

Abstract
A library of 91 heterocyclic compounds composed of 16 distinct scaffolds has been synthesized through a sequence of phosphine-catalyzed ring-forming reactions, Tebbe reactions, Diels–Alder reactions, and, in some cases, hydrolysis. This effort in diversity-oriented synthesis produced a collection of compounds that exhibited high levels of structural variation both in terms of stereochemistry and the range of scaffolds represented. A simple but powerful sequence of reactions thus led to a high-diversity library of relatively modest size with which to explore biologically relevant regions of chemical space. From this library, several molecules were identified that inhibit the migration and invasion of breast cancer cells and may serve as leads for the development of antimetastatic agents.
Keywords: antimigratory agents, chemical biology, diversity-oriented synthesis, heterocycles, synthesis design
Introduction
At its heart, modern chemistry is a quest to understand the relationship between structure and (biological or material) function. From a biological point of view, we do not yet understand the rules for predicting 1) whether a particular small molecule will interact with proteins, 2) what protein it will interact with if it does, and 3) which mode of interaction it will employ. Chemists have formulated only relatively limited empirical rules (such as Lipinski’s “rule of five”) to guide efforts to generate molecules of desirable biological function.[1] If we had such insight, designing small molecule modulators of proteins of known structures (crystallographically or spectroscopically) would be relatively easy. Our scientific understanding today is, however, quite far from allowing us to develop desired functions based on structure alone—without a priori information. One approach toward broadening our understanding of the relationship between structure and function would be to generate many new small molecules that modulate proteins’ functions and study the interactions between them.
In this context, collections of compound libraries have been established as common starting points for the study of chemical genetics and the discovery of new drugs.[2] The development of efficient methods for the construction of libraries encompassing the maximum amount of chemical space is a particularly challenging task for organic chemists.[3] Establishing the maximum amount of skeletal diversity is the key factor toward improving the efficiency for novel therapeutic leads screening.[4] Diversity-oriented synthesis (DOS) entails the development of pathways leading to the efficient synthesis of collections of small molecules exhibiting rich skeletal and stereochemical diversity.[5] Compared with libraries constructed from common scaffolds decorated with diverse substituents,[2b,6] examples of libraries of small molecules featuring high degrees of skeletal diversity are relatively limited.[7] In addition, although such exercises are undertaken based on the premise that a diverse range of scaffolds should provide a higher chance for discovery of small-molecule biological functional modulators, its actual realization is rarely reported. Herein, we report a highly efficient and modular synthesis of a library of 91 multicyclic heterocycles with 16 distinctive scaffolds and the resulting identification of new antimigratory agents.
Results and Discussion
Cycloaddition of electron-deficient allenes under phosphine catalysis is a robust method for obtaining a variety of carbo-and heterocycles in an atom-economical and environmentally friendly manner.[8] The phosphine-catalyzed reactions are typically high yielding and compatible with solid phase synthetic processes. For example, using methodology developed by Lu’s group[9] and our own,[10] we have previously generated a library of small-molecule protein geranylgeranyltransferase type I (GGTase-I) inhibitors through solid-phase split-pool synthesis.[6d,11] The pyrrolines and tetrahydropyridines resulting from the phosphine catalysis of allenoates and imines possess a common α,β-unsaturated ester group. In our previous synthesis of GGTase-I inhibitors, the α,β-enoate functionality provided a handle for highly diastereo-selective Michael additions of thiols. Although the conjugate addition of thiols provided only a relatively moderate level of skeletal variation, the pentasubstituted pyrrolidine products were identified as the first small-molecule RabGGTase-specific inhibitors.[11–12] We envisioned that the versatile chemistry of conjugated enoates might provide a handle for further scaffold diversification. When the C=O moiety of an α,β-unsaturated ester is methylenated, the resulting dienol ether (an electron-rich diene) can undergo Diels–Alder reactions with electron-deficient dienophiles to generate a variety of fused heterocyclic compounds possessing distinctive frameworks (Scheme 1).
Scheme 1.

Branched pathway for the construction of a library of 16 distinctive scaffolds; a) and b) phosphine-catalyzed ring-forming reactions; c) Tebbe reactions, d) Diels–Alder reactions. Reaction conditions: a) PBu3 (20 mol%), benzene, RT, 12 h; b) PBu3 (20 mol%), CH2Cl2, RT, 12 h; c) Tebbe reagent (3 equiv), pyridine (30 mol%), THF, RT, 12 h; d) for 7a–7i, maleimide (4 equiv), MeOH (5–15%), CH2Cl2, RT, 48 h; for 8a–8j, N-phenyltriazolinedione (2 equiv), CH2Cl2, 0 °C, 1 h; for 9a–9g, tetracyanoethylene (2 equiv), CH2Cl2, 0 °C, 1 h; for 10a–10d, dichlorobenzoquinone (4 equiv), toluene, 80°C, 5 h; for 10e, benzoquinone (4 equiv), toluene, 80°C, 5 h; then CHCl3, SiO2, Et3N, RT, 12 h; for 11a–11 f, maleimide (4 equiv), MeOH (5–15%), CH2Cl2, RT, 48 h; for 12a–12e, N-phenyltriazolinedione (2 equiv), CH2Cl2, −78 °C, 5 h; for 13a–13 f, tetracyanoethylene (2 equiv), CH2Cl2, −78 °C, 5 h; for 14a–14c, benzoquinone (4 equiv), toluene, 80°C, 10 h; for 15a–15j, imine (4 equiv), toluene, 65°C, 24 h. See the Supporting Information for the list of the dienophiles tested, the reaction details, and the compound structures.
To begin exploring this concept, we prepared, as starting materials, the pyrrolines 3a–3j and the tetrahydropyridines 4a–4 f, through phosphine-catalyzed ring-forming reactions between the allenoates 1 and the imines 2, in excellent yields (90–97%) on multigram scales (Scheme 1).[9–10] The methylenation of the α,β-unsaturated esters with the Tebbe reagent[13] gave the corresponding ethoxy dienes 5a–5j and 6a–6 f with good reaction efficiencies (50–82%). Although these enol ethers are slightly unstable on silica gel or in CDCl3 solution, some of them (e.g., 5b, 6a, and 6e) could be stored at −20°C for up to one month, without decomposition, after purification. All of the multiply substituted pyrrolines 3 and tetrahydropyridines 4 gave the desired products of their Tebbe reactions.
The Diels–Alder reactions between the Tebbe reaction products and electron-deficient dienophiles provided another key skeleton-diversifying branch in our DOS pathway (Scheme 2). Through dienophile screening, we identified maleimides, N-phenyl triazolinedione, tetracyanoethylene, imines, benzoquinone, and 2,6-dichlorobenzoquinone as very good reaction substrates for the Diels–Alder reactions of the ethoxy dienes. Although the reaction yields were only moderate to good (35–85%), the reaction stereoselectivities were excellent. Indeed, considering that the compounds in the library possess up to six stereogenic centers, the selectivities of the Diels–Alder reactions are quite remarkable.
Scheme 2.

Structures of 16 dienes and 12 dienophiles. Bn=benzyl; Bs= benzenesulfonyl; Ts=tosyl (p-toluenesulfonyl).
Specifically, we detected only single diastereoisomeric products from the cycloadditions of the pyrroline-derived dienes 5 with maleimides, N-phenyltriazolinedione, tetracyanoethylene, and 2,6-dichlorobenzoquinone and from the cycloadditions of the tetrahydropyridine-derived dienes 6 with N-phenyl triazolinedione, benzoquinone, and imines. The cycloadditions of the dienes 6 with maleimides and tetracyanoethylene exhibited diastereoselectivities of up to 6:1 and 10:1, respectively. When benzoquinones were used as the dienophiles, we detected no direct Diels–Alder reaction products; instead, we isolated the oxidized benzoquinones 10a and 14c and the naphthoquinones 10e and 14a (Scheme 3). Based on the isolation of dienophile-derived hydroquinone byproducts, we surmise that dehydrogenation or dehydrochlorination occurred, following the Diels–Alder reactions, in the presence of excesses of benzoquinone dienophiles.[14] It is also notable that the cyclohexene double bond, resulting from the Diels–Alder reaction, isomerized into conjugation with the benzoquinone in 10a.
Scheme 3.

Representative compounds possessing distinctive scaffolds in the library.
Given the complexity of the structures of the library compounds, determining their structures, especially their stereochemistry, was a challenge. The relative configuration of each scaffold obtained from the Diels–Alder reactions (Scheme 3), with the exception of 14c, was established unequivocally through X-ray crystallographic analysis (see the Supporting Information). The structure of 14c was established based on 1H, 13C, DEPT, and COSY NMR spectroscopic and mass spectrometric data. In addition, compound 14c, when left at room temperature for over 6 months, was converted into its isomer 14c′, which features its enol ether double bond in conjugation with the benzoquinone motif (compare with compound 10a); the X-ray diffraction data of the crystalline solid 14c′ provided the relative configuration of compound 14c. As verified by the X-ray crystallographic analysis, for the dienes 5, the chiral center(s) created in the phosphine-catalyzed [3+2] ring-forming reactions controlled the face from which the dienophile approached the diene in endo-selective Diels–Alder reactions (7d, 8a, 9a, 10a); specifically, dienophiles approached the dienes 5 from the face opposite the C2 substituent. The Diels–Alder reactions of the tetrahydropyridine-derived dienes 6 displayed relatively mixed facial selectivities. The reaction with maleimide, although endo-selective, produced the two dia-stereoisomers 11aa and 11ab in a 2:1 ratio, presumably because the C2 stereogenic center was too far removed from the reaction center to exert a significant steric bias. The dienophiles N-phenyltriazolinedione, tetracyanoethylene, and benzoquinone underwent [4+2] cycloadditions by adding to the diene 6 from the face opposite the C2 aryl group (yielding 12a, 13a, and 14c, respectively). Interestingly, for imine dienophiles, the endo-selective Diels–Alder reaction provided, for example, the octahydro-1,6-naphthyridine 15a, in which the tosyl group in the tetrahydropyridine ring of the diene 6 controlled the facial approach of the imine dienophile. X-ray crystallography revealed that the tetrahydropyridine[10a] and the octahydronaphthyridine featured anti relationships between their α-phenyl and N-tosyl groups.
Surprisingly, the Diels–Alder reaction products featuring enol ether units were quite stable during purification through silica gel. Most of them could be stored at −20°C for several months without detectable decomposition. The enol ether units were hydrolyzed to the corresponding ketone products (16–18) upon treatment with a solution of aqueous HCl (0.1M) in acetone (Scheme 4). Only a single isomer was detected in each case; the reaction yields ranged from good to excellent (75–95%). The ketones were obtained with cis-5,6-fused rings (compounds 16) or trans-6,6-fused rings (compounds 17).[15] It is noteworthy that the decahydronaphthyridinones 18 featured a cis-fused [4.4.0] bicyclic framework.
Scheme 4.
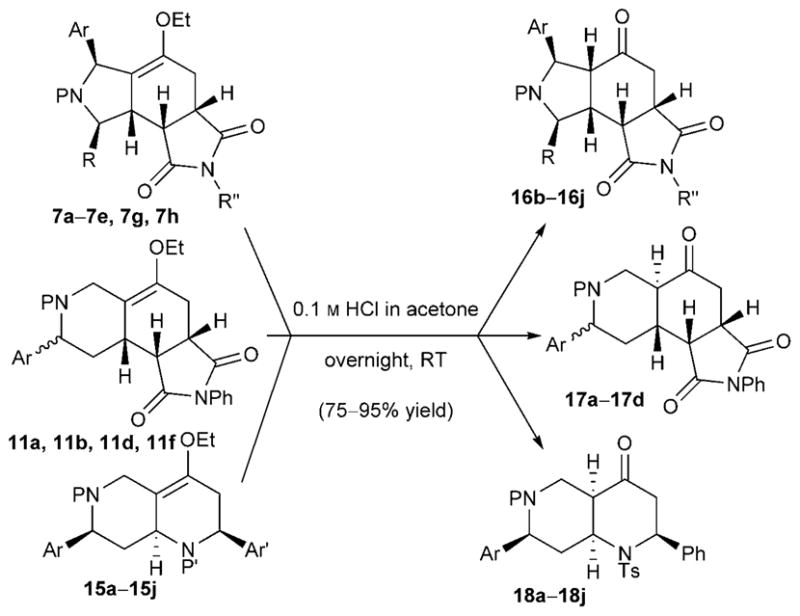
Hydrolysis of enol ethers to ketones.
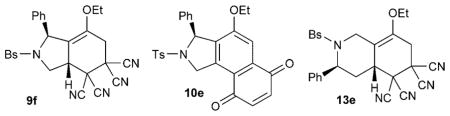
With the collection of 91 compounds in hand, we tested them in an assay for cancer cell migration. Although cell movement is a basic biological phenomenon involved in a range of normal and disease processes, including cancer cell invasion and metastasis, research probes and chemotherapeutics that function through known mechanisms that specifically target cell migration are rare.[16] Despite the relatively small number of compounds in our library, we were optimistic about the prospect of identifying compounds exhibiting antimigratory activity because of the broad chemical space covered by the products generated from the branched pathway (phosphine catalysis/Tebbe/Diels–Alder/(hydrolysis)). The compound library was screened in a medium-throughput wound closure assay to identify molecules that inhibited the migration of MDA-MB-231 human breast cancer cells, essentially as previously described.[17] To our delight, compounds 9 f, 10e, and 13e exhibited subtoxic antimigratory activity in the wound closure assay (Table 1).
Table 1.
Inhibition of migration of MDA-MB-231 human breast cancer
 | ||||
|---|---|---|---|---|
| Compound | MIC [μM][a] | IC50 [μM][b] | 95% CI [μM][c] | MLC [μM][d] |
| 9 f | 5 | 43.1 | 39.9–46.6 | 150 |
| 10e | 5 | 14.8 | 13.1–16.7 | 50 |
| 13e | 5 | 21.6 | 19.1–24.5 | 100 |
Minimum inhibitory concentration (MIC) represents the lowest concentration tested at which there was statistically significant (by unpaired, two-tailed Student’s t-tests (P <0.05)) inhibitory activity in the wound closure assay.
Half-maximal inhibitory concentration (IC50) values were calculated for inhibition of wound closure at 9 h post-wounding from concentration–response profiles for a range of subtoxic concentrations (n=7–9 wounds from three independent experiments).
The 95% confidence interval (CI) of the IC50.
Minimum lethal concentration (MLC) represents the lowest concentration tested at which there was statistically significant cell death, measured using the trypan blue dye exclusion assay, by the end of wound closure experiments.
These compounds inhibited cell migration over a range of concentrations below the threshold at which cell death first became apparent, suggesting that their antimigratory activity was separate from—and not just a secondary consequence of—general cytotoxic effects. Their IC50 values for inhibition of cell migration were comparable with that of the natural product migrastatin, which has an IC50 of 29 μM and has served as a lead for the subsequent development of more potent analogues.[18]
Cell migration is a necessary, but not sufficient, condition for the metastasis of solid tumors; a major determinant of a cancer cell’s metastatic potential is its ability to invade through extracellular matrix barriers. Therefore, we tested these compounds for their activities using a cell invasion assay. Of the three antimigratory compounds, compound 13e displayed the broadest range of subtoxic activity against cell invasion (Figure 1).
Figure 1.

Inhibition of invasion of MDA-MB-231 cells by 13e. Mean cell numbers (with standard error of the mean) invading through Matrigel in a transwell assay are shown (n=6–8 replicates from three independent experiments). Asterisks denote statistically significant differences from the control by unpaired, two-tailed Student’s t-tests (with P<0.0006 at all three concentrations). The minimum lethal concentration (MLC) for 13e under the conditions of the cell invasion assay was 50 μM. In the cell invasion assay, cells were at a lower density than in the wound closure assay, and compounds were added at the same time as the cells, rather than after attachment and growth.
Compounds 9 f, 10e, and 13e were also evaluated in a tetrazolium salt-based cell proliferation and viability assay. They did not inhibit cell growth at concentrations at which they inhibited cell migration and invasion, but did become cytotoxic at higher concentrations over a very narrow range, with minimum lethal concentration (MLC) values similar to those in Table 1 determined from the trypan blue dye exclusion assay conducted at the end of the wound closure experiments. Therefore, these compounds inhibit cell migration and invasion at subtoxic concentrations, but, interestingly, they do not exhibit subtoxic antiproliferative (cytostatic) activity separable from cytotoxicity. Our findings suggest that these compounds have specificity for targets involved in modulating cell migration and invasion over proliferation. These compounds may, therefore, serve as leads for the development of targeted inhibitors of cell migration and invasion to block breast cancer metastasis.
Conclusion
We have synthesized a library of fused heterocyclic compounds, with 16 distinct scaffolds, through a sequence of phosphine-catalyzed ring-forming reactions, Tebbe reactions, Diels–Alder reactions, and, in some cases, hydrolysis. Our aim for this DOS approach was to efficiently achieve sufficiently high levels of skeletal diversity to explore biologically relevant regions of chemical space. Indeed, our current library, despite its small number of components, allowed the identification of compounds that inhibit cell migration and cell invasion. We expect that this synthetic pathway can undergo further development and optimization, including the construction of solid phase libraries of the 16 distinct scaffolds for extensive chemical genetic screening. We are optimistic that this approach might lead to the development of effective tools for chemical biological studies and the identification of therapeutic lead compounds.
Experimental Section
Synthesis
Detailed synthetic procedures are available in the Supporting Information.
Statistical analyses
For all assays (wound closure; trypan blue dye exclusion; tetrazolium salt), statistical comparisons between the data obtained for the control and each treatment condition were performed by using unpaired, two-tailed Student’s t-tests, with statistically significant differences defined as P<0.05.
Wound closure
MDA-MB-231 cells (American Type Culture Collection number: HTB-26) were cultured in 75 cm2 tissue culture flasks with growth medium (Dulbecco’s minimum essential medium (DMEM) containing 10% fetal bovine serum) at 37°C under 5% CO2 in a humidified tissue culture incubator. The cells were grown to 80% confluence and then treated with trypsin, collected, and replated in 96-well tissue culture plates for initial screening. Once confluent, the cell cultures were treated with compounds at various concentrations or with DMSO carrier solvent alone with fresh growth medium. In all cases, the final DMSO concentration was 0.05%. After 30 min, the cell cultures were wounded with a sterile micropipette tip, and the wound closure experiments were performed essentially as described previously.[17] Compounds were first screened at 50 μM and evaluated for their antimigratory activity or cytotoxicity. This was accomplished by scoring wounds as opened or closed as a function of time following wounding to determine the time to closure. Cell viability was determined by the trypan blue dye exclusion assay conducted at the end of each experiment, with the ratio of dye-non-excluding (dead) to dye-excluding (live) cells compared with the control. The following compounds inhibited wound closure or were cytotoxic at 24 h after wounding: 9 f, 10e, 13b, and 13e (none of the other compounds exhibited any effect on the rate of wound closure, the morphology of the cells or their viability compared to the control). These four compounds were rescreened at 10 μM; only compounds 9 f, 10e, and 13e exhibited subtoxic antimigratory activity at this concentration (they were, however, inactive when rescreened at 2 μM). Therefore, compounds 9 f, 10e, and 13e were examined further in quantitative wound closure assays, over a more finely divided range of concentrations. Cell monolayers in 24-well tissue culture plates were treated with different concentrations of each compound 30 min before wounding and then wounded, as described above. Digital images of the wounds were captured immediately after wounding and at 3, 6, 9, 12, and 24 h post-wounding. The open wound areas, as a function of time after wounding, were determined with NIH Image J software (http://rsb.info.nih.gov/nih-image/) to establish mean percent wound closure over time for each treatment and parallel controls, as previously described.[17] The mean percent wound closure values at 9 h post-wounding for all concentrations up to the maximal subtoxic concentration were subjected to nonlinear regression to calculate IC50 values and 95% confidence intervals for inhibition of wound closure with GraphPad Prism software.
Cell invasion
Compounds 9 f, 10e, and 13e were also analyzed for their ability to inhibit invasion of cells through Matrigel in a cell invasion assay. The assay was performed in transwell chambers according to the manufacturer’s instructions (BD Biosciences). The polycarbonate membranes (8 μm pore size) of the upper chambers were coated with Matrigel (25 μg). Growth medium containing the tested compound or DMSO alone was added to the lower chambers (600 μLwell−1 in 24-well BD Falcon TC companion plates). MDA-MB-231 cells in serum-free DMEM containing the tested compound or DMSO alone were added to the upper chambers (100 μLchamber− of a 50104 cellsmL−1 suspension). In all cases, the final DMSO concentration was 0.05%. Cells were incubated at 37°C under 5% CO2 for 24 h. After removing noninvaded cells from the upper surface of the membrane with a cotton swab, the invaded cells on the lower surface were fixed with MeOH, stained with crystal violet solution, rinsed with water, air-dried, and then counted by using an inverted microscope. In parallel experiments, cell viability, determined using the trypan blue dye exclusion assay at various concentrations of the tested compounds, was evaluated under the conditions of the cell invasion assay.
Cell proliferation and cell viability
The assay was conducted according to the manufacturer’s instructions (CCK-8; Dojindo Molecular Technologies). MDA-MB-231 cells were plated in 96-well tissue culture plates (125 μLwell−1 of a 40104 cellsmL−1 suspension in growth medium) and incubated for 24 h at 37°C under 5% CO2. The cells were then treated with the tested compounds at various concentrations or with DMSO alone. In all cases, the final DMSO concentration was 0.05%. At the same time, other cells from parallel plates were analyzed using the CCK-8 assay to establish the cell density at the start of the experiment. After 48 h of incubation, the cell numbers for the experimental samples were determined and compared with the initial cell numbers. A cytostatic effect was defined as a reduction in cell number relative to the 48 h control that did not fall significantly below the mean initial cell number. A cytotoxic effect was defined as a reduction in cell number that was significantly below that of the mean initial cell number.
Supplementary Material
Acknowledgments
We are grateful to the U.S. National Institutes of Health (O.K.: R01M071779, P41M081282; G.F.: R01M077622) for financial support. S.C. thanks the Italian Government (MIUR) for a research grant; S.S.K. thanks the Nederlandse Organisatie voor Wetenschappelijk Onderzoek for a TALENT fellowship. We thank Dr. Saeed Khan for performing the X-ray crystallographic analyses.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201002195.
Contributor Information
Prof. Gabriel Fenteany, Email: gabriel.fenteany@uconn.edu.
Prof. Ohyun Kwon, Email: ohyun@chem.ucla.edu.
References
- 1.For a review, see: Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0.
- 2.a) Walsh DP, Chang YT. Chem Rev. 2006;106:2476–2530. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]; b) Dolle RE, Bourdonnec BL, Goodman AJ, Morales GA, Thomas CJ, Zhang W. J Comb Chem. 2009;11:739–790. doi: 10.1021/cc9000828. [DOI] [PubMed] [Google Scholar]
- 3.For reviews, see: Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964.Spring DR. Org Biomol Chem. 2003;1:3867–3870. doi: 10.1039/b310752n.
- 4.Bemis GW, Murcko MA. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 5.a) Burke MD, Berger EM, Schreiber SL. Science. 2003;302:613–618. doi: 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]; b) Burke MD, Schreiber SL. Angew Chem. 2004;116:48–60. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:46–58. [Google Scholar]; c) Burke MD, Berger EM, Schreiber SL. J Am Chem Soc. 2004;126:14095–14104. doi: 10.1021/ja0457415. [DOI] [PubMed] [Google Scholar]; d) Nielsen TE, Schreiber SL. Angew Chem. 2008;120:52–61. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:48–56. [Google Scholar]
- 6.For recent examples, see: Dothager RS, Putt KS, Allen BJ, Leslie BJ, Nesterenko V, Hergenrother PJ. J Am Chem Soc. 2005;127:8686–8696. doi: 10.1021/ja042913p.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J Am Chem Soc. 2005;127:12762–12763. doi: 10.1021/ja0530321.Leßmann T, Waldmann H. Chem Commun. 2006:3380–3389. doi: 10.1039/b602822e.Castellano S, Fiji HDG, Kinderman SS, Watanabe M, Leon Pd, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843–5845. doi: 10.1021/ja070274n.Wang N, Xiang J, Ma Z, Quan J, Chen J, Yang Z. J Comb Chem. 2008;10:825–834. doi: 10.1021/cc800025n.Verdié P, Subra G, Averland-Petit MC, Amblard M, Martinez J. J Comb Chem. 2008;10:869–874. doi: 10.1021/cc800085d.
- 7.a) Ding S, Gray NS, Wu X, Ding Q, Schultz PG. J Am Chem Soc. 2002;124:1594–1596. doi: 10.1021/ja0170302. [DOI] [PubMed] [Google Scholar]; b) Kwon O, Park SB, Schreiber SL. J Am Chem Soc. 2002;124:13402–13404. doi: 10.1021/ja028086e. [DOI] [PubMed] [Google Scholar]; c) Couladouros EA, Strongilos AT. Angew Chem. 2002;114:3829–3832. [Google Scholar]; Angew Chem Int Ed. 2002;41:3677–3680. [Google Scholar]; d) Taylor SJ, Taylor AM, Schreiber SL. Angew Chem. 2004;116:1713–1717. [Google Scholar]; Angew Chem Int Ed. 2004;43:1681–1685. doi: 10.1002/anie.200353466. [DOI] [PubMed] [Google Scholar]; e) Spiegel DS, Schroeder FC, Duvall JR, Schreiber SL. J Am Chem Soc. 2006;128:14766–14767. doi: 10.1021/ja065724a. [DOI] [PubMed] [Google Scholar]; f) Mitchell JM, Shaw JT. Angew Chem. 2006;118:1754–1758. [Google Scholar]; Angew Chem Int Ed. 2006;45:1722–1726. doi: 10.1002/anie.200503341. [DOI] [PubMed] [Google Scholar]; g) Ko KS, Jang HJ, Kim E, Park SB. Chem Commun. 2006:2962–2964. doi: 10.1039/b606341a. [DOI] [PubMed] [Google Scholar]; h) Wyatt EE, Fergus S, Plowright AT, Jessiman AS, Welch M, Spring DR. Chem Commun. 2006:3296–3298. doi: 10.1039/b607710b. [DOI] [PubMed] [Google Scholar]; i) Comer E, Rohan E, Deng L, Porco JA. Org Lett. 2007;9:2123–2126. doi: 10.1021/ol070606t. [DOI] [PubMed] [Google Scholar]; j) Shang S, Iwadare H, Macks DE, Ambronisi LM, Tan DS. Org Lett. 2007;9:1895–1898. doi: 10.1021/ol070405p. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) An H, Eum SJ, Koh M, Lee SK, Park SB. J Org Chem. 2008;73:1752–1761. doi: 10.1021/jo702196f. [DOI] [PubMed] [Google Scholar]; l) Thomas GL, Spandl RJ, Glansdorp FG, Welch M, Bender A, Cockfield J, Lindsay JA, Bryant C, Brown DFJ, Loiseleur O, Rudyk H, Ladlow M, Spring DR. Angew Chem. 2008;120:2850–2854. doi: 10.1002/anie.200705415. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:2808–2812. doi: 10.1002/anie.200705415. [DOI] [PubMed] [Google Scholar]; m) Morton D, Leach S, Cordier C, Warriner S, Nelson A. Angew Chem. 2008;121:110–115. doi: 10.1002/anie.200804486. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;48:104–109. [Google Scholar]
- 8.For reviews, see: Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535–544. doi: 10.1021/ar000253x.Ye LW, Zhou J, Tang Y. Chem Soc Rev. 2008;37:1140–1152. doi: 10.1039/b717758e.Cowen BJ, Miller SJ. Chem Soc Rev. 2009;38:3102–3116. doi: 10.1039/b816700c.
- 9.a) Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461–3464. [Google Scholar]; b) Xu Z, Lu X. J Org Chem. 1998;63:5031–5041. [Google Scholar]
- 10.a) Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716–4717. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; b) Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276–6282. [Google Scholar]
- 11.Watanabe M, Fiji HDG, Guo L, Umetsu A, Kinderman SS, Slamon DJ, Kwon O, Tamanoi F. J Biol Chem. 2008;283:9571–9579. doi: 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Wetzel S, Klein K, Renner S, Rauh D, Oprea TI, Mutzel P, Waldmann H. Nat Chem Biol. 2009;5:581–583. doi: 10.1038/nchembio.187. [DOI] [PubMed] [Google Scholar]; b) Tan KT, Guiu-Rozas E, Bon RS, Guo Z, Delon C, Wetzel S, Arndt S, Alexandrov K, Waldmann H, Goody RS, Wu YW, Blankenfeldt W. J Med Chem. 2009;52:8025–8037. doi: 10.1021/jm901117d. [DOI] [PubMed] [Google Scholar]
- 13.Cannizzo LF, Grubbs RH. J Org Chem. 1985;50:2386–2387. [Google Scholar]
- 14.Kaliappan KP, Ravikumar V. Org Biomol Chem. 2005;3:848–851. doi: 10.1039/b418659a. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Nakagawa M, Torisawa Y, Hino T. Heterocycles. 1994;38:1609–1618. [Google Scholar]
- 16.For a review, see: Fenteany G, Zhu S. Curr Top Med Chem. 2003;3:593–616. doi: 10.2174/1568026033452348.
- 17.Mc Henry KT, Ankala SV, Ghosh AK, Fenteany G. ChemBio-Chem. 2002;3:1105–1111. doi: 10.1002/1439-7633(20021104)3:11<1105::AID-CBIC1105>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 18.a) Nakae K, Yoshimoto Y, Sawa T, Homma Y, Hamada M, Takeuchi T, Imoto M. J Antibiot. 2000;53:1130–1136. doi: 10.7164/antibiotics.53.1130. [DOI] [PubMed] [Google Scholar]; b) Njardarson JT, Gaul C, Shan D, Huang XY, Danishefsky SJ. J Am Chem Soc. 2004;126:1038–1040. doi: 10.1021/ja039714a. [DOI] [PubMed] [Google Scholar]; c) Gaul C, Njardarson JT, Shan D, Dorn DC, Wu KD, Tong WP, Huang XY, Moore MA, Danishefsky SJ. J Am Chem Soc. 2004;126:11326–11337. doi: 10.1021/ja048779q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.