)), 8.82 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 34.5, 43.5, 44.7, 45.9, 47.7, 101.7, 111.9, 114.5, 117.3, 119.0, 120.8, 132.9, 135.2, 138.2, 145.1, 146.0, 151.5; LRMS (ESI) m/z 358.2 [C19H21N5O + Na]. This secondary amine (34 mg, 0.1 mmol) was reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A to yield the title compound 1ah as a white foam (43 mg, 90%): δH (500 MHz, MeOH-d4) 3.38 (obscured t, 2H, ((furan-3-ylmethyl)-NHCH2CH2N)), 3.56 (t, J = 7.0 Hz, 2H, ((furan-3-ylmethyl)-NHCH2CH2N)), 3.81 (s, 3H, CH3 (Im)), 3.89 (s, 3H, CH3 (Im)), 4.19 (s, 2H, CH2-furan), 4.70 (s, 2H, CH2Im), 6.57 (s, 1H, CH (furan)), 6.77 (d, J = 8.94 Hz, 2H, 2 CH (benzonitrile)), 7.25 (s, 1H, CH (furan)), 7.46–7.52 (m, 4H, 2 CH (benzonitrile), 2 CH (Im)), 7.78 (s, 1H, CH (furan)), 7.81 (s, 1H, CH (Im)), 8.91 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 34.3, 34.4, 45.3, 45.5, 45.9, 51.0, 100.4, 111.9, 113.8, 119.3, 120.8, 122.0, 126.8, 132.9, 134.8, 137.9, 139.1, 141.5, 143.2, 145.3, 151.7; HRMS (ESI) m/z calcd for [C23H25N7O3S + H] 480.1818, obsd 480.1809; HPLC (I) tR = 12.62 min (100%).
[N-(Thiophen-3-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1ai, 4.39)
Thiophene-3-carbaldehyde was reductively aminated with amine 9 (38 mg, 0.15 mmol) according to general procedure C to give, after flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1), 4-((3-methyl-3H-imidazol-4-ylmethyl)-{2-[(thiophen-3-ylmethyl)-amino]-ethyl}-amino)-benzonitrile as a glassy film (40 mg, 76%): δH (400 MHz, MeOH-d4) 3.20 (obscured t, 2H, NHCH2CH2N), 3.70–3.75 (m, 5H, NHCH2CH2N, CH3 (Im)), 4.16 (s, 2H, CH2-thiophene), 4.69 (s, 2H, CH2Im), 6.74 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 7.06 (s, 1H, CH (Im)), 7.09 (d, J = 3.9 Hz, 1H, CH (thiophene)), 7.41–7.48 (m, 4H, 2CH (benoznitrile), 2 CH (thiophene)), 8.77 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 34.7, 45.0, 46.1, 47.4, 47.9, 102.0, 114.7, 119.2, 120.9, 129.0, 129.1, 129.5, 133.1, 133.2, 135.4, 138.4, 151.6; LRMS (ESI) m/z 374.2 [C19H21N5S + Na]. This secondary amine (35 mg, 0.1 mmol) was then reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A to give, after purification by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), compound 1ai as a white foam (46 mg, 93%): δH (400 MHz, MeOH-d4)3.29(obscured t, 2H, ((thiophen-3-ylmethyl)NCH2CH2N)), 3.39 (t, J =6.9Hz, 2H, ((thiophen-3-ylmethyl)NCH2CH2N)), 3.69 (s, 3H, CH3 (Im)), 3.75 (s, 3H, CH3 (Im)), 4.20 (s, 2H, 2 CH (CH2-thiophene)), 4.50 (s, 2H, CH2Im), 6.57 (d, J = 9.1 Hz, 2H, 2 CH (benoznitrile)), 6.95 (d, J = 3.8 Hz, 1H, CH (thiophene)), 7.09 (s, 1H, CH (thiophene)), 7.22 (s, 1H, CH (Im)), 7.31 (m, 1H, CH (thiophene)), 7.36 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.65 (s, 1H, CH (Im)), 7.70 (s, 1H, CH (Im)), 8.79 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 34.7, 34.8, 45.5, 46.7, 50.3, 51.5, 100.7, 114.1, 119.6, 121.2, 126.2, 127.2, 128.3, 129.7, 133.2, 135.2, 138.2, 139.0, 139.5, 141.9, 152.0; HRMS (ESI) m/z calcd for [C23H25N7O2S2 + H] 496.1589, obsd 496.1576; HPLC (I) tR = 12.64 min (99.0%), (II) tR = 18.84 min (98.2%).
[N-(Furan-2-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1aj, 4.41)
Furan-2-carbaldehyde was reductively aminated with amine 9 according to general procedure C, on a 0.15 mmol scale. Purification by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) afforded 4-[{2-[(furan-2-ylmethyl)-amino]-ethyl}-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile as a film (39 mg, 78%): δH (500 MHz, MeOH-d4) 3.31 (obscured t, 2H, NHCH2CH2N), 3.84 (t, J = 7.4 Hz, 2H, NHCH2CH2N), 3.87 (s, 3H, CH3 (Im)), 4.33 (s, 2H, CH2-furan), 4.81 (s, 2H, CH2Im), 6.48 (m, 1H, CH (furan)), 6.61 (d, J = 3.19 Hz, 1H, CH (furan)), 6.95 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 7.18, (s, 1H, CH (Im)), 7.54 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 7.60 (m, 1H, CH (furan)), 8.88 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 32.8, 43.0, 43.1, 44.2, 46.0, 100.1, 110.7, 112.4, 113.0, 117.4, 119.0, 131.2, 133.5, 136.5, 144.5, 144.8, 149.7; LRMS (ESI) m/z 358.2 [C19H21N5 O + Na]. This secondary amine was reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A, on a 0.1 mmol scale. After workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish the title compound 1aj as a white foam (43 mg, 90% 47.91818): δH (500 MHz, MeOH-d4) 3.34 (t, J = 6.9 Hz, 2H, ((furan-3-ylmethyl)NCH2CH2N)), 3.46 (t, J = 6.9 Hz, 2H, ((furan-3-ylmethyl)NCH2CH2N)), 3.67 (s, 3H, CH3 (Im)), 3.79 (s, 3H, CH3 (Im)), 4.23 (s, 2H, CH2-furan), 4.63 (s, 2H, CH2Im), 6.18 (m, 1H, CH (furan)), 6.23 (m, 1H, CH (furan)), 6.73 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.15 (s, 1H, CH (Im)), 7.29 (s, 1H, CH (furan)), 7.41 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.59 (s, 1H, CH (Im)), 7.64 (s, 1H, CH (Im)), 8.79 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 34.3, 45.4, 46.5, 47.1, 49.6 (obscured), 50.8, 100.4, 111.3, 111.7, 113.9, 119.3, 120.9, 126.8, 132.9, 134.8, 137.8, 139.1, 141.4, 144.4, 150.9, 151.8; HRMS (ESI) m/z calcd for [C23H25N7O3S + H] 480.1818, obsd 480.1833; HPLC (I) tR = 12.51 min (100%).
[N-(Thiophen-2-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1ak, 4.42)
Thiophene-2-carbaldehyde was reductively aminated with amine 9 (38 mg, 0.15 mmol) according to general procedure C, giving, after flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1), 4-((3-methyl-3H-imidazol-4-ylmethyl)-{2-[(thiophen-2-ylmethyl)-amino]-ethyl}-amino)-benzonitrile as a colorless film (38 mg, 72%): δH (500 MHz, MeOH-d4)3.37 (obscured t, 2H, NHCH2CH2N), 3.8–3.92 (m, 5H, NHCH2-CH2N, CH3 (Im)), 4.54 (s, 2H, CH2-thiophene), 4.86, (s, 2H, CH2Im), 6.87 (m, 1H, CH (thiophene)), 7.00 (d, J = 8.8 Hz, 2H, 2 CH (benzonitrile)), 7.13 (m, 1H, CH (thiophene)), 7.23 (s, 1H, CH (Im)), 7.34 (m, 1H, CH (thiophene)), 7.59 (d, J = 8.8 Hz, 2H, 2 CH (benzonitrile)), 8.93 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 32.8, 42.9, 44.2, 45.0, 45.9, 100.2, 113.0, 117.4, 119.0, 127.3, 128.2, 130.6, 131.2, 131.5, 133.5, 136.6, 149.7; LRMS (ESI) m/z 374.2 [C19H21N5S + Na]. This secondary amine (35 mg, 0.1 mmol) was reactedwith1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A. The usual workup and purification by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) gave 1ak as a glassy film (44 mg, 89%): δH (400 MHz, MeOH-d4)3.31 (t, J = 7.1 Hz, 2H, ((thiophen-2-yl)NCH2-CH2N)), 3.47 (t, J = 7.1 Hz, 2H, ((thiophen-2-yl)NCH2CH2N)), 3.69 (s, 3H, CH3 (Im)), 3.75 (s, 3H, CH3 (Im)), 4.41 (s, 2H, CH2-thiophene), 4.55 (s, 2H, CH2Im), 6.61, (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 6.85 (m, 1H, CH (thiophene)), 6.89 (d, J = 2.70 Hz, 1H), 7.11 (s, 1H, CH (Im)), 7.29 (m, 1H, CH (thiophene)), 7.37 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 7.67 (s, 1H, CH (Im)), 7.69 (s, 1H, CH (Im)), 8.78 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 34.7, 34.8, 45.7, 46.6, 50.3 (obscured), 51.0, 100.9, 114.2, 119.6, 121.2, 126.5, 127.2, 128.5, 130.0, 133.2, 135.2, 138.2, 139.5, 140.7, 141.9, 152.0; HRMS (ESI) m/z calcd for [C23H25N7O2S2 + H] 496.1589, obsd 496.1585; HPLC (I) tR = 12.90 min (100%).
[N-(3,5-Dimethyl-isoxazol-4-ylmethyl), N-{2-[(4-Cyanophenyl)(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1al, 4.36)
Amine 9 was reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A, on a 0.15 mmol scale. Flash column chromatography of the crude material over silica gel (eluent CH2Cl2/MeOH/ NH4OH, 92:7:1) afforded 1-methyl-1H-imidazole-4-sulfonic acid {2-[(4-cyano-phenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}-amide as a white powder (55 mg, 92%): δH (400 MHz, MeOH-d4)3.19(t, J = 6.3 Hz, 2H, NHCH2CH2N), 3.64 (t, J = 6.3 Hz, 2H, NHCH2CH2N), 3.72 (s, 3H, CH3 (Im)), 3.88 (s, 3H, CH3 (Im)), 4.81 (s, 2H, CH2Im), 6.89 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 7.22 (s, 1H, CH (Im)), 7.50 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 7.62 (s, 1H, CH (Im)), 7.71 (s, 1H, CH (Im)), 8.87 (s, 1H, CH (Im)); δC (100 MHz, MeOH-d4) 33.8, 33.9, 40.7, 45.4, 50.9, 99.5, 113.5, 118.5, 120.5, 125.3, 132.3, 134.2, 137.2, 139.9, 140.7, 151.2; HRMS (ESI) m/z calcd for [C18H21N7O2S + H] 400.1556, obsd 400.1545. This secondary sulfonamide was then alkylated with 4-chloromethyl-3,5-dimethyl-isoxazole according to general procedure B, on a 0.1 mmol scale. After workup, purification was accomplished by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish the title compound 1al as a white foam (40 mg, 79%): δH (500 MHz, MeOH-d4)2.15 (s, 3H, CH3 (isoxazole)), 2.20 (s, 3H, CH3 (isoxazole)), 3.23 (t, J = 6.7 Hz, 2H, ((isoxazol-4-ylmethyl)NCH2-CH2N)), 3.53 (t, J = 6.7 Hz, 2H, ((isoxazol-4-ylmethyl)NCH2-CH2N)), 3.77 (s, 3H, CH3 (Im)), 3.82 (s, 3H, CH3 (Im)), 4.02 (s, 2H, CH2-isoxazole), 4.56 (s, 2H, CH2Im), 6.66 (d, J = 8.7 Hz, 2H, 2 CH (benzonitrile)), 7.17 (s, 1H, CH (Im)), 7.45 (d, J =8.7 Hz, 2H, 2 CH (benzonitrile)), 7.74 (s, 1H, CH (Im)), 7.78 (s, 1H, CH (Im)), 8.85 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 10.2, 10.9, 34.3, 34.4, 44.4, 45.0, 47.0, 51.2, 100.6, 110.7, 113.7, 119.3, 120.8, 127.1, 132.6, 134.9, 137.9, 138.2, 141.7, 151.4, 161.6, 169.6; HRMS (ESI) m/z calcd for [C24H28N8O3S H] 509.2083, obsd 509.2072; HPLC (I) tR = 12.27 min (100%), (II) tR = 17.76 min (100%).
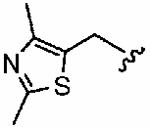
[N-(2,4-Dimethyl-thiazol-5-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1am, 4.40)
2,4-Dimethyl-thiazole-5-carbaldehyde was reductively aminated with amine 9 (38 mg, 0.15 mmol) according to general procedure C to give, after flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1), 4-[{2-[(2,4-dimethyl-thiazol-5-ylmethyl)-amino]-ethyl}-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile as a glassy film (42 mg, 74%): δH (500 MHz, MeOH-d4)2.34(s,3H,CH3 (thiazole)), 2.60 (s, 3H, CH3 (thiazole)), 3.32 (t, J = 7.06 Hz, 2H, NHCH2CH2N), 3.83 (m, 5H, NHCH2CH2N, CH3 (Im)), 4.39 (s, 2H, CH2-thiazole), 4.78 (s, 2H, CH2Im), 6.92 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 7.14 (s, 1H, CH (Im)), 7.51 (d, J = 9.0 Hz, 2H, 2 CH (benzonitrile)), 8.85 (s, 1H, CH (Im)); LRMS (ESI) m/z 403.2 [C20H24N6S + Na]. The secondary amine (38 mg, 0.1 mmol) was then treated with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A. Workup and purification (flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1)) as usual yielded 1am as a white foam (47 mg, 90%): δH (500 MHz, MeOH-d4)2.21 (s, 3H, CH3 (thiazole)), 2.55 (s, 3H, CH3 (thiazole)), 3.39 (t, J = 6.5 Hz, 2H, ((thiazol-5-ylmethyl)-NCH2CH2N)), 3.64 (t, J = 6.5 Hz, 2H, ((thiazol-5-ylmethyl)-NCH2CH2N)), 3.77 (s, 3H, CH3 (Im)), 3.86 (s, 3H, CH3 (Im)), 4.38 (s, 2H, CH2-thiazole), 4.73 (s, 2H, CH2Im), 6.71 (d, J =9.1 Hz, 2H, 2 CH (benzonitrile)), 7.20 (s, 1H, CH (Im)), 7.46 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.76 (s, 1H, CH (Im)), 7.79 (s, 1H, CH (Im)), 8.87 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 14.8, 18.9, 34.7, 34.8, 45.9, 47.3, 47.6, 51.3, 100.8, 114.0, 119.6, 121.1, 127.5, 128.6, 133.2, 135.2, 138.3, 138.8, 142.0, 142.1, 150.9, 151.8; HRMS (ESI) m/z calcd for [C24H28N8O2S2 + H] 525.1855, obsd 525.1828; HPLC (I) tR = 12.14 min (96.61%), (II) tR = 17.00 min (96.03%).
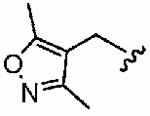
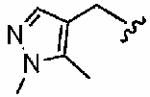
[N-(1,3,5-Trimethyl-1H-pyrazol-4-ylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1an, 4.49)
Trimethyl-1H-pyrazole-4-carbaldehyde was reductively aminated with amine 9, according to general procedure C, on a 0.15 mmol scale. The crude material was chromatographed over silica gel (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give 4-((3-methyl-3H-imidazol-4-ylmethyl)-{2-[(1,3,5-trimethyl-1H-pyrazol-4-ylmethyl)-amino]-ethyl}-amino)-benzonitrile as a film (46 mg, 81%): δH (400 MHz, MeOH-d4)2.28(s, 3H, CH3 (pyrazole)), 2.34 (s, 3H, CH3 (pyrazole)), 3.39 (t, J = 7.5 Hz, 2H, NHCH2CH2N), 3.75 (s, 3H, N–CH3 (pyrazole)), 3.88–3.95 (m, 5H, NHCH2CH2N, CH3 (Im)), 4.15 (s, 2H, CH2-pyrazole), 4.88 (s, 2H, CH2Im), 7.01 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 7.22 (s, 1H, CH (Im)), 7.59 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 8.94 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 9.8, 11.4, 34.3, 36.1, 42.3, 44.7, 45.6, 47.5, 99.9, 108.3, 114.3, 118.8, 120.6, 132.8, 135.0, 138.0, 142.6, 148.5, 151.3; LRMS (ESI) m/z 400.2 [C21H27N7 + Na]. This secondary amine (38 mg, 0.1 mmol) was then reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A. Workup then the usual purification (silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1)) furnished the title compound 1an as a white foam (44 mg, 84%): δH (500 MHz, MeOH-d4)2.00 (s, 3H, CH3 (pyrazole)), 2.06 (s, 3H, CH3 (pyrazole)), 3.15 (t, J = 6.5 Hz, 2H, ((pyrazole-4-ylmethyl)-NCH2CH2N)), 3.44 (t, J = 6.5 Hz, 2H, ((pyrazole-4-ylmethyl)-NCH2CH2N)), 3.51 (s, 3H, N-CH3 (pyrazole)), 3.73 (s, 3H, CH3 (Im)), 3.78 (s, 3H, CH3 (Im)), 3.92 (s, 2H, CH2-pyrazole), 4.48 (s, 2H, CH2Im), 6.50 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 7.11 (s, 1H, CH (Im)), 7.39 (d, J = 8.9 Hz, 2H, 2 CH (benzonitrile)), 7.72 (s, 1H, CH (Im)), 7.75 (s, 1H, CH (Im)), 8.80 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4)9.5, 11.7, 34.3, 34.4, 36.0, 45.1, 45.4, 46.5, 51.2, 100.2, 111.8, 113.4, 119.2, 120.8, 127.1, 132.7, 134.8, 137.9, 138.1, 141.2, 141.7, 147.9, 151.4; HRMS (ESI) m/z calcd for [C25H31-N9O2S + H] 522.2400, obsd 522.2377; HPLC (I) tR = 12.49 min (95.83%), (II) tR = 17.78 min (96.33%).
[N-(Cyclopropylmethyl), N-{2-[(4-Cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (1aw, 4.34)
First, cyclopropanecarbaldehyde was reductively aminated with amine 9 (38 mg, 0.15 mmol), according to general procedure C. Flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) purified the material to give 4-[[2-(cyclopropylmethyl-amino)-ethyl]-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzonitrile as a colorless film (39 mg, 84%): δH (400 MHz, CDCl3)0.07 (m, 2H, 2 CH (cyclopropyl)), 0.46 (m, 2H, 2 CH (cyclopropyl)), 0.78 (m, 1H, CHCH2NHCH2CH2N), 2.41 (m, 2H, CH2-cyclopropyl), 2.75 (s, 2H, NHCH2CH2N), 3.50 (m, 2H, NHCH2CH2N), 3.55 (s, 3H, CH3 (Im)), 4.88 (s, 2H, CH2Im), 6.68 (d, J =8.8 Hz, 2H, 2 CH (benzonitrile)), 6.86 (s, 1H, CH (Im)), 7.26 (s, 1H, CH (Im)), 7.46 (d, J = 8.8 Hz, 2H, 2 CH (benzonitrile); LRMS (ESI) m/z 332.2 [C18H23N5 + Na]. This secondary amine was then reacted with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A, on a 0.1 mmol scale. After workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 1aw as a white powder (39 mg, 86%): δH (500 MHz, MeOH-d4) 0.14 (m, 2H, 2 CH (cyclopropyl)), 0.43 (m, 2H, 2 CH (cyclopropyl)), 0.84 (m, 1H, CHCH2NCH2-CH2N), 2.50 (d, J =6.9,2H, CH2-cyclopropyl), 2.99 (t, J = 6.9 Hz, 2H, ((cyclopropylmethyl)NCH2CH2N)), 3.73 (s, 3H, CH3 (Im)), 3.81 (t, J = 6.9 Hz, 2H, ((cyclopropylmethyl)-NCH2CH2N)), 3.88 (s, 3H, CH3 (Im)), 4.85 (s, 2H, CH2Im), 6.97 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.28 (s, 1H, CH (Im)), 7.62 (d, J = 9.1 Hz, 2H, 2 CH (benzonitrile)), 7.67 (s, 1H, CH (Im)), 7.71 (s, 1H, CH (Im)), 8.87 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 4.6, 11.0, 34.3, 34.4, 45.8, 46.8, 51.6, 55.9, 100.4, 114.1, 119.3, 120.9, 126.5, 133.1, 134.9, 137.9, 139.5, 141.4, 151.9; HRMS (ESI) m/z calcd for [C22H27N7-O2S + H] 454.2025, obsd 454.2009; HPLC (I) tR = 12.55 min (99.05%), (II) tR = 18.46 min (98.47%).
tert-Butyl 4-[{2-[benzyl-(1-methyl-1H-imidazole-4-sulfonyl)-amino]-ethyl}-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzoate (10)
tert-Butyl p-fluorobenzoate (750 mg, 3.93 mmol, 1 equiv) was dissolved in ethylenediamine (9;4mL,59.8mmol,15.2equiv)and heated to 120 °C for 24 h. All solvent was evaporated, and then the residue was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to furnish tert-butyl 4-(2-amino-ethylamino)-benzoate as a white powder (891 mg, 96%): δH (400 MHz, CD2Cl2)1.59 (s, 9H, CO2-(CH3)3), 2.98 (t, J = 5.8 Hz, 2H, H2NCH2CH2NH), 3.23 (q, J = 5.8 Hz, 2H, H2NCH2CH2NH), 4.67 (br, 1H, H2NCH2CH2NH), 6.62 (d, J = 8.6 Hz, 2H, 2 CH (Ar)), 7.81 (d, J = 8.6 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CD2Cl2) 28.9, 41.7, 46.6, 80.3, 112.1, 120.9, 131.9, 152.9, 166.7; HRMS (EI+) m/z calcd for [C13H20N2O2] 236.1525, obsd 236.1527. tert-Butyl 4-(2-amino-ethylamino)-benzoate (700 mg, 3.03 mmol, 1 equiv) was then sulfonylated with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A. The crude material was dry-loaded onto silica gel, andthenpurifiedbyflash column chromatography (eluent CH2-Cl2/MeOH/NH4OH, 192:7:1) to give tert-butyl 4-[2-(1-methyl-1H-imidazole-4-sulfonylamino)-ethylamino]-benzoate as a white powder (1.04 g, 90%): δH (500 MHz, CDCl3) 1.56 (s, 9H, CO2-(CH3)3), 3.17 (m, 2H, SO2NHCH2CH2NH), 3.28 (m, 2H, SO2NHCH2CH2NH), 3.67 (s, 3H, CH3 (Im)), 6.51 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.46 (s, 1H, CH (Im)), 7.51 (s, 1H, CH (Im)), 7.75 (d, J = 8.5 Hz, 2H, 2 CH (Ar)); δC (125 MHz, MeOH-d4) 28.7, 34.6, 42.4, 43.1, 80.8, 111.9, 120.5, 125.2, 131.8, 140.1, 140.3, 152.4, 167.4; HRMS (ESI+) m/z calcd for [C17H24N4O4S + H] 381.1597, obsd 381.1573. tert-Butyl 4-[2-(1-methyl-1H-imidazole-4-sulfonylamino)-ethylamino]-benzoate (610 mg, 1.63 mmol, 1 equiv) was then chemoselectively benzylated on the sulfonamide NH with benzyl bromide according to general procedure B. The crude material was then flash chromatographed over silica gel to furnish tert-butyl 4-{2-[benzyl-(1-methyl-1H-imidazole-4-sulfonyl)-amino]-ethylamino}-benzoate as a white solid (703 mg, 92%): δH (400 MHz, CDCl3)1.52(s,9H,CO2(CH3)3), 3.13 (t, J = 6.2 Hz, 2H, (Bn)NCH2CH2NH), 3.43 (t, J = 6.2 Hz, 2H, (Bn)NCH2CH2NH), 3.68 (s, 3H, CH3 (Im)), 4.32 (s, 2H, CH2Ph), 6.30 (d, J = 8.8 Hz, 2H, 2 CH (Ar)), 7.23–7.30 (m, 5H, 5 CH (Ph)), 7.42 (s, 1H, CH (Im)), 7.49 (s, 1H, CH (Im)), 7.70 (d, J = 8.8 Hz, 2H, 2 CH (Ar)); δC (500 MHz, CDCl3) 28.1, 33.9, 41.6, 46.6, 52.6, 79.6, 111.1, 119.9, 124.4, 127.9, 128.3, 128.6, 131.1, 136.0, 139.0, 139.8, 151.1, 166.1; HRMS (ESI ) m/z calcd for [C24H30N4O4S + H] 471.2066, obsd 471.2053. tert-Butyl 4-{2-[benzyl-(1-methyl-1H-imidazole-4-sulfonyl)-amino]-ethyl-amino}-benzoate (220 mg, 0.473 mmol, 1 equiv) was alkylated on the aniline NH with 5-chloromethyl-1-methyl-1H-imidazole 3 HCl31 according to general procedure E. The crude material was flash chromatographed over silica gel (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford the title compound 11 as a colorless foam (200 mg, 76%): δH (500 MHz, CDCl3) 1.53 (s, 9H, C(CH3)3), 3.09–3.15 (m, 2H, CH2CH2NSO2), 3.29–3.34 (m, 2H, CH2CH2-NSO2), 3.43 (s, 3H, CH3), 3.73 (s, 3H, CH3), 4.26 (s, 2H, CH2Ph), 4.30 (s, 2H, CH2Im), 6.35 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.69 (s, 1H, CH (Im)), 7.31–7.37 (m, 5H, 5 CH (Ph)), 7.39 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 7.46 (s, 1H, CH (Im)), 7.68 (d, J = 9.0 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 28.2, 31.6, 33.9, 44.1, 45.0, 49.7, 54.3, 79.8, 111.0, 120.0, 124.4, 127.5, 128.1, 128.6, 128.7, 128.9, 131.1, 136.2, 138.5, 139.0, 139.1, 150.3, 165.8; HRMS (ESI+) m/z calcd for [C29H36N6O4S + H] 565.2597, obsd 565.2576; HPLC (I) tR = 12.53 min (100%),(II) tR = 18.77 min (100%).
4-[{2-[Benzyl-(1-methyl-1H-imidazole-4-sulfonyl)-amino]-ethyl}(3-methyl-3H-imidazol-4-ylmethyl)-amino]-benzoic Acid (12)
TFA (1.5 mL) was added to a solution of 11 (150 mg, 0.268 mmol, 1 equiv) in CH2Cl2 (1.5 mL), and the resulting mixture was stirred for 3 h at room temperature, after which time all solvents were evaporated. Residual TFA was removed by repeated coevaporation with a 2:1 mixture of CHCl3/MeOH, then the residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 25:7:1) to afford the title compound as a sticky foam (131 mg, 96%): δH (500 MHz, MeOH-d4)3.34–3.39 (m, 2H, CH2CH2NSO2), 3.42–3.46 (m, 2H, CH2CH2NSO2), 3.81 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.30 (s, 2H, CH2Ph), 4.55 (s, 2H, CH2Im), 6.53 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 7.15 (s, 1H, CH (Im)), 7.34–7.38 (m, 5H, 5 CH (Ph)), 7.76–7.79 (m, 3H, 2 CH (Ar), CH (Im)), 7.82 (s, 1H, CH (Im)), 8.87 (s, 1H, CH (Im)); δC (125 MHz, MeOH-d4) 34.3, 34.4, 45.0, 46.5, 51.2, 55.2, 112.8, 119.3, 120.3, 126.8, 129.3, 129.9 (2 CH), 132.7, 133.1, 137.7, 137.9, 139.1, 141.5, 152.0, 170.1; HRMS (ESI+) m/z calcd for [C25H28N6O4S + H] 509.1971, obsd 509.1953; HPLC (I) tR = 11.24 min (100%), (II) tR = 15.21 min (100%).
4-[{2-[Benzyl-(1-methyl-1H-imidazole-4-sulfonyl)-amino]-ethyl}(1-methyl-1H-imidazol-4-ylmethyl)-amino]-benzamide (13)
To a solution of acid 12 (50 mg, 0.0984 mmol, 1 equiv), NH4Cl (10.5 mg, 0.197 mmol, 2 equiv), and DIPEA (34 μL) in DMF (1 mL) was added HBTU (48.5 mg, 0.128 mmol, 1.3 equiv). The reaction mixture was stirred for 1 h, after which time TLC indicated the reaction was complete. Water (25 mL) was added, and then the crude product was extracted into EtOAc (5 × 5mL). The EtOAc extractions were combined, washed with water (3 × 5 mL), brine (5 mL), dried (Na2SO4), filtered, and concentrated. The residue was dry-loaded onto silica gel, then flash chromatographed (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to furnish primary amide 13 as a white powder (44 mg, 89%): δH (500 MHz, CDCl3) 3.15 (m, 2H, (Bn)NCH2CH2N), 3.31 (m, 2H, (Bn)NCH2CH2N), 3.41 (s, 3H, CH3 (Im)), 3.71 (s, 3H, CH3 (Im)), 4.23 (s, 2H, CH2Ph), 4.29 (s, 2H, CH2Im), 6.02 (br, 2H, NH2), 6.42 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.66 (s, 1H, CH (Im)), 7.29–7.34 (m, 6H, 5 CH (Ph), CH (Im)), 7.41 (s, 1H, CH (Im)), 7.45 (s, 1H, CH (Im)), 7.56 (d, J = 9.0 Hz, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 31.3, 33.7, 43.9, 44.8, 49.4, 50.2, 54.0, 111.1, 121.0, 124.2, 127.2, 127.9, 128.4, 128.6, 128.9, 136.0, 138.4, 138.8, 138.9, 149.8, 168.9; HRMS (ESI+) m/z calcd for [C25H29N7O3S + H] 508.2137, obsd 508.2131; HPLC (I) tR = 11.72 min (100%), (II) tR = 16.29 min (100%).
[N-Benzyl, N-{2-[(biphenyl-4-yl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}] 1-methyl-1H-imidazole-4-sulfonamide (14)
Previously reported 1-tert-butoxycarbonylamino-{2-[biphenyl-4-yl-(3-methyl-3H-imidazol-4-ylmethyl)-amino]}-ethane (compound 9a in ref 31) (30 mg, 0.0739 mmol) was deprotected by stirring in a 1:1 mixture of TFA-CH2Cl2 (1 mL) for 30 min. After removal of all solvent in vacuo, the residue (23 mg, 0.0739 mmol) was sulfonylated with 1-methyl-1H-imidazole-4-sulfonyl chloride according to general procedure A, but with 5 equiv of DIPEA. After the usual workup, the residue was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to afford 1-methyl-1H-imidazole-4-sulfonic acid benzyl-[2-(biphenyl-4-ylamino)-ethyl]-amide as a colorless film (30 mg, 91%) Next, the secondary aniline of 1-methyl-1H-imidazole-4-sulfonic acid benzyl-[2-(biphenyl-4-ylamino)-ethyl]-amide (30 mg, 0.067 mmol, 1 equiv) was alkylated with the HCl salt of 5-chloromethyl-1-methyl-1H-imidazole31 according to general procedure E. After the usual workup, the crude material was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give 14 as a glassy film (24 mg, 65%): δH (500 MHz, CDCl3) 3.16 (m, 2H, (Bn)NCH2-CH2N), 3.33 (m, 2H, (Bn)NCH2CH2N), 3.44 (s, 3H, CH3 (Im)), 3.75 (s, 3H, CH3 (Im)), 4.27 (s, 2H, CH2Ph), 4.38 (s, 2H, CH2Im), 6.56 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.79 (s, 1H, CH (Im)), 7.34 – 7.51 (m, 15 H, 10 CH (Ph), 3 CH (Im), 2 CH (Ar)); δC (125 MHz, CDCl3) 31.9, 34.2, 44.9, 45.4, 50.0, 54.5, 113.5, 124.6, 126.4, 126.5, 128.1, 128.3 (2), 128.9, 129.0, 129.2, 129.4, 130.5, 136.8, 138.8, 139.3, 139.9, 141.1, 147.2; HRMS (ESI+) m/z calcd for [C30H32N6-O2S + H] 541.2380, obsd 541.2386; HPLC (I) tR = 13.01 min (100%), (II) tR = 20.24 min (100%).
N-(2-tert-Butoxycarbonylaminoethyl) 1-Methyl-1H-imidazole-4-sulfonamide (15)
Mono-N-Boc-ethylenediamine (6; 5 g, 31.2 mmol, 1 equiv) was sulfonylated with 1-methyl-1H-imidazole-4-sulfonyl chloride (6.76 g, 37.4 mmol, 1.2 equiv), according to general procedure A. After 16 h, the solvent was reduced and then the reaction mixture was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 1092:7:1) to give the title compound as a white powder (9 g, 95%): δH (500 MHz, CD2Cl2) 1.37 (s, 9H, C(CH3)3), 2.99 (t, J = 5.8 Hz, 2H, CH2NHSO2), 3.11 (q, J = 5.8 Hz, 2H, CH2NHBoc), 3.71 (s, 3H, CH3), 5.53 (s, 1H, NHSO2), 5.68 (s, 1H, NHBoc), 7.48 (s, 1H, Im), 7.56 (s, 1H, Im); δC (125 MHz, CD2Cl2) 27.6, 33.5, 39.8, 42.6, 78.9, 124.0, 127.2, 139.2, 156.1; HRMS (ESI+) m/z calcd for [C11H20N4O4S + H] 305.1284, obsd 305.1294.
[N-(2-tert-Butoxycarbonylaminoethyl), N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (16)
N-(2-tert-Butoxycarbonylaminoethyl) 1-methyl-1H-imidazole-4-sulfonamide (5.94 g, 19.5 mmol, 1 equiv) was chemoselectively alkylated on the sulfonamide NH with N-(2-pyrimidinyl)-piperidin-4-ylmethyl bromide (6 g, 23.4 mmol, 1.2 equiv), according to general procedure B, but the reaction was stirred for 4 d at room temperature. [N-(2-Pyrimidinyl)-piperidin-4-ylmethyl bromide was made from the reaction of N-(2-pyrimidinyl)-piperidin-4-ylmethyl alcohol (5 g, 25.9 mmol, 1 equiv) with PPh3Br2 (14.2 g, 33.7 mmol, 1.3 equiv) in CH2Cl2 (130 mL) at 0°C to room temperature over 6 h. At this point, the reaction was complete. Water (100 mL) was added to quench, and then the CH2Cl2 layer was diluted further (500 mL total volume). The organic layer was collected, and the aqueous layer was extracted twice further (2 × 50 mL). The organic layers were collected, dried (Na2SO4), filtered, and concentrated. The residue was redissolved in CH2Cl2, dry-loaded onto silica gel, then purified by flash column chromatography (eluent Hex/EtOAc, 7:1) to furnish N-(2-pyrimidinyl)-piperidin-4-ylmethyl bromide as a white powder (6.04 g, 91%).] After workup, the crude material was dry-loaded onto silica gel and purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), affording the title compound as a white solid (8.81 g, 94%): δH (500 MHz, CDCl3)1.09(qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.36 (s, 9H, C(CH3)3), 1.69–1.78 (m, 2H, 2 CH (piperidinylmethyl)), 1.84–1.92 (m, 1H, CH (piperidinylmethyl)), 2.77–2.83 (m, 2H, 2 CH (piperidinylmethyl)), 2.97 (br d, J = 8.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.25–3.34 (m, 4H, CH2CH2NHBoc), 3.67 (s, 3H, CH3), 4.64–4.69 (m, 2H, 2 CH (piperidinylmethyl)), 5.53 (m, 1H, NHBoc), 6.34 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.36 (s, 1H, CH (Im)), 7.40 (s, 1H, CH (Im)), 8.20 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 28.3, 29.4, 34.8, 35.4, 39.6, 43.4, 49.1, 55.2, 79.0, 109.2, 124.2, 138.9, 140.6, 155.9, 157.5, 161.4; HRMS (ESI+) m/z calcd for [C21H33N7O4S + H] 480.2393, obsd 480.2409.
[N-(2-Aminoethyl), N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (17)
To a cooled (0 °C), stirring solution of 16 (8.67 g, 18.1 mmol) in CH2Cl2 (47.5 mL) was added triisopropylsilane (TIPS; 2.5 mL), H2O (2.5 mL) and TFA (47.5 mL). After 5 min, the reaction was removed from the ice bath and allowed to stir for 1 h at room temperature, and then all solvents were evaporated. The residue was redissolved in CH2Cl2 and the minimal required volume of MeOH, then dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give 17 as a white, sticky foam (7.2 g, 100%): δH (500 MHz, CDCl3)1.10 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.73–1.78 (m, 2H, 2 CH (piperidinylmethyl)), 1.87–1.95 (m, 1H, CH (piperidinyl)), 2.77–2.83 (m, 2H, 2 CH (piperidinylmethyl)), 2.92 (t, J = 6.5 Hz, 2H, CH2NH2) 3.00 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.32 (t, J = 6.4 Hz, 2H, CH2CH2NH2), 3.46–3.51 (br s, NH2), 3.69 (s, 3H, CH3 (Im)), 4.66–4.72 (m, 2 H, 2 CH (piperidinylmethyl)), 6.38 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 7.40 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)), 8.22 (t, J = 4.7 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.5, 33.8, 35.5, 40.1, 43.4, 52.5, 54.9, 109.2, 124.2, 139.0, 139.4, 158.5, 161.5; HRMS (ESI+) m/z calcd for [C16H25N7O2S + H] 380.1869, obsd 380.1886.
[N-{2-(2-Chloro-4-cyanophenyl)-aminoethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18a)
Compound 17 was reacted with 2-chloro-4-fluorobenzonitrile on a 0.338 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a colorless, viscous oil (158 mg, 91%): δH (500 MHz, CDCl3)1.08 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.68–1.74 (m, 2H, 2 CH (piperidinylmethyl)), 1.77–1.86 (m, 1H, CH (piperidinylmethyl)), 2.72 (td, J = 12.5, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.00 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.48–3.55 (m, 4H, CH2CH2NHAr), 3.71 (m, 3H, CH3 (Im)), 4.63–4.69 (m, 2 H, 2 CH (piperidinylmethyl)), 5.77 (t, J = 5.3 Hz, 1H, CH2NHAr), 6.40 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 6.66 (d, J = 8.5 Hz, 1H, CH (Ar)), 7.37 (dd, J = 8.5, 2.0 Hz, 1H, CH (Ar)), 7.43 (s, 1H, CH (Im)), 7.45 (d, J = 2.0 Hz, 1H, CH (Ar)), 7.46 (s, 1H, CH (Im)), 8.22 (t, J = 4.7 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 33.9, 36.0, 42.2, 43.4, 48.7, 56.1, 98.7, 109.4, 110.0, 118.6, 119.0, 124.4, 132.4, 132.5, 139.1, 139.5, 147.0, 157.5, 161.4; HRMS (ESI+) m/z calcd for [C23H27N8O2SCl + H] 515.1744, obsd 515.1754.
N-(2-(2-Cyanopyridin-5-ylamino)ethyl)-1-methyl-N-((1-(pyrimidin-2-yl)piperidin-4-yl)methyl)-1H-imidazole-4-sulfonamide (18b)
Compound 17 was reacted with 2-cyano-5-fluoropyridine on a 0.791 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a colorless, viscous oil (320 mg, 84%): δH (500 MHz, CDCl3) 0.95 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.63–1.71 (m, 2H, 2 CH (piperidinylmethyl)), 1.74–1.83 (m, 1H, CH (piperidinylmethyl)), 2.72 (td, J = 12.5, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.93 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.23–3.27 (m, 2H, CH2CH2NHAr), 3.34–3.36 (m, 2H, CH2CH2NHAr), 3.65 (m, 3H, CH3 (Im)), 4.55–4.59 (m, 2 H, 2 CH (piperidinylmethyl)), 6.54 (m, 1H, CH2NHAr), 6.96 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.04 (m, 1H, CH (Ar)), 7.62 (s, 1H, CH (Im)), 7.77–7.78 (m, 2H, CH (Im), CH (Ar)), 8.02 (m, 1H, CH (Ar)), 8.28 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, DMSO-d6) 28.96, 33.34, 34.88, 40.33, 41.20, 43.03, 47.87, 54.82, 109.46, 117.39, 118.92, 124.99, 129.69, 136.90, 137.56, 139.75, 146.98, 157.72, 161.02; HRMS (ESI+) m/z calcd for [C22H27N9O2S + H] 482.2087, obsd 482.2086.
[N-{2-(5-Bromopyridin-2-yl)-aminoethyl}, N-{N-(2-pyrimidinyl)-piperidin-4-ylmethyl}] 1-methyl-1H-imidazole-4-sulfonamide (18c)
Compound 17 was reacted with 5-bromo-2-fluoropyridine on a 0.512 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a colorless, viscous oil (200 mg, 73%): δH (500 MHz, CDCl3) 1.11 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.72–1.78 (m, 2H, 2 CH (piperidinylmethyl)), 1.87–1.97 (m, 1H, CH (piperidinylmethyl)), 2.78 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.03 (br d, J = 8.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.49–3.57 (m, 4H, CH2CH2-NHAr), 3.72 (m, 3H, CH3 (Im)), 4.67–4.73 (m, 2 H, 2 CH (piperidinylmethyl)), 5.62 (t, J = 5.3 Hz, 1H, CH2NHAr), 6.34 (dd, J = 9.0 Hz, 1H, CH (Ar)), 6.41 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.40 (dd, J = 9.0, 2.5 Hz, 1H, CH (Ar)), 7.42 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)), 8.05 (d, J = 2.5 Hz, 1H, CH (Ar)), 8.25 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.7, 33.9, 35.6, 40.5, 43.6, 48.5, 55.0, 106.8, 109.3, 109.7, 124.4, 138.9, 139.3, 140.0, 148.4, 156.9, 157.6, 161.5; HRMS (ESI+) m/z calcd for [C21H27N8O2SBr + H] 535.1239, obsd 535.1259.
[N-{2-(5-Bromopyrimidin-2-yl)-aminoethyl},N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18d)
Compound 17 was reacted with 5-bromo-2-fluoropyrimidine on a 0.528 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a white foam (255mg, 90%): δH (500 MHz, CDCl3) 1.08 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.70–1.77 (m, 2H, 2 CH (piperidinylmethyl)), 1.83–1.92 (m, 1H, CH (piperidinylmethyl)), 2.78 (td, J = 12.8, 2.5Hz, 2H, 2 CH (piperidinylmethyl)), 3.03 (br d, J = 8.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.43 (t, J = 6.3 Hz, 2H ,CH2CH2NHAr), 3.57 (t, J = 6.3 Hz, 2H, CH2CH2NHAr), 3.68 (m, 3H, CH3 (Im)), 4.60–4.66 (m, 2 H, 2 CH (piperidinylmethyl)), 6.39 (t, J = 4.6 Hz, 1H, CH (pyrimidine)), 7.40 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)), 8.19 (s, 2H, 2CH(Ar)), 8.21 (t, J = 4.6 Hz, 2H, 2CH (pyrimidine)); δC (125 MHz, CDCl3) 29.5, 33.8, 35.5, 40.4, 43.6, 48.3, 55.0, 106.1, 109.3, 124.3, 139.1, 139.6, 157.6, 158.1, 160.3, 161.3; HRMS (ESI+) m/z calcd for [C20H26N9O2SBr + H] 536.1192, obsd 536.1213.
[N-{2-(3-Fluoro-4-cyanophenyl)-aminoethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18e)
Compound 17 was reacted with 2,4-difluorobenzonitrile on a 0.417 mmol scale, according to general procedure D, to give a separable mixture of the two expected isomers. After the usual workup, the crude material was purified by prepTLC (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to give the title compound as a white foam (99 mg, 48%): δH (500 MHz, CDCl3) 1.15 (qd, J = 12.5, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.73–1.79 (m, 2H, 2 CH (piperidinylmethyl)), 1.87–1.95 (m, 1H, CH (piperidinylmethyl)), 2.80 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.03 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.36 (q, J = 5.5 Hz, 2H, CH2CH2NHAr), 3.65 (t, J = 5.8 Hz, 2H, CH2CH2NHAr), 3.77 (m, 3H, CH3 (Im)), 4.70–4.75 (m, 2 H, 2 CH (piperidinylmethyl)), 6.10–6.13 (m, 1H, CH2NHAr), 6.30 (dd, J = 12.0, 2.0 Hz, 1H, CH (Ar)), 6.39 (dd, J = 8.8, 2.0 Hz, 1H, CH (Ar)), 6.44 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.32 (dd, J = 8.8, 7.3 Hz, 1H, CH (Ar)), 7.47 (s, 1H, CH (Im)), 7.52 (s, 1H, CH (Im)), 8.27 (t, J = 4.9 Hz, 2H, CH (pyrimidine)); δC (125 MHz, CDCl3) 29.5, 34.0, 35.5, 41.5, 43.5, 47.7, 54.4, 87.0 (d, JCF = 15.5 Hz), 98.1 (d, JCF = 22.8 Hz), 108.8 (br), 109.4, 115.7, 124.6, 133.9 (d, JCF = 2.75 Hz), 139.0, 139.6, 153.4 (d, JCF = 10.9 Hz), 157.6, 161.5, 165.0 (d, JCF = 252 Hz); HRMS (ESI+) m/z calcd for [C23H27N8O2FS + H] 499.2040, obsd 499.2057.
[N-{2-(2-Fluoro-4-cyanophenyl)-aminoethyl}, N-{N-(2-pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18f)
Compound 17 was reacted with 3,4-difluorobenzonitrile on a 0.343 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a white foam (138 mg, 82%): δH (500 MHz, CDCl3)1.13 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.73–1.78 (m, 2H, 2 CH (piperidinylmethyl)), 1.83–1.91 (m, 1H, CH (piperidinylmethyl)), 2.78 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.04 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.51 (q, J = 5.7 Hz, 2H, CH2CH2NHAr), 3.59 (t, J = 6.0 Hz, 2H, CH2CH2NHAr), 3.75 (m, 3H, CH3 (Im)), 4.69–4.75 (m, 2 H, 2 CH (piperidinylmethyl)), 5.74–5.78 (m, 1H, CH2NHAr), 6.43 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 6.68 (t, J = 8.5 Hz, 1H, CH (Ar)), 7.19 (dd, J = 11.5, 1.5 Hz, 1H, CH (Ar)), 7.31 (br dd, J = 8.5, 1.5 Hz, 1H, CH (Ar)), 7.44 (s, 1H, CH (Im)), 7.49 (s, 1H, CH (Im)), 8.27 (t, J = 4.7 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.5, 33.9, 35.7, 41.6, 43.4, 48.3, 55.1, 97.4 (d, JCF = 9.13 Hz), 109.4, 110.8 (d, JCF = 4.5 Hz), 117.5 (d, JCF = 21.9 Hz), 119.2 (d, JCF = 2.75 Hz), 124.3, 130.1 (d, JCF = 2.75 Hz), 139.1, 139.6, 140.5 (d, JCF = 11.9 Hz), 149.8 (d, JCF = 241 Hz), 157.6, 161.4; HRMS (ESI+) m/z calcd for [C23H27N8O2FS + H] 499.2040, obsd 499.2055.
[N-{2-(2,6-Difluoro-4-cyanophenyl)-aminoethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18g)
Compound 17 was reacted with 3,4,5-trifluorobenzonitrile on a 0.359 mmol scale, according to general procedure D. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a white foam (179 mg, 97%): δH (500 MHz, CDCl3) 1.09 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.68–1.75 (m, 2H, 2 CH (piperidinylmethyl)), 1.81–1.90 (m, 1H, CH (piperidinylmethyl)), 2.75 (td, J = 12.5, 2.5 Hz, 2H, 2 CH(piperidinylmethyl)), 2.98 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.51 (t, J = 5.8 Hz, 2H, CH2CH2NHAr), 3.72 (m, 5H, CH2CH2-NHAr, CH3 (Im)), 4.65–4.73 (m, 2H, 2 CH (piperidinylmethyl)), 5.67 (m, 1H, CH2NHAr), 6.40 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.05 (dd, J = 7.5, 2.5 Hz, 2H, CH (Ar)), 7.43 (s, 1H, CH (Im)), 7.47 (s, 1H, CH (Im)), 8.23 (t, J = 4.9 Hz, 2H, 2CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 33.9, 35.7, 43.5, 43.7, 50.2, 55.4, 96.7 (t, JCF = 11.4 Hz), 109.3, 115.8 (dd, JCF = 18.3, 8.13 Hz), 117.9 (t, JCF = 3.19 Hz), 124.4, 130.8 (t, JCF = 12.3 Hz), 139.0, 139.7, 150.8 (dd, JCF = 241, 10.0 Hz), 157.5, 161.4; HRMS (ESI+) m/z calcd for [C23H26N8O2F2S + H] 517.1946, obsd 517.1953.
[N-{2-(2,3,5,6-Tetrafluoro-4-cyanophenyl)-aminoethyl},N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (18h)
Compound 17 was reacted with 2-chloro-4-fluorobenoznitrile on a 0.338 mmol scale, according to general procedure D, although the reaction was complete within 12 h at room temperature. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give the title compound as a white foam (158 mg, 85%): δH (500 MHz, CDCl3) 1.13 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.72–1.79 (m, 2H, 2 CH (piperidinylmethyl)), 1.88–1.97 (m, 1H, CH (piperidinylmethyl)), 2.81 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.97 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.63 (t, J = 5.0 Hz, 2H, CH2CH2NHAr), 3.77 (s, 3H, CH3 (Im)), 3.78–3.84 (m, 2H, CH2-CH2NHAr), 4.71–4.77 (m, 2H, 2CH (piperidinylmethyl)), 6.44 (t, J = 4.8 Hz, 1H, CH (pyrimidine)), 6.81–6.86 (m, 1H, CH2NHAr), 7.47 (s, 1H, CH (Im)), 7.51 (s, 1H, CH (Im)), 8.27 (t, J = 4.8 Hz, 2H, CH (pyrimidine)); δC (125 MHz, CDCl3) 29.7, 34.1, 35.9, 43.4, 43.5, 50.2, 55.0, 78.4 (t, JCF = 18.3 Hz), 109.0 (m), 109.5, 124.7, 133.5 (m), 135.9 (dm), 139.0, 140.1, 148.1 (dm), 157.7, 161.6; HRMS (ESI+) m/z calcd for [C23H24N8O2F4S + H] 553.1757, obsd 553.1774.
[N-{2-[(2-Chloro-4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19a)
As per general procedure E with 18a on a 0.068 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/ MeOH/NH4OH, 192:7:1) to afford the title compound as a white foam (37 mg, 90%): δH (500 MHz, CDCl3) 1.04 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.63–1.68 (m, 2H, 2 CH (piperidinylmethyl)), 1.70–1.76 (m, 1H, CH (piperidinylmethyl)), 2.76 (td, J = 12.5, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.94 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.31–3.36 (m, 2H, SO2NCH2CH2N), 3.39–3.44 (m, 2H, SO2-NCH2CH2N), 3.51 (s, 3H, CH3 (Im)), 3.73 (m, 3H, CH3 (Im)), 4.41 (s, 2H, CH2Im), 4.65–4.71 (m, 2 H, 2 CH (piperidinylmethyl)), 6.42 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 6.97 (s, 1H, CH (Im)), 7.23 (d, J = 8.5 Hz, 1H, CH (Ar)), 7.36 (s, 1H, CH (Im)), 7.41 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.48 (dd, J = 8.5, 2.0 Hz, 1H, CH (Ar)), 7.63 (d, J = 2.0 Hz, 1H, CH (Ar)), 8.26 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 31.7, 33.9, 36.0, 43.5, 45.7, 47.1, 50.7, 55.6, 107.1, 109.4, 117.6, 123.7, 124.1, 126.6, 129.0, 129.9, 131.4, 134.3, 138.9, 139.0, 139.7, 151.0, 157.6, 161.5; HRMS (ESI+) m/z calcd for [C28H33-N10O2SCl + H] 609.2275, obsd 609.2296; HPLC (I) tR = 12.90 min (99.13%), (II) tR = 19.65 min (98.61%).
N-(2-((2-Cyanopyridin-5-yl)((1-methyl-1H-imidazol-5-yl)methyl)-amino)ethyl)-1-methyl-N-((1-(pyrimidin-2-yl)piperidin-4-yl)methyl)-1H-imidazole-4-sulfonamide (19b)
As per general procedure E with 18b on a 0.415 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford the title compound as a white foam (186 mg, 78%): δH (500 MHz, DMSO-d6) 1.05–1.13 (m, 4H, 4 CH (piperidinylmethyl)), 1.69–1.74 (m, 1H, CH (piperidinylmethyl)), 2.79 (td, J = 12.5, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.95 (br d, J = 7.0 Hz, 2H, 2 CH(piperidinylmethyl)), 3.28 (s, 3H, CH3 (Im)), 3.34–3.39 (m, 2H, SO2NCH2CH2N), 3.41–3.42 (m, 2H, SO2NCH2CH2N), 3.68 (s, 3H, CH3 (Im)), 4.38 (s, 2H, CH2Im), 4.60–4.64 (m, 2 H, 2 CH (piperidinylmethyl)), 6.41 (t, J = 4.8 Hz, 1H, CH (pyrimidine)), 6.86 (s, 1H, CH (Ar)), 7.02 (s, 1H, CH (Im)), 7.20 (m, 1H, CH (Ar)), 7.41–7.50 (m, 2H, CH (Im), CH (Ar)), 7.65 (s, 1H, CH (Im)), 7.80 (s, 1H, CH (Im)), 8.26 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, DMSO-d6) 29.5, 32.0, 32.8, 34.0, 35.9, 43.5, 44.2, 48.9, 49.2, 49.4, 49.7, 53.2, 56.2, 109.5, 118.1, 120.8, 122.3, 124.6, 129.4, 135.8, 139.3, 145.5, 157.7, 161.4; HRMS (ESI+) m/z calcd for [C27H33N11O2S + H] 576.2618, obsd 576.2617.
[N-{2-[(5-Cyanopyridin-2-yl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19c)
As per general procedure E with 18c on a 0.178 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish [N-{2-[(5-bromopyridin-2-yl)-(3-methyl-3H-imidazol-4-ylmethyl)amino]ethyl},N-{N-(2-pyrimidinyl)-piperidin-4-ylmethyl}] 1-methyl-1H-imidazole-4-sulfonamide as a white foam (107 mg, 96%): δH (500 MHz, CDCl3) 1.12 (qd, J = 12.5, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.72–1.80 (m, 3H, 3 CH (piperidinylmethyl)), 2.84 (td, J = 13.0, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.98 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.14–3.18 (m, 2H, NSO2CH2CH2N), 3.55 (s, 3H, CH3 (Im)), 3.62–3.67 (m, 2H, NSO2CH2CH2N), 3.73 (m, 3H, CH3 (Im)), 4.69–4.78 (m, 2H, 2 CH (piperidinylmethyl)), 4.80 (s, 2H, CH2Im), 6.43 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 6.69 (d, J = 9.5 Hz, 1H, CH (Ar)), 6.96 (s, 1H, CH (Im)), 7.38 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.46 (s, 1H, CH (Im)), 7.55 (dd, J = 9.0, 2.5 Hz, 1H, CH (Ar)), 8.15 (d, J = 2.5 Hz, 1H, CH (Ar)), 8.27 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 31.8, 33.9, 35.6, 41.0, 43.5, 46.2, 47.0, 55.9, 107.1, 107.6, 109.3, 124.2, 128.2, 129.1, 138.8, 138.9, 139.3, 140.1, 148.2, 155.9, 157.6, 161.5; HRMS (ESI+) m/z calcd for [C26H33N10O2SBr + H] 629.1770, obsd 629.1780; HPLC (I) tR = 12.78 min (100%), + (II) tR = 18.88 min (100%). The aryl bromide was then converted to the corresponding aryl nitrile on a 0.115 mmol scale, according to general procedure F. After workup, the crude material was dry-loaded onto silica gel, and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 19c as a white foam (51 mg, 77%): δH (500 MHz, CDCl3) 1.11 (qd, J = 12.5, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.67–1.77 (m, 3H, 3 CH (piperidinylmethyl)), 2.78–2.87 (m, 2H, 2 CH (piperidinylmethyl)), 2.93 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.15–3.22 (m, 2H, SO2NCH2CH2N), 3.54 (s, 3H, CH3 (Im)), 3.69–3.78 (m, 5H, SO2NCH2CH2N, CH3 (Im)), 4.68–4.76 (m, 2H, 2 CH (piperidinylmethyl)), 4.91 (s, 2H, CH2Im), 6.43 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 6.85 (d, J = 9.5 Hz, 1H, CH (Ar)), 6.97 (s, 1H, CH (Im)), 7.39 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.66 (dd, J = 9.0, 2.5 Hz, 1H, CH (Ar)), 8.27 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)), 8.41 (d, J = 2.5 Hz, 1H, CH (Ar)); δC (125 MHz, CDCl3) 29.7, 31.9, 34.0, 35.7, 41.0 (br), 43.6, 46.3, 47.1, 56.0, 97.1, 106.1, 109.4, 118.3, 124.3, 127.3, 129.7, 139.0, 139.2, 139.4, 140.3, 152.4, 157.6, 158.5, 161.6; HRMS (ESI+) m/z calcd for [C27H33N11O2S + H] 576.2618, obsd 576.2628; HPLC (I) tR = 12.43 min (100%), (II) tR = 17.87 min (100%).
[N-{2-[(5-Cyanopyrimidin-2-yl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19d)
As per general procedure E with 18d on a 0.187 mmol scale to afford [N-{2-[(5-bromopyrimidin-2-yl)-(3-methyl-3H-imidazol-4-ylmethyl)amino]ethyl},N-{N-(2-pyrimidinyl)-piperidin-4-ylmethyl}] 1-methyl-1H-imidazole-4-sulfonamide as a white foam (109 mg, 93%): δH (500 MHz, CDCl3) 1.17 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.78–1.84 (m, 2H, 2 CH (piperidinylmethyl)), 1.92–1.99 (m, 1H, CH (piperidinylmethyl)), 2.82 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.10 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.19–3.23 (m, 2H, SO2NCH2-CH2N), 3.58 (s, 3H, CH3 (Im)), 3.71–3.75 (m, 5H, SO2NCH2-CH2N, CH3 (Im)), 4.71–4.77 (m, 2 H, 2 CH (piperidinylmethyl)), 4.89 (s, 2H, CH2Im), 6.43 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 7.05 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 7.43 (s, 1H, CH (Im)), 7.46 (s, 1H, CH (Im)), 8.28 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)) 8.30 (s, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 29.7, 31.9, 33.9, 35.6, 40.7, 43.7, 46.0, 46.2, 55.8, 106.4, 109.3, 124.4, 128.1, 129.5, 138.5, 138.9, 139.3, 157.6, 158.0, 159.3, 161.6; HRMS (ESI ) m/z calcd for [C25H32N11O2SBr + H] 630.1723, obsd 630.1748; +HPLC (I) tR = 13.05 min (100%), (II) tR = 19.52 min (100%). The aryl bromide was then converted to the corresponding aryl nitrile on a 0.127 mmol scale, according to general procedure F. After workup, the crude material was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to give 19d as a white foam (50 mg, 69%): δH (500 MHz, CDCl3) 1.16 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.75–1.82 (m, 2H, 2 CH (piperidinylmethyl)), 1.86–1.94 (m, 1H, CH (piperidinylmethyl)), 2.80 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.07 (br d, J = 7.5 Hz, 2H, 2CH (piperidinylmethyl)), 3.26–3.30 (m, 2H, SO2NCH2CH2N), 3.58 (s, 3H, CH3 (Im)), 3.74 (s, 3H, CH3 (Im)), 3.83–3.87 (m, 2H, SO2NCH2-CH2N), 4.70–4.76 (m, 2 H, 2 CH (piperidinylmethyl)), 4.99 (s, 2H, CH2Im), 6.44 (t, J = 4.8 Hz, 1H, pyrimidine), 7.07 (s, 1H, CH (Im)), 7.39–7.44, (s, 3H, 3 CH (Im)), 8.28 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)), 8.54 (br s, 2H, 2 CH (Ar)); δC (125 MHz, CDCl3) 29.7, 32.0, 34.0, 35.8, 40.9, 43.7, 46.3, 46.5, 55.9, 96.8, 109.5, 116.2, 124.5, 127.2, 129.8, 138.8, 139.0, 139.3, 157.7, 160.6, 161.0, 161.6; HRMS (ESI+) m/z calcd for [C26H32N12O2S + H] 577.2570, obsd 577.2569; HPLC (I) tR = 12.45 min (100%), (II) tR = 17.94 min (100%).
[N-{2-[(3-Fluoro-4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19e)
As per general procedure E with 18e on a 0.0683 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to furnish 19e as a white foam (36 mg, 89%): δH (500 MHz, CDCl3) 1.10 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.60–1.67 (m, 1H, CH (piperidinylmethyl)), 1.69–1.75 (m, 2H, 2 CH (piperidinylmethyl)), 2.80 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.95 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.20–3.24 (m, 2H, SO2NCH2CH2N), 3.56 (s, 3H, CH3 (Im)), 3.62–3.66 (m, 2H, SO2NCH2CH2N), 3.72 (m, 3H, CH3 (Im)), 4.52 (s, 2H, CH2Im), 4.67–4.73 (m, 2H, 2 CH (piperidinylmethyl)), 6.43 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 6.60 (dd, J = 13.0, 2.5 Hz, 1H, CH (Ar)), 6.67 (dd, J = 9.0, 2.5 Hz, 1H, CH (Ar)), 6.87 (s, 1H, CH (Im)), 7.37–7.40 (m, 2H, CH (Im), CH (Ar)), 7.43 (s, 1H, CH (Im)), 7.48 (s, 1H, CH (Im)), 8.26 (t, J = 4.7 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.5, 31.7, 34.0, 35.9, 43.5, 44.5, 46.5, 49.3, 56.3, 88.1 (d, JCF = 15.5 Hz), 99.3 (d, JCF = 24.6 Hz), 108.4 (d, JCF = 1.9Hz), 109.5, 115.2, 124.4, 126.3, 129.1, 134.3 (d, JCF = 2.75 Hz), 139.0, 139.1, 139.2, 152.7 (d, JCF = 10.9 Hz), 157.6, 161.5, 164.9 (d, JCF = 252 Hz); HRMS (ESI+) m/z calcd for [C28H33N10O2SF + H] 593.2571, obsd 593.2588; HPLC (I) tR = 12.54 min (98.51%), (II) tR = 18.51 min (98.09%).
[N-{2-[(2-Fluoro-4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19f)
As per general procedure E with 18f on a 0.060 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 19f as a white foam (33 mg, 93%): δH (500 MHz, CDCl3) 1.09 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.70–1.78 (m, 3H, 3 CH (piperidinylmethyl)), 2.76–2.83 (m, 2H, 2 CH (piperidinylmethyl)), 2.95 (br d, J = 7.0 Hz, 2H, 2 CH (piperidinylmethyl)), 3.31 (br t, J =7.5Hz,2H, SO2NCH2CH2N), 3.53 (br t, J =7.5Hz, 2H, SO2NCH2CH2N), 3.58 (m, 3H, CH3 (Im)), 3.73 (m, 3H, CH3 (Im)), 4.50 (s, 2H, CH2Im), 4.67–4.73 (m, 2H, 2 CH (piperidinylmethyl)), 6.43 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 6.93 (s, 1H, CH (Im)), 7.06 (t, J = 8.8 Hz, 1H, CH (Ar)), 7.28 (dd, J = 13.0, 1.8 Hz, 1H, CH (Ar)), 7.31 (br dd, J = 8.5, 1.8 Hz, 1H, CH (Ar)), 7.37 (s, 1H, CH (Im)), 7.41 (s, 1H, CH (Im)), 7.48 (s, 1H, CH (Im)), 8.26 (t, J = 4.7 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 31.8, 34.0, 35.9, 43.5, 45.9, 47.5, 50.5, 55.8, 103.0 (d, JCF = 10 Hz), 109.4, 118.2 (d, JCF = 1.8 Hz), 119.5 (d, JCF = 4.6 Hz), 120.2 (d, JCF = 25.5 Hz), 124.2, 126.9, 129.3, 129.5 (d, JCF = 2.8 Hz), 138.9, 139.0, 139.6, 141.5 (d, JCF = 8.1 Hz), 152.9 (d, JCF =244 Hz), 157.6, 161.5; HRMS (ESI+) m/z calcd for [C28H33N10O2-FS + H] 593.2571, obsd 593.2568; HPLC (I) tR = 12.75 min (100%), (II), tR = 19.09 (99.22%).
[N-{2-[(2,6-Difluoro-4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19g)
As per general procedure E with 18g on a 0.068 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to yield 19g as a white foam (40 mg, 97%): δH (500 MHz, CDCl3) 1.06 (qd, J = 12.3, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.67–1.77 (m, 3H, 3 CH (piperidinylmethyl)), 2.78 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.96 (br d, J = 6.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.26–3.30 (m, 2H, SO2NCH2CH2N), 3.39–3.43 (m, 2H, SO2NCH2CH2N), 3.57 (s, 3H, CH3 (Im)), 3.73 (s, 3H, CH3 (Im)), 4.38 (s, 2H, CH2Im), 4.66–4.73 (m, 2H, 2 CH (piperidinylmethyl)), 6.42 (t, J = 4.9 Hz, 1H, CH (pyrimidine)), 6.92 (s, 1H, CH (Im)), 7.12–7.17 (m, 2H, 2 CH (Ar)), 7.35 (s, 1H, CH (Im)), 7.40 (s, 1H, CH (Im)), 7.42 (s, 1H, CH (Im)), 8.26 (t, J = 4.9 Hz, 2H, 2 CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 31.4, 33.9, 36.0, 43.5, 46.3 (t, JCF = 4.5 Hz), 47.8, 51.9 (t, JCF = 2.6 Hz), 55.6, 106.9 (t, JCF = 11.9 Hz), 109.4, 116.6 (m), 116.6 (t, JCF = 2.8 Hz), 124.1, 126.9, 129.9, 131.1 (t, J = 13.3 Hz), 138.9, 139.2, 139.7, 157.6, 158.3 (dd, JCF = 249, 8.3 Hz), 161.5; HRMS (ESI+) m/z calcd for [C28H32N10O2F2S + H] 611.2477, obsd 611.2494; + HPLC (I) tR = 12.93 min (100%), (II) tR = 19.59 min (100%).
[N-{2-[(2,3,5,6-Tetrafluoro-4-cyanophenyl)-(3-methyl-3H-imidazol-4-ylmethyl)-amino]-ethyl}, N-{N-(2-Pyrimidinyl)-piperidin-4-ylmethyl}] 1-Methyl-1H-imidazole-4-sulfonamide (19h)
As per general procedure E with 18h on a 0.0688 mmol scale. After the usual workup, the crude material was purified by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1) to afford 19h as a white foam (28 mg, 63%): δH (500 MHz, CDCl3) 1.08 (qd, J = 12.2, 4.0 Hz, 2H, 2 CH (piperidinylmethyl)), 1.69–1.81 (m, 3H, 3 CH (piperidinylmethyl)), 2.80 (td, J = 12.8, 2.5 Hz, 2H, 2 CH (piperidinylmethyl)), 2.96 (br d, J = 7.5 Hz, 2H, 2 CH (piperidinylmethyl)), 3.34–3.40 (m, 2H, SO2NCH2CH2N), 3.51–3.56 (m, 2H, SO2NCH2CH2N), 3.57 (s, 3H, CH3 (Im)), 3.74 (s, 3H, CH3 (Im)), 4.53 (s, 2H, CH2Im), 4.68–4.74 (m, 2 H, 2 CH (piperidinylmethyl)), 6.43 (t, J = 4.7 Hz, 1H, CH (pyrimidine)), 6.98 (s, 1H, CH (Im)), 7.38 (s, 1H, CH (Im)), 7.41–7.46 (m, 2H, 2 CH (Im)), 8.27 (t, J = 4.7 Hz, 2H, CH (pyrimidine)); δC (125 MHz, CDCl3) 29.6, 31.5, 33.9, 36.0, 43.5, 46.2, 47.9, 51.9 (m), 55.7, 86.9 (t, JCF = 17.4 Hz), 107.8 (m), 109.5, 124.2, 126.2, 130.2, 133.9 (m), 139.0, 139.4, 139.6, 142.1 (dm), 147.8 (dm), 157.6, 161.5; HRMS (ESI+) m/z calcd for [C28H30N10O2F4S + H] 647.2288, obsd 647.2314; HPLC (I) tR = 12.74 min (98.92%), (II) tR = 19.07 min (98.14%).
N-(2-Aminoethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21)
Mono-N-Boc-ethylenediamine (6; 860 mg, 5.37 mmol, 1 equiv) was reacted with pyridine-2-sulfonyl chloride according to general procedure A. After workup, the crude material was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to afford tert-butyl 2-(pyridine-2-sulfonamido)ethylcarbamate as a white powder (1.26 g, 80%): δH (400 MHz, DMSO-d6) 0.89 (s, 9H, C(CH3)3), 2.04 (m, 2H, SO2NHCH2CH2NHBoc), 2.86 (m, 2H, SO2NHCH2CH2NHBoc), 6.30 (br, 1H, NHSO2), 7.21 (m, 1H, CH (Py)), 7.414 (m, 1H, NHBoc), 7.47 (m, 1H, CH (Py)), 7.63, (m, 1H, CH (Py)), 8.27 (m, 1H, CH (Py)); δC (125 MHz, DMSO-d6) 28.0, 39.9, 42.5, 77.6, 121.4, 126.8, 138.5, 149.8, 155.3, 157.7; HRMS (ESI+) m/z calcd for [C12H19N3O4S + H] 302.1175, obsd 302.1174. The sulfonamide NH of tert-butyl 2-(pyridine-2-sulfonamido)ethylcarbamate (1.1 g, 3.65 mmol, 1 equiv) was then chemoselectively alkylated with cyclohexylmethyl bromide (840 mg, 4.75 mmol, 1.3 equiv) according to general procedure B, but the reaction was stirred for 4 days at room temperature. After workup, the crude material was dry-loaded onto silica gel and purified by flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 92:7:1) to furnish tert-butyl 2-(N-(cyclohexylmethyl)pyridine-2-sulfonamido)ethylcarbamate in quantitative yield (1.53 g) as a white solid: δH (500 MHz, DMSO-d6) 0.77–0.83 (m, 2H, 2 CH (cyclohexylmethyl)), 1.09–1.19 (m, 3H, 3 CH (cyclohexylmethyl)), 1.36 (s, 9H, C(CH3)3), 1.54–1.65 (m, 6H, 6 CH (cyclohexylmethyl)), 3.03–3.07 (m, 4H, SO2NCH2CH2-NHBoc, NCH2CH (cyclohexylmethyl)), 3.24 (t, J = 7.0 Hz, 2H, SO2NCH2CH2NHBoc), 6.79 (br, 1H, NHBoc)), 7.67 (m, 1H, CH (Py)), 7.92 (m, 1H, CH (Py)), 8.09 (m, 1H, CH (Py)), 8.72 (m, 1H, CH (Py)); δC (125 MHz, DMSO-d6) 25.1, 25.8, 28.0, 29.9, 35.6, 39.9, 48.4, 55.4, 77.6, 122.1, 127.0, 138.6, 150.0, 155.3, 157.1; HRMS (ESI+) m/z calcd for [C19H31N3O4S + H] 398.2114, obsd 398.2113. tert-Butyl 2-(N-(cyclohexylmethyl)pyridine-2-sulfonamido)ethyl-carbamate (1.28 g, 3.22 mmol, 1 equiv) was dissolved in propan-2-ol (15 mL). Upon complete dissolution of the solid, 4 M HCl (15 mL) was added, and the reaction was stirred for one hour. All solvents were then evaporated. The residue was redissolved in CH2Cl2 (250 mL) and carefully washed with saturated NaHCO3 (25 mL × 3), dried (Na2SO4), filtered, and concentrated to afford N-(2-aminoethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21) as a viscous oil (945 mg, 97%): δH (500 MHz, DMSO-d6) 0.78–0.84 (m, 2H, 2 CH (cyclohexylmethyl)), 1.09–1.17 (m, 3H, 3 CH (cyclohexylmethyl)), 1.57–1.68 (m, 6H, 6 CH (cyclohexylmethyl)), 2.75 (t, J = 7.0 Hz, 2H, NCH2CH (cyclohexylmethyl)), 3.04 (t, J = 7.4 Hz, 2H, SO2NCH2CH2NH2), 3.34 (t, J = 7.4 Hz, 2H, SO2NCH2CH2NH2), 5.15 (br, 2H, NH2), 7.68 (m, 1H, CH (Py)), 7.93 (m, 1H, CH (Py)), 8.10 (m, 1H, CH (Py)), 8.73 (m, 1H, CH (Py)); δC (125 MHz, DMSO) 25.1, 25.8, 29.9, 35.7, 49.4, 55.5, 66.2, 122.2, 127.1, 138.7, 150.0, 157.1; HRMS (ESI+) m/z calcd for [C14H23N3O2S + H] 298.1589, obsd 298.1589.
(2-((4-Cyanophenyl)((1-methyl-1H-imidazol-5-yl)methyl)amino)-ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21a)
The primary amine of N-(2-aminoethyl)-N-(cyclohexylmethyl)-pyridine-2-sulfonamide (21) was arylated with p-fluorobenzonitrile on a 0.336 mmol scale, according to general procedure D. After workup and purification by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), N-(2-(4-cyanophenylamino)ethyl)-N-(cyclohexylmethyl)-pyridine-2-sulfonamide was obtained as a colorless gum (119 mg, 89%): δH (500 MHz, DMSO-d6)0.77–0.83 (m, 2H, 2 CH (cyclohexylmethyl)), 1.09 (br, 3H, 3 CH (cyclohexyl)), 1.48 1.63 (m, 6H, 6 CH (cyclohexylmethyl)), 3.06 (d, J = 6.4 Hz, 2H, NCH2CH (cyclohexylmethyl)), 3.28–3.38 (m, 4H, SO2NCH2CH2NHAr), 6.61 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 6.70 (t, J = 6.4 Hz, 1H, NH), 7.45 (d, J = 8.5 Hz, 2H, 2 CH (Ar)), 7.66 (m, 1H, CH (Py)), 7.94 (m, 1H, CH (Py)), 8.07 (m, 1H, CH (Py)), 8.73 (m, 1H, CH (Py)); δC (125 MHz, DMSO) 25.1, 25.8, 29.9, 35.8, 41.5, 47.7, 55.6, 95.9, 111.6, 120.3, 122.2, 127.1, 133.3, 138.6, 150.1, 151.6, 157.0; HRMS (ESI+) m/z calcd for [C21H26N4O2S + H] 399.1855, obsd 399.1857. N-(2-(4-Cyanophenylamino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was then alkylated with 5-chloromethyl-1H-imidazole·3 HCl according to general procedure E on a 0.117 mmol scale. After workup and chromatography over silica gel (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), the title compound N-(2-((4-cyanophenyl)((1-methyl-1H-imidazol-5-yl)methyl)amino)ethyl)-N-(cyclohexylmethyl)-pyridine-2-sulfonamide (21a) was furnished as a glassy film (46 mg, 80%): δH (500 MHz, CDCl3)0.84–0.78 (m, 2H, 2 CH (cyclohexylmethyl)), 1.06–1.14 (m, 3H, 3 CH (cyclohexylmethyl)), 1.35–1.27 (m, 1H, CH (cyclohexylmethyl)), 1.66–1.59 (m, 5H, 5 CH (cyclohexylmethyl)), 2.95 (d, J = 7.0 Hz, 2H, NCH2CH (cyclohexyl)), 3.36 (t, J = 7.9 Hz, 2H, SO2NCH2CH2N), 3.61 (s, 3H, CH3 (Im)), 3.65 (t, J = 7.9 Hz, 2H, SO2NCH2CH2N), 4.55 (s, 2H, CH2Im), 6.85 (d, J = 9.0 Hz, 2H, 2 CH (Ar)), 6.88 (s, 1H, CH (Im)), 7.47–7.44 (m, 3H, 2 CH (Ar), CH (Im)), 7.71 (s, 1H, CH (Py)), 7.91–7.86 (m, 2H, 2 CH (Py)), 8.61 (m, 1H, CH (Py)); δC (125 MHz, CDCl3) 25.6, 26.2, 30.6, 32.0, 36.9, 44.3, 46.8, 49.4, 57.1, 99.5, 112.3, 119.9, 122.5, 126.6, 127.2, 127.7, 133.8, 137.9, 138.7, 149.9, 150.4, 157.5; HRMS (ESI+) m/z calcd for [C26H32N6O2S + H] 493.2386, obsd 493.2364.
N-(2-((4-Cyano-2-fluorophenyl)((1-methyl-1H-imidazol-5-yl)-methyl)amino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21b)
The primary amine of N-(2-aminoethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21) was arylated with 3,4-difluorobenzonitrile on a 0.337 mmol scale, according to general procedure D. After workup and purification by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), N-(2-(4-cyano-2-fluorophenylamino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was obtained as a white foam (111 mg, 79%): δH (500 MHz, DMSO-d6) 0.83–0.77 (m, 2H, 2 CH (cyclohexylmethyl)), 1.09 (br, 3H, 3 CH (cyclohexylmethyl)), 1.62–1.51 (m, 6H, 6 CH (cyclohexylmethyl)), 3.06 (d, J = 7.5 Hz, 2H, NCH2CH (cyclohexyl)), 3.42–3.34 (m, 4H, SO2NCH2CH2N), 6.54 (br, 1H, NHAr), 6.80 (t, J = 8.5 Hz, 1H, CH (Ar)), 7.45 (dd, J = 11.5, 1.5 Hz, 1H, CH (Ar)), 7.53 (dd, J = 8.5, 1.5 Hz, 1H, CH (Ar)), 7.66 (m, 1H, CH (Py)), 7.93 (m, 1H, CH (Py)), 8.07 (m, 1H, CH (Py)), 8.71 (m, 1H, CH (Py)); δC (125 MHz, DMSO-d6) 25.1, 25.9, 30.0, 35.7, 37.1, 41.3, 47.5, 55.5, 61.8, 94.4, 122.2, 127.1, 130.4, 138.7, 140.7, 142.5, 150.1, 156.9, 179.7; HRMS (ESI+) m/z calcd for [C21H25FN4O2S + H] 417.1760, obsd 417.1749. N-(2-(4-Cyano-2-fluorophenylamino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was then alkylated with 5-chloromethyl-1H-imidazole·HCl according to general procedure E on a 0.216 mmol scale. After workup and chromatography over silica gel (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), the title compound N-(2-((4-cyano-2-fluorophenyl)((1-methyl-1H-imidazol-5-yl)methyl)amino)ethyl)-N-(cyclohexylmethyl)-pyridine-2-sulfonamide was afforded as a white foam (90 mg, 81%): δH (500 MHz, CDCl3) 0.68–0.75 (m, 2H, 2 CH (cyclohexylmethyl)), 1.03 (br, 3H, 3 CH (cyclohexylmethyl)), 1.23–1.32 (m, 1H, CH (cyclohexylmethyl)), 1.50–1.60 (m, 5H, 5 CH (cyclohexylmethyl)), 2.80 (s, 2H, NCH2CH (cyclohexylmethyl)), 3.34–3.47 (m, 4H, SO2NCH2CH2N), 3.54 (s, 3H, CH3 (Im)), 4.46 (s, 2H, CH2Im), 6.61 (br, 1H, CH (Ar)), 6.95 (s, 1H, CH (Im)), 7.11 (m, 1H, CH (Ar)), 7.26 (br, 1H, CH (Ar)), 7.53 (m, 2H, CH (Py), CH (Im)) 7.65 (br, 1H, CH (Py)), 8.26 (m, 1H, CH (Py)), 8.69 (m, 1H, CH (Py)); δC (125 MHz, CDCl3) 25.5, 26.1, 30.5, 31.3, 31.8, 36.6, 45.7, 47.3, 50.4, 56.4, 102.9, 118.1, 119.5, 120.2, 122.4, 126.5, 128.7, 129.3, 138.8, 141.3, 149.8, 153.9, 157.6, 162.4; HRMS (ESI+) m/z calcd for [C26H31FN6O2S + H] 511.2291, obsd 511.2265; + HPLC (I) tR = 18.69 min (99.3%), (II) tR = 36.03 min (99.7%).
N-(2-((5-Cyanopyridin-2-yl)((1-methyl-1H-imidazol-5-yl)-methyl)amino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21c)
The primary amine of N-(2-aminoethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide (21) was arylated with 5-cyano-2-fluoropyridine on a 0.350 mmol scale, according to general procedure D. After workup and purification by silica gel flash column chromatography (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), N-(2-(5-cyanopyridin-2-ylamino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was obtained as a colorless gum (115 mg, 82%): δH (500 MHz, DMSO-d6) 0.78–0.82 (m, 2H, 2 CH (cyclohexylmethyl)), 1.09 (br, 3H, 3 CH (cyclohexylmethyl)), 1.57–1.63 (m, 6H, 6 CH (cyclohexylmethyl)), 3.06 (d, J = 7.5 Hz, 2H, NCH2CH (cyclohexylmethyl)), 3.37–3.47 (m, 4H, SO2-NCH2CH2N), 6.50 (t, J = 8.5 Hz, NH), 6.87 (d, J = 9.0 Hz, 1H, CH (Ar)), 7.61–7.66 (m, 3H, 2 CH (Ar), CH (Py)), 7.92 (d, J = 7.5 Hz, 1H, CH (Py)), 8.06 (m, 1H, CH (Py)), 8.36 (m, 1H, CH (Ar)), 8.71 (m, 1H, CH (Py)); δC (125 MHz, DMSO-d6) 25.1, 25.8, 30.0, 35.6, 47.7, 54.7, 55.5, 69.3, 94.7, 108.7, 118.7, 122.1, 126.9, 138.5, 150.0, 152.8, 157.0, 159.6; HRMS (ESI+) m/z calcd for [C20H25N5O2S + H] 400.1807, obsd 400.1795. N-(2-(5-Cyanopyridin-2-ylamino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was then alkylated with 5-chloromethyl-1H-imidazole·HCl according to general procedure E on a 0.240 mmol scale. After workup and chromatography over silica gel (eluent CH2Cl2/MeOH/NH4OH, 192:7:1), the title compound N-(2-((5-cyanopyridin-2-yl)((1-methyl-1H-imidazol-5-yl)methyl)-amino)ethyl)-N-(cyclohexylmethyl)pyridine-2-sulfonamide was afforded as a white foam (110 mg, 93%): δH (500 MHz, CDCl3) 0.75–0.83 (m, 2H, 2 CH (cyclohexylmethyl)), 1.07–1.16 (m, 3H, 3 CH (cyclohexylmethyl)), 1.33–1.43 (m, 1H, CH (cyclohexylmethyl)), 1.59–1.65 (m, 5H, 5 CH (cyclohexylmethyl)), 2.94 (d, J = 7.0 Hz, 2H, NCH2CH (cyclohexylmethyl)), 3.28 (t, J = 8.1 Hz, 2H, SO2NCH2CH2N), 3.55 (s, 3H, CH3 (Im)), 3.70 (t, J = 8.1 Hz, 2H, SO2NCH2CH2N), 4.89 (s, 2H, CH2Im), 6.81 (d, J = 9.0 Hz, 1H, CH (Ar)), 6.97 (s, 1H, CH (Im)), 7.45 (m, 1H, CH (Im)), 7.57 (s, 1H, CH (Ar)), 7.64 (m, 1H, CH (Ar)), 7.89–7.85 (m, 2H, 2 CH (Py)), 8.39–8.38 (m, 1H, CH (Ar)), 8.60 (m, 1H, CH (Py)); δC (125 MHz, CDCl3) 25.6, 26.2, 30.5, 32.0, 36.6, 40.9, 47.2, 56.8, 97.1, 105.9, 118.2, 122.5, 126.6, 127.4, 128.8, 137.9, 138.9, 140.2, 149.9, 152.3, 157.5, 158.4, 162.4; HRMS (ESI ) m/z calcd for [C25H31N7O2S + H] 494.2338, obsd 494.2313; HPLC + (I) tR = 18.15 min (99.5%), (II) tR = 34.66 min (97.4%).
In Vitro and Whole Cell Inhibition Assays of Protein Prenylation
In vitro inhibition assays for FTase and GGTase-I were conducted by measuring the incorporation of [3H]FPP and [3H]GGPP into recombinant H-Ras-CVLS and H-Ras-CVLL, respectively, as previously described.36 The in vivo inhibition of farnesylation and geranylgeranylation was determined based on the level of inhibition by synthetic compounds of H-Ras and Rap1A processing, respectively.37 Briefly, oncogenic H-Ras-transformed NIH3T3 cells were treated with various concentrations of inhibitors, and the cell lysates were isolated and proteins separated on a 12.5% SDS-PAGE gel. The separated proteins were transferred to nitrocellulose and immunoblotted using an anti-Ras antibody (Y13–258) or an anti-Rap1A antibody (SantaCruz Biotechnology, Santa Cruz, CA), respectively. Antibody reactions were visualized using either peroxidase-conjugated goat antirat IgG or goat antirabbit IgG (Jackson ImmunoResearch, West Grove, PA) and an enhanced chemiluminescence detection system.
Acknowledgment
Financial support for this work was provided by the National Institutes of Health (CA67771 & GM52382) and the Medicines for Malaria Venture (MMV). We also thank Vijay M. Shahani for additional assistance in the flexible ligand GOLD docking experiments.
Footnotes
Abbreviations: FTase, farnesyltransferase; hFTase, human FTase; rFTase, rat FTase; PfFTase, Plasmodium falciparum FTase; FTI, farnesyltransferase inhibitor; FPP, farnesyl pyrophosphate; GGTase-I, geranylgeranyltransferase-I; IC50, inhibitor concentration that inhibits 50% of enzyme activity; rt, room temperature.
References
- 1.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyltransferase inhibitors. J. Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Bell I. Inhibitors of farnesyltransferase: a rational approach to cancer chemotherapy? J. Med. Chem. 2004;47:1869–1878. doi: 10.1021/jm0305467. [DOI] [PubMed] [Google Scholar]
- 3.Doll RJ, Kirschmeier P, Bishop WR. Farnesyltransferase inhibitors as anticancer agents: critical crossroads. Curr. Opin. Drug Discovery Dev. 2004;7:478–486. [PubMed] [Google Scholar]
- 4.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: “It ain't over 'til it's over”. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 5.Bos JL. Ras oncogenes in human cancer: A review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 6.Clark GJ, Der CJ. Ras proto-oncogene activation in human malignancy. In: Garrett CT, Sell S, editors. Cellular Cancer Markers. Humana Press; Totowa, NJ: 1995. pp. 17–52. [Google Scholar]
- 7.Willumsen BM, Norris K, Papageorge AG, Hubbert NL, Lowy DR. Harvey murine sarcoma virus p21 Ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J. 1984;3:2581–2585. doi: 10.1002/j.1460-2075.1984.tb02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey PJ, Solski PA, Der C, Buss JE. p21 Ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. U.S.A. 1989;86:8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sousa SF, Fernandes PA, Ramos MJ. Farnesyltransferase inhibitors: a detailed chemical view on an elusive biological problem. Curr. Med. Chem. 2008;15:1478–1492. doi: 10.2174/092986708784638825. [DOI] [PubMed] [Google Scholar]
- 10.Asoh K, Kohchi M, Hyoudoh I, Ohtsuka T, Masubuchi M, Kawasaki K, Ebiike H, Shiratori Y, Fukami TA, Kondoh O, Tsukaguchi T, Ishii N, Aoki Y, Shimma N, Sakaitani M. Synthesis and structure-activity relationships of novel benzofuran farnesyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:1753–1757. doi: 10.1016/j.bmcl.2009.01.074. [DOI] [PubMed] [Google Scholar]
- 11.Lethu S, Ginisty M, Bosc D, Joelle D. Discovery of a new class of protein farnesyltransferase inhibitors in the arylthiophene series. J. Med. Chem. 2009;55:6205–6208. doi: 10.1021/jm901280q. [DOI] [PubMed] [Google Scholar]
- 12.Bell IM. Inhibitors of protein prenylation 2000. Exp. Opin. Ther. Patents. 2000;10:1813–1831. [Google Scholar]
- 13.Venet M, End D, Angibaud P. Farnesyl Protein Transferase Inhibitor ZARNESTRA R115777–History of a Discovery. Curr. Top. Med. Chem. 2003;3:1095–1102. doi: 10.2174/1568026033452050. [DOI] [PubMed] [Google Scholar]
- 14.Rao S, Cunningham D, de Gramont A, Shceithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T, Dostalova J, Illes A, Belly R, Perez-Ruixo JJ, Park YC, Palmer PA. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 2004;22:3950–3957. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 15.Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL, III, Giguere JK, Abbruzzese JL. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest. New Drugs. 2005;23:485–487. doi: 10.1007/s10637-005-2908-y. [DOI] [PubMed] [Google Scholar]
- 16.Sparano JA, Moulder S, Kazi A,A, Coppola D, Negassa A,A, Vahdat L, Li T, Pellegrino C, Fineberg S, Munster P, Malafa M, Lee D, Hoschander S, Hopkins U, Hershman D, Wright JJ, Kleer C, Merajver S, Sebti SM. Phase II trial of tipifarnib plus neoadjuvant doxorubicin-cyclophosphamide in patients with clinical stage IIB–IIIC breast cancer. Clin. Cancer Res. 2009;15:2942–2948. doi: 10.1158/1078-0432.CCR-08-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sparano JA, Moulder S, Kazi A, Vahdat L, Li T, Pellegrino C, Munster P, Malafa M, Lee D, Hoschander S, Hopkins U, Hershman D, Wright JJ, Sebti SM. Targeted inhibition of farnesyltransferase in locally advanced breast cancer: a phase I and II trial of tipifarnib plus dose-dense doxorubicin and cyclophosphamide. J. Clin. Oncol. 2006;24:3013–3018. doi: 10.1200/JCO.2005.04.9114. [DOI] [PubMed] [Google Scholar]
- 18.Falsetti SC, Wang DA, Peng H, Carrico D, Cox AD, Der CJ, Hamilton AD, Sebti SM. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol. Cell. Biol. 2007;27:8003–8014. doi: 10.1128/MCB.00057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prendergast GC, Rane N. Farnesyltransferase Inhibitors: Mechanism and Applications. Expert Opin. Invest. Drugs. 2001;10:2105–2116. doi: 10.1517/13543784.10.12.2105. [DOI] [PubMed] [Google Scholar]
- 20.Liu A-X, Du W, Liu J-P, Jessell TM, Prendergast GC. RhoB Alteration Is Necessary for Apoptotic and Antineoplastic Responses to Farnesyltransferase Inhibitors. Mol. Cell. Biol. 2000;20:6105–6113. doi: 10.1128/mcb.20.16.6105-6113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Sun J, Pradines A, Favre G, Adnane J, Sebti SM. Both Farnesylated and Geranylgeranylated RhoB Inhibit Malignant Transformation and Suppress Human Tumor Growth in Nude Mice. J. Biol. Chem. 2000;275:17974–17978. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 22.Casey PJ. Biochemistry of Protein Prenylation. Lipid Res. 1992;33:1731–1740. [PubMed] [Google Scholar]
- 23.Zhang FL, Casey PJ. Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annu. Rev. Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 24.Der CJ, Cox AD. Isoprenoid Modification and Plasma-Membrane Association: Critical Factors for Ras Oncogenicity. Cancer Cells. 1991;3:331–340. [PubMed] [Google Scholar]
- 25.Qian Y, Blaskovich MA, Saleem M, Seong CM, Wathen SP, Hamilton AD, Sebti SM. Design and structural requirements of potent peptidomimetic inhibitors of p21ras farnesyltransferase. J. Biol. Chem. 1994;269:12410–12413. [PubMed] [Google Scholar]
- 26.Qian Y, Marugan JJ, Fossum RD, Vogt A, Sebti SM, Hamilton AD. Probing the hydrophobic pocket of farnesyltransferase: aromatic substitution of CAAX peptidomimetics leads to highly potent inhibitors. Bioorg. Med. Chem. 1999;7:3011–3024. doi: 10.1016/s0968-0896(99)00252-7. [DOI] [PubMed] [Google Scholar]
- 27.Ohkanda J, Lockman JW, Kothare MA, Qian Y, Blaskovich MA, Sebti SM, Hamilton AD. Design and synthesis of potent nonpeptidic farnesyltransferase inhibitors based on a terphenyl scaffold. J. Med. Chem. 2002;45:177–188. doi: 10.1021/jm0103099. [DOI] [PubMed] [Google Scholar]
- 28.Ohkanda J, Strickland CL, Blaskovich MA, Carrico D, Lockman JW, Vogt A, Bucher CJ, Sun J, Qian Y, Knowles D, Pusateri EE, Sebti SM, Hamilton AD. Structure-based design of imidazole-containing peptidomimetic inhibitors of protein farnesyltransferase. Org. Biomol. Chem. 2006;4:482–492. doi: 10.1039/b508184j. [DOI] [PubMed] [Google Scholar]
- 29.(a) Glenn MP, Chang SY, Hucke O, Verlinde CL, Rivas K, Hornéey C, Yokoyama K, Buckner FS, Pendyala PR, Chakrabarti D, Gelb M, Van Voorhis WC, Sebti SM, Hamilton AD. Structurally simple farnesyltransferase inhibitors arrest the growth of malaria parasites. Angew. Chem., Int. Ed. 2005;44:4903–4906. doi: 10.1002/anie.200500674. [DOI] [PubMed] [Google Scholar]; (b) Glenn MP, Chang SY, Hornéey C, Rivas K, Yokoyama K, Pusateri EE, Fletcher S, Cummings CG, Buckner FS, Pendyala PR, Chakrabarti D, Sebti SM, Gelb M, Van Voorhis WC, Hamilton AD. Structurally simple, potent, Plasmodium selective farnesyltransferase inhibitors that arrest the growth of malaria parasites. J. Med. Chem. 2006;49:5710–5727. doi: 10.1021/jm060081v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fletcher S, Cummings CG, Rivas K, Katt WP, Hornéey C, Buckner FS, Chakrabarti D, Sebti SM, Gelb MH, Van Voorhis WC, Hamilton AD. Potent, Plasmodium-selective farnesyltransferase inhibitors that arrest the growth of malaria parasites: structure-activity relationships of ethylenediamine-analogue scaffolds and homology model validation. J. Med. Chem. 2008;51:5176–5197. doi: 10.1021/jm800113p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eastman RT, White J, Hucke O, Bauer K, Yokoyama K, Nallan L, Chakrabarti D, Verlinde CL, Gelb MH, Rathod PK, Van Voorhis WC. Resistance to a protein farnesyltransferase inhibitor in Plasmodium falciparum. J. Biol. Chem. 2005;280:13554–13559. doi: 10.1074/jbc.M413556200. [DOI] [PubMed] [Google Scholar]
- 32.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 33.Hunt JT, Ding CZ, Batorsky R, Bednarz M, Bhide R, Cho Y, Chong S, Chao S, Gullo-Brown J, Guo P, Kim SH, Lee FY, Leftheris K, Miller A, Mitt T, Patel M, Penhallow BA, Ricca C, Rose WC, Schmidt R, Slusarchyk WA, Vite G, Manne V. Discovery of (R)-7-cyano-2,3,4,5-tetrahydro-1-(1H-imidazol-4-ylmethyl)-3-(phenylmethyl)-4-(2-thienylsulfonyl)-1H-1,4-benzodiazepine (BMS-214662), a farnesyltransferase inhibitor with potent preclinical antitumor activity. J. Med. Chem. 2000;43:3587–3595. doi: 10.1021/jm000248z. [DOI] [PubMed] [Google Scholar]
- 34.Lombardo LJ, Camuso A, Clark J, Fager K, Gullo-Brown J, Hunt JT, Inigo I, Kan D, Kolowitz B, Lee F, McGlinchey K, Qian L, Ricca C, Rovnyak G, Traeger S, Tokarski J, Williams DK, Wu LI, Zhao Y, Manne V, Bhide RS. Design, synthesis and structure-activity relationships of tetrahydroquinoline-based farnesyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2005;15:1895–1899. doi: 10.1016/j.bmcl.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Long SB, Hancock PJ, Kral AM, Hellinga HW, Beese LS. The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12948–12953. doi: 10.1073/pnas.241407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid TS, Beese LS. Crystal structures of the anticancer clinical candidates R115777 (Tipifarnib) and BMS-214662 complexed with protein farnesyltransferase suggest a mechanism of FTI selectivity. Biochemistry. 2004;43:6877–6884. doi: 10.1021/bi049723b. [DOI] [PubMed] [Google Scholar]
- 37.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 38.Hast MA, Fletcher S, Cummings CG, Pusateri EE, Blaskovich MA, Rivas K, Gelb MH, Van Voorhis WC, Sebti SM, Hamilton AD, Beese LS. Structural basis for binding and selectivity of antimalarial and anticancer ethylenedia-mine inhibitors to protein farnesyltransferase. Chem Biol. 2009;16:181–192. doi: 10.1016/j.chembiol.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogt A, Qian Y, McGuire TF, Hamilton AD, Sebti SM. Protein geranylgeranylation, not farnesylation, is required for G1 to S phase transition in mouse fibroblasts. Oncogene. 1996;13:1991–1999. [PubMed] [Google Scholar]
- 40.Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene. 1998;16:1467–1473. doi: 10.1038/sj.onc.1201656. [DOI] [PubMed] [Google Scholar]