

Abstract
The ovarian steroid hormones estradiol and progesterone regulate a wide variety of non-reproductive functions in the central nervous system by interacting with molecular and cellular processes. A growing literature from studies using rodent models suggests that 17β-estradiol, the most potent of the biologically relevant estrogens, enhances synaptic transmission and the magnitude of long-term potentiation recorded from in vitro hippocampal slices. In contrast, progesterone has been shown to decrease synaptic transmission and reduce hippocampal long-term potentiation in this model system. Hippocampal long-term depression, another form of synaptic plasticity, occurs more prominently in slices from aged rats. A decrease in long-term potentiation magnitude has been recorded in hippocampal slices from both adult and aged rats behaviorally stressed just prior to hippocampal slice tissue preparation and electrophysiological recording. 17β-estradiol modifies synaptic plasticity in both adult and aged rats, whether behaviorally stressed or not by enhancing long-term potentiation and attenuating long-term depression. The studies discussed in this review provide an understanding of new approaches used to investigate the protective effects of ovarian hormones against aging and stress, and how these hormones impact age and stress-related learning and memory dysfunction.
Keywords: Hippocampus, synaptic plasticity, rat, estrogen, progesterone, aging, stress
I. Introduction
Richard Thompson’s area of research and scholarly interest is the broad field of psychobiology, with a focus on the neurobiological substrates of learning and memory function. Three major research topics have been the primary focus of his work spanning the past several years: 1) brain substrates of basic associative learning and memory, 2) essential role of the cerebellum in classical conditioning of discrete responses, and 3) role of the hippocampus in basic processes of synaptic plasticity and memory. It is this third research topic that I will focus on in this review. One of the key issues in analyzing brain substrates of memory is the nature and localization of memory traces. There are several different forms of memory involving different brain systems, and learning-induced changes in neuronal activity can be used to identify memory traces. One strategy for the study of memory storage is to pick a mechanism of neural plasticity such as long-term potentiation (LTP) in a structure known to be important for memory (e.g., hippocampus), and attempt to show that it is a substrate for memories. LTP of synaptic transmission in the hippocampus (and neocortex) is considered to be a cellular model of memory trace formation in the brain, at least for certain forms of memory (Baudry, Davis, and Thompson, 2000). The extensive body of work on the molecular and synaptic mechanisms underlying hippocampal LTP is now matched with a growing number of studies suggesting the critical role of LTP in behavioral learning and memory. Because the ovarian hormone estrogen seems to promote changes in synaptic plasticity within the nervous system, much effort in Dick’s lab has been devoted to identifying the role of ovarian hormons on hippocampal synaptic plasticity. In this article, I review the collaborative work that Dick and I have been engaged in most recently, and focused on the influence that ovarian hormones, aging and behavioral stress have on hippocampal synaptic plasticity.
Steroid hormones play an essential role in a variety of key biological functions including reproduction, sexual differentiation, brain development, cognition, memory, and behavior. The nervous system is a major target of hormone action, and contains specific receptors for circulating hormones secreted from peripheral organs such as the adrenal cortex, testis and ovary. Gonadal hormones, such as the estrogens, androgens and progestins, function not only at the genomic level through classic receptors that belong to the superfamily of nuclear receptors (Guerriero, 2009), but also non-genomically through G protein-coupled steroid receptors and membrane-localized steroid receptors (Hammes and Levin, 2007). While many of the effects of estrogen and other hormones that have a prolonged latency and duration of action in the brain can be readily explained by the genomic mechanism of action (McEwen and Alves, 1999), ovarian hormones have also been found to produce rapid, short-term effects on electrophysiological properties of neurons with latencies and durations on the scale of seconds to minutes (Foy, 2001; Smith, Waterhouse, and Woodward, 1987; 1988; Teyler, Vardaris, Lewis, and Rawitch, 1980). Since the activation of transcriptional and translational mechanisms by intracellular steroid hormone receptors requires a longer latency, physiological responses occurring within extremely short latencies have been presumed to involve non-nuclear, membrane-mediated effects. This review will focus on the ovarian hormones 17β-estradiol (E2) and progesterone (P4), as they are the primary hormones that have been shown to regulate hippocampal synaptic plasticity.
Synaptic plasticity is a term used to describe several forms of long-lasting, activity-dependent changes in synaptic strength. It also refers to the ability of neuronal circuits to undergo functional or organizational changes due to previous activity, and is strongly associated with forebrain structures including the hippocampal formation. Since different cell types are neatly orgainzed into layers within the hippocampus, it makes it an ideal model system for the studying the neurophysiology of synaptic plasticity. In the late 1940’s, Hebb postulated that synaptic plasticity was an essential way for the brain to be modified by experience and practice, a concept that has since then been associated with the notion of Hebbian synapses (Berlucchi and Buchtel, 2009). A considerable body of work has now demonstrated that hippocampal long-term potentiation (LTP) and long-term depression (LTD) of synaptic transmission are two forms of synaptic plasticity considered to be cellular models of memory trace formation in the brain (Baudry et al., 2000; Bear and Malenka, 1994; Bliss and Collingridge, 1993). While the molecular and synaptic mechanisms underlying LTP have been studied extensively, there is a relative paucity of studies demonstrating the critical role of LTP in behavioral learning and memory (Shors and Matzel, 1997); but see (Lee and Silva, 2009). Nonetheless, whether LTP (and/or LTD) is or is not the substrate of synaptic modifications that occur during learning in forebrain structures of vertebrates, studies of its mechanisms have revealed the existence of a number of processes that undoubtedly play critical roles in memory formation (Bi and Poo, 2001; Fanselow and Poulos, 2005), including biochemical intracellular cascades involving NMDA/AMPA receptor function (Kramar, Chen, Brandon, Rex, Liu, Gall, and Lynch, 2009; Zadran, Qin, Bi, Zadran, Kim, Foy, Thompson, and Baudry, 2009).
In area CA1 of hippocampus, LTP, the most widely studied form of synaptic plasticity, represents an activity-dependent modification of synaptic efficacy that results in a long-lasting increase in synaptic transmission following a brief burst of high-frequency stimulation of the Schaffer collateral input to pyramidal neurons, and may act as a potential mechanism for learning and memory function. Hippocampal LTP requires N-methly-d-aspartate (NMDA) receptor activation for its induction, and an increase in α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate (AMPA) receptor function for its expression and maintenance (Lisman, 2003; Malenka and Nicoll, 1999; Stanton, 1996). Conversely, hippocampal LTD is a form of activity-dependent synaptic plasticity where the repeated activation of specific hippocampal inputs results in a decrease in responsiveness of activated neurons, requires activation of NMDA and metabotropic glutamate receptors, and is due to internalization of AMPA receptors (Malenka and Bear, 2004; Stanton, 1996). In this review, the relationship between gonadal steroid hormone action and hippocampal synaptic plasticity will be described by analyzing the effects of E2 and P4 on hippocampal synaptic plasticity, along with findings that demonstrate the effects of aging and behavioral stress on synaptic plasticity modified by gonadal hormone action.
II. Estrogen and Hippocampus
Over 40 years ago, estrogen was found to promote changes in synaptic plasticity within the nervous system. In one pioneering study, decreased hippocampal seizure thresholds were found in animals primed with estrogen and also during proestrus, the time of the estrous cycle when estrogen levels are at their highest levels (Terasawa and Timiras, 1968). In humans, changes in electrical activity of nervous system tissue correlate with hormonal factors that appear to play a role in catamenial epilepsy, a form of epilepsy in which the likelihood of seizures varies during the menstrual cycle. Many women with catamenial epilepsy experience a sharp increase in seizure frequency immediately before menstruation, when estrogen concentrations relative to those of progesterone are at their highest levels (Backstrom, 1976; Reddy, 2009). Changes in hippocampal responsiveness correlate with estrogen activity, as LTP (recorded in vivo) is maximal in female rats during the afternoon of proestrus, when endogenous estrogen concentrations are highest (Warren, Humphreys, Juraska, and Greenough, 1995). Furthermore, induction of in vivo hippocampal LTP is facilitated in ovariectomized rats treated with E2 as compared to untreated ovariectomized rats (Cordoba Montoya and Carrer, 1997).
Due in large part to allow more control of biological tissues and experimental variables, the development of in vitro models to study the mechanisms of synaptic plasticity have provided researchers better tools to investigate how hormones regulates synaptic excitability in the nervous system, and, in particular, in hippocampus (Teyler, 1980). It should be noted, however, that the binding of 3H-estradiol in hippocampus is less than that seen in hypothalamus and related diencephalic structures (McEwen and Alves, 1999; McEwen, Gerlach, and Micco, 1975). Nonetheless, studies by Teyler and colleagues using the in vitro hippocampal slice preparation showed that gonadal hormones dramatically affected neuronal excitability in specific pathways of the rodent hippocampus (Teyler et al., 1980; Vardaris and Teyler, 1980). In the initial series of experiments, extracellular monosynaptic population field responses recorded from area CA1 of hippocampal slices from male and female rats were monitored before and after the addition of E2 (100 pM) to the slice incubation medium (artificial cerebrospinal fluid; aCSF). In male rats, E2 produced a rapid (< 10 min) enhancement of population field responses evoked by stimulation of the afferents to CA1 pyramidal cells (Fig. 1). This was the first published report demonstrating that picomolar concentrations of the gonadal hormone E2 directly enhanced what is now known to be glutamatergic synaptic transmission in hippocampus (Teyler et al., 1980).
Figure 1.

Effects of Gonadal Hormones on Hippocampal Synaptic Activity in Rats.
(A) Diagram of a transverse hippocampal slice. Stimulating electrodes were located in the afferent pathway, which contains the Schaffer (Sch.) collaterals. Recording micropipettes were situated in the pyramidal cell body layer in CA1. Cells of this subfield receive monosynaptic input from the CA3 pyramids via the Schaffer collateral system.
(B) Representative field potentials from slice preparations in the various experimental conditions. Extracellular population spike responses to a given stimulus intensity are shown from the control period (before steroid administration) and after the administration of 10−10 M 17β-estradiol (E) or 10−10 M testosterone (T). Potentials from slices obtained from males and from proestrus and diestrus females are shown for purposes of comparison. All potentials are single sweeps recorded at the same voltage and time scales.
(C) Bar graph summarizing the major experimental outcomes. Values on the ordinate are mean percentages of spike amplitudes after steroid administration. Data for each condition are from 6 to 10 animals, each contributing one slice. Cursors representing magnitude of variability (standard error of the mean) are shown for each bar.
Reprinted with permission from (Teyler et al., 1980). Copyright 2010 American Association for the Advancement of Science.
III. Estrogen, NMDA and AMPA Receptor Regulation
Although the mechanism of action of estrogen in hippocampus is not entirely understood, it is likely to be receptor-mediated via the DNA-binding domains of estrogen receptor α and β (ERα, ERβ), and plasma membrane estrogen receptors (mER). Electrophysiological experiments conducted in hippocampus found there was no facilitation of field responses when the inactive estrogen, 17α-estradiol, was added to the hippocampal slice medium (Foy and Teyler, 1983), and the further addition of E2 to slices pretreated with 17α-estradiol no longer resulted in an increased response as observed in the presence of E2 alone (Foy and Teyler, 1983; Wong and Moss, 1991; 1992). Similar results were found when the estrogen receptor antagonist tamoxifen was applied to hippocampal slices before addition of E2 (Foy, 1983). The ability of 17α-estradiol and tamoxifen to block the effects of E2 on hippocampal excitability provides strong evidence that the rapid physiological modulation of estrogen is most likely due to the activation of mER.
In vitro intracellular recordings of CA1 neurons from adult ovariectomized female rats have shown that addition of E2 increases synaptic excitability in part by enhancing the magnitude of AMPA receptor-mediated synaptic responses (Wong and Moss, 1992). The rapid onset of the increased excitability, and its blockade by 6-cyano-7-nitroquinaxaline (CNQX, an AMPA receptor antagonist) but not by D-2-amino-5-phosphonovalerate (D-APV a competitive NMDA receptor antagonist), supported a postsynaptic membrane site of action resulting in enhanced non-NMDA glutamate receptor function. Later studies using whole cell recordings found that acute E2 application also potentiated kainate-induced currents in a subpopulation of CA1 cells (Gu and Moss, 1996), although a direct interaction between estrogen and the receptor channel was not indicated (Wong and Moss, 1994). A presynaptic mechanism of E2’s effect on synaptic plasticity has been somewhat ruled out because the paired-pulse facilitation ratio (PPF), an indirect measure of presynaptic glutamate release probability, was measured in E2 vs. vehicle-treated control hippocampal slices, and there were no differences found at any of the tested intervals (Smith and McMahon, 2005). However, the authors state that a lack of change of the PPF does not rule out all potential presynaptic effects of estradiol.
In addition to reseach supporting an E2-increased activation of AMPA receptor-mediated synaptic responses, a large body of evidence demonstrates that E2-mediated regulation of synapse formation is dependent on NMDA receptor activation. Morphological studies during the course of neuronal development conducted in cultured neurons prepared from embryonic day 18 rat fetuses have shown that estrogenic steroids exert a growth-promoting, neurotrophic effect on hippocampal and cortical neurons via a mechanism that requires NMDA receptor activation (Brinton, Proffitt, Tran, and Luu, 1997; Brinton, Tran, Proffitt, and Kihil, 1997). In vivo studies using adult ovariectomized female rats have also revealed a proliferation of dendritic spines in hippocampal CA1 pyramidal cells after E2 treatment that could be prevented by blockade of NMDA receptors, but not of AMPA or muscarinic receptors (Woolley and McEwen, 1994). Other reports using adult ovariectomized female rats provided evidence that chronic E2 treatment increases the number of NMDA receptor binding sites and NMDA receptor-mediated responses (Gazzaley, Weiland, McEwen, and Morrison, 1996; Woolley, Weiland, McEwen, and Schwartzkroin, 1997). Collectively, these studies indicate that estrogen and NMDA receptors are heavily involved in synapse formation.
Because of the voltage-dependent blockade of the NMDA receptor channel by Mg2+ and the slow kinetics of the channel opening relative to that of the AMPA receptor, there is only a minor NMDA receptor-mediated component of the excitatory postsynaptic potential (EPSP) evoked by low-frequency stimulation of glutamatergic afferents. This NMDA receptor component can be enhanced with low Mg2+ concentrations or the high-frequency stimulation patterns used to induce LTP, which eliminate the Mg2+ blockade due to membrane depolarization resulting from the summation of overlapping EPSPs (Xie, Berger, and Barrionuevo, 1992). In experiments using low Mg2+ concentrations and in the presence of the AMPA receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX), an acute application of E2 in adult male rat hippocampal slices resulted in a rapid increase in the amplitude of NMDA receptor-mediated EPSPs evoked by stimulation of the Schaffer collaterals (Foy, Xu, Xie, Brinton, Thompson, and Berger, 1999). The effect of E2 on pharmacologically isolated NMDA receptor-mediated synaptic responses was such that concentrations of E2 greater than 10 nM induced seizure activity in hippocampal neurons, and lower concentrations (1 nM) markedly increased the amplitude of NMDA receptor-mediated EPSPs.
IV. Estrogen and Hippocampal LTP
To investigate the effect of estrogen on synaptic plasticity, hippocampal slices from adult male rats were treated with E2 before applying high-frequency stimulation to induce LTP at the Schaffer collateral inputs to CA1. When LTP magnitude was assessed after high-frequency stimulation, fEPSP values were increased significantly in E2-treated slices as compared to vehicle-treated slices (Fig. 2). fEPSP mean increases in slope was 192% (experimental) vs. 154% (control). Thus, hippocampal slices from adult male rats treated with E2 exhibited a pronounced, persisting and significant increase in LTP as measured by both population fEPSP slope and fEPSP amplitude (Foy, Baudry, Foy, and Thompson, 2008b; Foy et al., 1999).
Figure 2.

17β-estradiol Enhances Hippocampal LTP in Adult Male Rats
All hippocampal slices were perfused with aCSF for 10 min to obtain fEPSP slope and amplitude percentage baseline data. After 10 min of baseline recording, experimental slices were perfused with 100 pM 17β-estradiol (E2). Control slices continued to be perfused with aCSF. After 30 min of either E2 or aCSF perfusion, all slices received high-frequency stimulation, designed to induce long-term potentiation. Data points represent averaged fEPSP slope ± SEM (taken at each 20 sec sweep) for experimental (E2-treated) and control (aCSF) hippocampal slices.
Reprinted with permission from (Foy et al., 1999). Copyright 2010 American Physiology Society.
To further evaluate the effects of E2 on the magnitude of hippocampal LTP, the intensity of afferent stimulation to Schaffer collaterals in slices perfused with E2 was decreased in order to reduce baseline values to pre-E2 levels immediately before the delivery of the high-frequency stimulation train used to elicit LTP (Bi, Broutman, Foy, Thompson, and Baudry, 2000). Under these conditions, E2 still produced an increase in the magnitude of LTP in adult male rat hippocampal slices as compared to that obtained in vehicle-treated slices (Fig. 3). When similar experiments were performed in the presence of the src inhibitor PP2, E2 no longer increased basal EPSP amplitude, nor did it increase LTP magnitude, indicating that E2-induced enhancement of hippocampal LTP is not due to simply a change in basal EPSP level, but is more likely due to biochemical activation of an intracellular cascade, presumably mediated by activation of a src tyrosine pathway that enhances NMDA receptor function. More recent studies have provided evidence that the effects of E2 on LTP induction are due to the activation of an intracellular cascade leading to actin polymerization and increased numbers of AMPA receptors (Kramar, Chen, Brandon, Rex, Liu, Gall, and Lynch, 2009; Zadran, Qingyu, Bi, Zadran, Kim, Foy, Thompson, and Baudry, 2009).
Figure 3.

Effects of an src inhibitor (PP2) on 17β-estradiol (E2)-mediated enhancement of EPSP amplitude and degree of LTP in acute hippocampal slices. A stimulating electrode was located in CA3, and a recording electrode was located in stratum radiatum of CA1. Extracellular EPSPs were evoked by stimulation every 30 sec, and the amplitude of the EPSP was measured. After a stable baseline was recorded, PP2 (10 µM) was added at the indicated time. Likewise, E2 (1 nM) was added at the indicated time. After resetting the stimulation intensity to obtain an EPSP of the same amplitude as before treatment with PP2 or E2, high-frequency stimulation was delivered, and low-frequency stimulation was resumed. PP2 blocked the estrogen enhancement of LTP, but had no effect on LTP itself. Results are expressed as percentages of pre-drug values and are means ± SEM of 6–10 experiments. HFS, high-frequency stimulation.
Reprinted with permission from Bi et al. (2000). Copyright 2010 National Academy of Sciences, USA.
In another series of animal studies, hormonal changes during the estrous cycle were correlated with changes in synaptic plasticity in rats. Hippocampal slices were prepared from cycling female rats in diestrus (low estrogen concentration) or proestrus (high estrogen concentration), and LTP was elicited by high-frequency stimulation. The difference in LTP magnitude between these groups following high-frequency stimulation was dramatic: slices from rats in proestrus exhibited LTP representing about a 50% increase over baseline, whereas slices from rats in diestrus had LTP values representing about a 25% increase over baseline (Bi, Foy, Vouimba, Thompson, and Baudry, 2001) (Fig. 4). These findings support the original observations of Teyler et al (1980) who identified changes in baseline synaptic transmission that were correlated with the phase of the estrous cycle in female rats at the time of hippocampal slice preparation.
Figure 4.

Changes in LTP in field CA1 of hippocampal slices from female rats in proestrus and diestrus. Hippocampal slices from female rats in proestrus or diestrus were prepared and fEPSP amplitude and slope values were obtained for each slice and averaged across slices to produce one average before and after the train of high-frequency stimulation (HFS). fEPSP amplitudes and slopes were normalized for the 10 min pre-HFS period for each slice. Separate ANOVAs and planned two-tailed t tests for the pre-HFS and post-HFS periods were used to evaluate the effects of estrous cycle on fEPSP slope and amplitude.
(A) Representative waveforms from female rats in proestrus and diestrus for pre-HFS (1) and post-HFS (2) periods.
(B) Means ± SEM of fEPSP slopes recorded in slices from female rats in proestrus (filled circles; n=6) and diestrus (open circles; n=5).
Reprinted with permission from (Bi et al., 2001). Copyright 2010 National Academy of Sciences, USA.
The electrophysiological work described above has shown that female rats in proestrus exhibited an increased magnitude of hippocampal LTP compared to females in diestrus. Another study reexamined the effects of E2 on hippocampal LTP during these two critical time periods in the rat estrous cycle: proestrus and diestrus (Foy, Baudry, and Thompson, 2004). Estrous cycles of adult (3–5 mo) Sprague-Dawley rats were monitored for 10 days prior to any physiological experiments, and hippocampal slices were prepared from rats that were either in proestrus or diestrus. Recording and stimulating electrodes were positioned in the dendrites of area CA1 and Schaffer collaterals, respectively. Baseline stimulation was adjusted to elicit 50% of the maximum fEPSP amplitude. After 10 min of stable baseline stimulation, aCSF or E2 at a concentration of 100 pM (experimental group) was perfused for 30 min, and LTP was induced by a brief period of high-frequency stimulation. Subsequent synaptic responses were monitored for 30 min post-LTP induction. The magnitude of LTP induced in area CA1 was larger in vehicle-treated slices from proestrus rats, compared to slices from diestrus rats, as previously reported (Bi et al., 2001). Surprisingly, the addition of E2 increased LTP in slices from diestrus rats, while it decreased LTP in slices from proestrus rats (Fig. 5). These observations indicate that E2 alters hippocampal LTP in female rats, depending on the state of their estrous cycle (i.e., on the levels of circulating estrogen). In cycling female rats, when endogenous circulating levels of estrogen are at their highest levels (i.e., during proestrus), LTP magnitude is high, and exogenously applied estrogen during proestrus decreases LTP magnitude, possibly through the activation of an inhibitory or ceiling effect. When endogenous circulating levels of estrogen are at their lowest levels (i.e., during diestrus), the situation is completely reversed from that observed in the proestrus state. Here, LTP magnitude is low, and exogenously applied estrogen increases LTP magnitude under this condition.
Figure 5.

Changes in LTP in field CA1 of hippocampal slices from female rats in proestrus and diestrus, and with 17β-estradiol (experimental) or aCSF (control) treatment. Hippocampal slices from female rats in proestrus or diestrus were prepared as described.
(A) Means ± SEM of fEPSP amplitudes recorded following tetanus in slices from female rats in diestrus. 17β-estradiol (filled circles) enhanced LTP relative to control aCSF (open circles).
(B) Means ± SEM of fEPSP amplitudes recorded following tetanus in slices from female rats in proestrus. 17β-estradiol (filled circles) impaired LTP relative to control aCSF (open circles). Reprinted with permission from (Foy et al., 2004). Copyright 2010 Cambridge University Press.
These results indicate that cyclic changes in estrogen levels occurring during the estrous cycle in female rats are associated with changes in the magnitude of LTP recorded from hippocampal CA1 neurons. They also corroborate several results mentioned earlier indicating the facilitation of LTP induction by estrogen in ovariectomized female rats (Cordoba Montoya and Carrer, 1997) and the increased LTP in the afternoon of proestrus of female rats (Warren et al., 1995), and are in agreement with results of other studies showing improved memory performance with high estrogen levels in female rats (Acosta, Mayer, Talboom, Zay, Scheldrup, Castillo, Demers, Enders, and Bimonte-Nelson, 2009; Leuner, Mendolia-Loffredo, and Shors, 2004).
V. Estrogen, Aging and Hippocampal LTD
During aging, the decline in cognitive function has been attributed to alterations in cellular structure and function, presumably and ultimately affecting mechanisms of synaptic plasticity. In many cases, changes in synaptic plasticity during aging are quite subtle; however, in other cases such as animal models that emulate age-associated disorders, they are more apparent. It has been noted that when memory function declines, the processes of synaptic plasticity in hippocampus are altered. More specifically, while LTP may become impaired, LTD is enhanced (Barnes, 1979; 1994; Barnes, Rao, Foster, and McNaugton, 1992; Foster, 1999; Foster and Norris, 1997; Geinisman, Detoledo-Morell, and Heller, 1995; Landfield and Lynch, 1977; Landfield, Pitler, and Applegate, 1986; Norris, Halpain, and Foster, 1998; Norris, Korol, and Foster`, 1996). The effect of aging on LTD was replicated in a group of aged male rats, and a profound action of estrogen on this process in aged male rats was discovered (Foy et al., 2008b; Vouimba, Foy, and Thompson, 2000). Female rats were not studied in aged LTD experiments because of their lack of hormonal cycling. LTD was induced using standard conditions in CA1 region of hippocampal slices prepared from adult (3–5 mo) and aged (18–24 mo) rats. In agreement with earlier studies (Foster, 1999; Foster and Norris, 1997), the standard protocol for inducing LTD (1Hz for 15 min) resulted in little or no LTD in slices from adult animals, but LTD is marked in slices from aged animals (Fig. 6A) (Foy et al., 2008b). Infusion of E2 in slices caused a slight increase in synaptic transmission (baseline), as in previous studies. It had little effect on LTD in slices from adult animals, but markedly attenuated LTD in slices from aged animals (Fig. 6B). Thus, the prevention by E2 of age-related LTD enhancement may account, in part, for the protective effects of estrogen on memory functions in aged organisms.
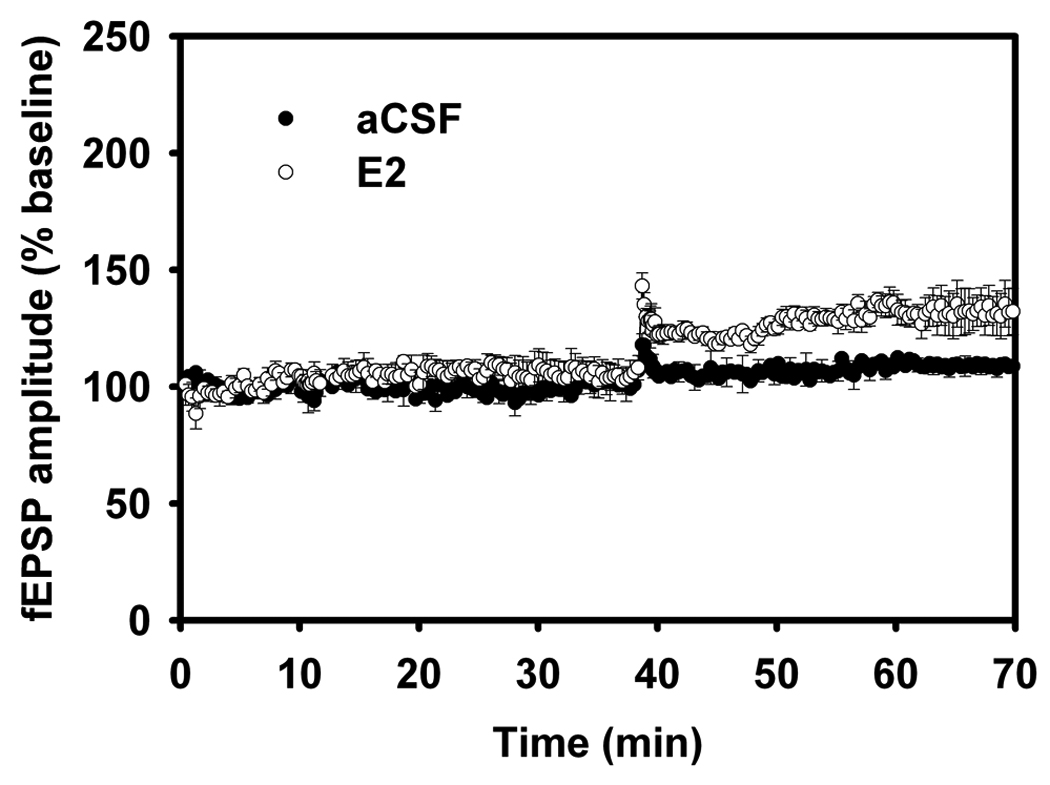
Figure 6.
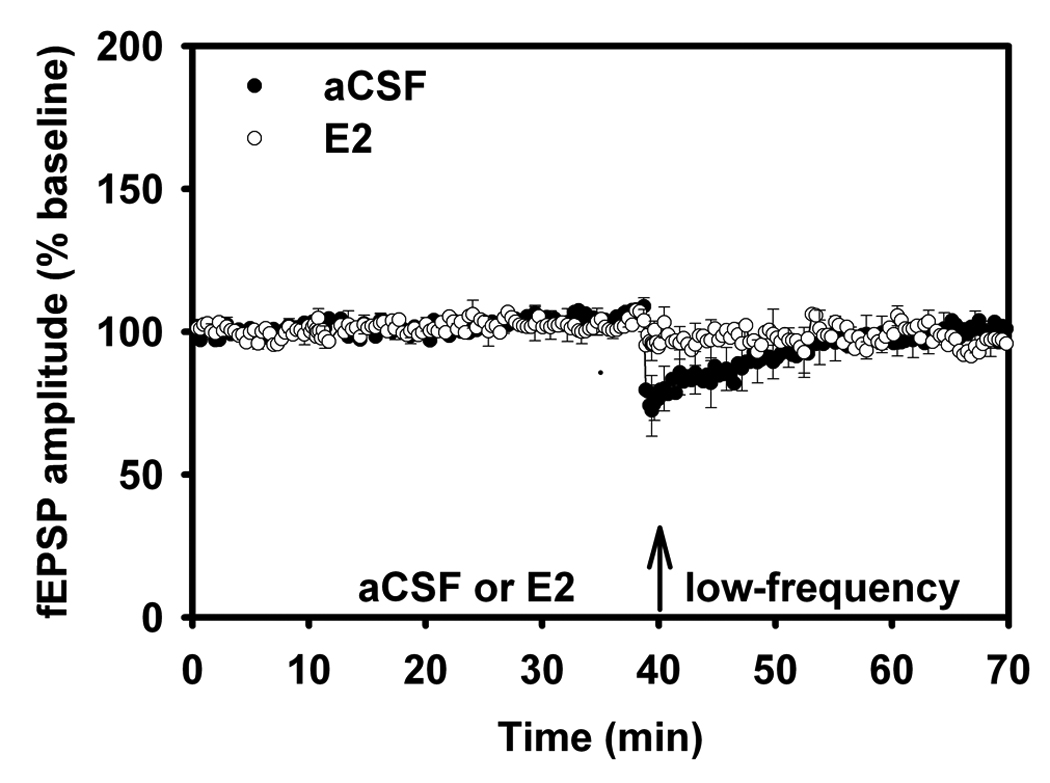
Long-term depression in Adult and Aged Male Rats.
(A) LTD. Adult (3–5 mo) aCSF versus E2. In this figure, following baseline and drug/aCSF periods, slices received low-frequency stimulation (low-frequency) to elicit long-term depression. LTD that was initially induced in the aCSF adult males quickly diminished. The arrows indicate the time at which aCSF/E2 was applied, and when low-frequency stimulation (900 pulses at 1 Hz) was delivered.
(B) LTD. Aged (18–24 mo) aCSF versus E2. LTD was examined in slices from aged rats, with fEPSP comparisons between aCSF versus E2. LFS delivered to aged rat slices perfused with 17β-estradiol failed to induce robust LTD. Reprinted with permission from (Foy et al., 2008b). Copyright 2010 American Psychological Association.
VI. Estrogen, Behavioral Stress, Aging and Hippocampal Synaptic Plasticity
A number of hormones secreted from the pituitary-adrenal system during behavioral stress are known to affect learning and memory processes (Foy, Kim, Shors, and Thompson, 2005). In rats, an in vivo uncontrollable acute stress experience (30-min body restraint + 1 sec tailshock/min) impaired the subsequent development of hippocampal LTP in the slice preparation (Foy, Stanton, Levine, and Thompson, 1987). This was the first demonstration that behavioral stress impairs a form of synaptic plasticity in the rodent hippocampus (Fig. 7). Subsequent to this report, LTP was found to be much less impaired in rats that could control stress than in rats that could not exert control, even though both groups received identical physical stress (Shors, Seib, Levine, and Thompson, 1989). The same behavioral stress (restraint + tailshock) facilitated hippocampal LTD in adult rats (Kim, Foy, and Thompson, 1996). In contrast, electrophysiological recordings from nonstressed adult controls failed to display significant LTD. Although robust LTD has been found to occur in hippocampus of very young rats (< 2 weeks old), smaller decreases in LTD magnitude have been reported in adult (5 weeks old) rats (Dudek and Bear, 1993). As mentioned earlier, in aged rats, more robust LTD can be elicited, possibly because of age-related changes in synaptic plasticity mechanisms that result in a lower threshold for LTD induction (Norris, Korol, and Foster, 1996; Rosenzweig and Barnes, 2003). More recently, it has been found that the stress impairment of LTP from adult animals exposed to behavioral stress can be reversed through acute application of E2 to hippocampal slices from which LTP is recorded (Foy et al., 2008b) (Fig. 8). It is important to note that adult animals not exposed to behavioral stress (both aCSF and E2-treated slices) show enhanced fEPSP values compared to those behaviorally stressed (compare Fig. 2 with Fig. 8). In aged rats, the stress effect on LTP and LTD has also been investigated, which is also reversed following acute administration of E2 (Foy et al., 2008b) (Fig. 9). In hippocampal slices obtained from aged rats in the behaviorally stressed condition, E2 enhances LTP magnitude and decreases LTD magnitude. In summary, E2 has now been shown to reverse the effects of both age and behavioral stress on hippocampal synaptic plasticity.
Figure 7.

Stress Impairs Long-term Potentiation in Adult Male Rats.
(A) Diagram of a transverse section through the hippocampus illustrating the lamellar organization of the slice. To activate CA1 pyramidal cells monosynaptically, a bipolar concentric stimulating electrode was positioned in the afferent pathway that contains the Schaffer collateral branches (Sch.) of the CA3 pyramidal cell axons. Field potentials were recorded by means of a glass micropipet filled with 2 M NaCl and placed in the cell body layer of the CA1 cell field. A dissecting microscope was used to facilitate positioning of the electrodes in the transilluminated hippocampal slices.
(B) Representative field potentials from slice preparations in the various experimental conditions. Extracellular field potential responses to a given stimulus intensity before and 30 min following high-frequency stimulation are shown from the Control, Restraint, and Restraint + Shock groups. High-frequency stimulation enhanced the population field potential by 205% for the Control group, but did not affect the field potenials of the Restraint or the Restraint + Shock groups 30 min post-tetanus.
(C) A post-tetanus curve shows the change of CA1 hippocampal excitability (expressed as a percentage of pretetanus control) for Control (O), Restraint (▲), and Restraint + Shock (●) groups. Tetanizing stimulation shows a short post-tetanic (PTP) effect in slices from all groups; a long-term potentiation (LTP) effect is seen only in the Control group and not in the Restraint and Restraint + Shock groups while monitoring to 30 min post-tetanus. Reprinted with permission from (Foy et al., 1987). Copyright 2010 Academic Press.
Figure 8.

E2 Reverses Stress Impairment of LTP in Adult Male Rats.
Plot of normalized field EPSP (fEPSP) recordings in area CA1. All hippocampal slices were perfused with aCSF for 10 min to obtain fEPSP amplitude percent baseline data. After 10 min of baseline recording, experimental slices were perfused with 100 pM 17β-estradiol (E2). Control slices continued to be perfused with aCSF. After 30 min of either E2 or aCSF, slices then received high-frequency stimulation to elicit long-term potentiation. Hippocampal slices were obtained from adult (3–5 mo) male rats exposed to behavioral stress immediately before tissue preparation. Reprinted with permission from (Foy et al., 2008b). Copyright 2010 American Psychological Association.
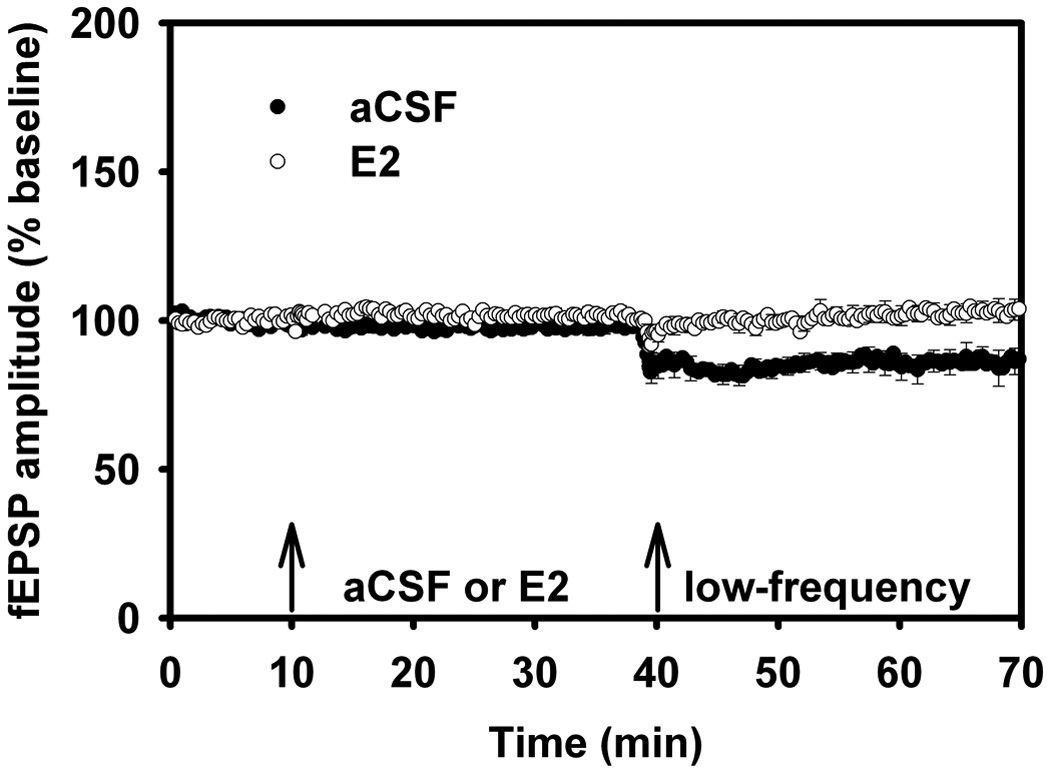
Figure 9.
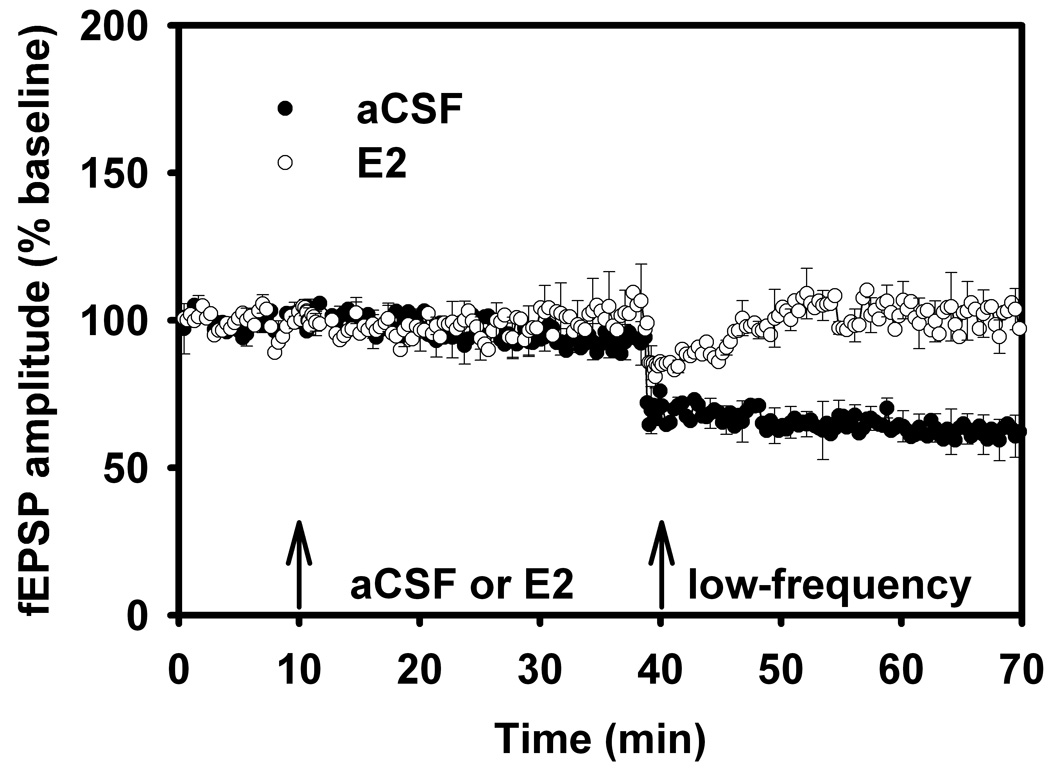
Long-term Potentiation in Aged Male Rats, aCSF vs. 17β-estradiol, Behavioral Stress.
(A) LTP was examined in slices from aged (18–24 mo) male rats exposed to behavioral stress, with fEPSP comparisons between aCSF versus E2. Behavioral stress impairs LTP in the aged hippocampal slice preparation, but is reversed with E2.
(B) LTD was examined in slices from aged (18–24 mo) male rats exposed to behavioral stress, with fEPSP comparisons between aCSF versus E2. LTD is prominent and persistent (enhanced) for up to 30 min in the aged hippocampal slice preparation, but is reversed (decreased magnitude) with E2. Reprinted with permission from (Foy et al., 2008b). Copyright 2010 American Psychological Association.
VII. Progesterone and Hippocampus
In contrast to the extensive studies examining the physiological role of estrogen in hippocampus, the effects of P4 on hippocampal synaptic transmission and plasticity have yet to be fully investigated. One study reported that P4 (10−5 M) had no effect on LTP recorded in vitro from hippocampal CA1 slices in adult rats, but no non-drug control was provided to compare the experimental (P4) condition, and the experimental subjects were a combined group of gonadally-intact male and female rats (Ito, Skinkle, and Hicks, 1999). Another study reported that P4 (10−8 M) significantly enhanced synaptic transmission in CA1, but following seizure-inducing tetanus, P4 decreased both field potential and population spike responses, and decreased the duration of after-discharges (Edwards, Epps, Carlen, and MacLusky, 2000). In a study using whole cell patch clamps of pyramidal neurons from slices of prelimbic cortex, it was reported that P4 (100 µM) had no effect on the frequency of spontaneous excitatory postynaptic currents (EPSCs), but inhibited dopamine-induced increases in the frequency of EPSCs (Feng, Dong, Fu, Zhu, Sun, Wang, Sun, and Zheng, 2004). P4 dose-response functions were not performed in any of these studies, and no previous work had investigated the effect of P4 on hippocampal LTD. In summary, the existing data support the hypothesis that progestagens and hormone therapy modify certain properties of hippocampal physiology and function.
VIII. Progesterone and Progesterone Receptors
The classical nuclear progesterone receptor (cPR) was characterized in the 1970s and has been localized to many regions of the CNS, including hippocampus, cortex, hypothalamus and cerebellum (Camacho-Arroyo, Guerra-Araiza, and Cerbon, 1998; Guerra-Araiza, Villamar-Cruz, Gonzalez-Arenas, Chavira, and Camacho-Arroyo, 2003; Hagihara, Hirata, Osada, Hirai, and Kato, 1992). P4 exerts its effects by binding and activating specific cellular receptors through its cognate receptors (PR), classically defined as ligand-activated transcription factors (Brinton, Thompson, Foy, Baudry, Wang, Finch, Morgan, Pike, Mack, Stanczyk, and Nilsen, 2008). Two major isoforms of cPR are known to exist, the full-length B isoform (PRB) and the N-terminal-truncated A isoform (PRA) (Conneely, Maxwell, Toft, Schrader, and O'Malley, 1987). While many of P4’s effects can be explained by the classical nuclear DNA-binding model of steroid action, P4 can also exert rapid effects on diverse signaling pathways that are independent of transcriptional or genomic regulation (Losel and Wehling, 2003). The discovery of plasma membrane progestagen receptors occurred in 2003, and included the progesterone receptor membrane component 1 (PGRMCI), and a family of membrane progesterone receptors (mPR), e.g., mPRα, β, and γ (Gellersen, Fernandes, and Brosens, 2009; Guennoun, Meffre, Labombarda, Gonzalez, Deniselle, Stein, De Nicola, and Schumacher, 2008). It is through activation of these membrane receptors (PGRMCI and mPR) that P4 is suspected of rapidly regulating basic cellular metabolism and homeostasis.
IX. Progesterone and Hippocampal LTP and LTD
A recent study examined the effects of several concentrations of P4 (10−9, 10−8, 10−7 and 10−6 M) on hippocampal basal synaptic transmission, LTP and LTD (Foy et al., 2008a). P4 resulted in a rapid (~30 min) decrease in baseline synaptic transmission only at the 10−6 M concentration (Fig. 10A). In addition, P4 concentrations of 10−7 and 10−6 M markedly decreased LTP in CA1 neurons from adult, ovariectomized rats (Fig. 10B). No changes in synaptic responses were found following low-frequency stimulation to induce hippocampal LTD at any of the P4 concentrations tested (Fig. 11A). Thus, in contrast to P4’s effect on LTP, P4 did not affect LTD, suggesting that the high-frequency stimulation used to induce hippocampal LTP might increase the inhibitory action of P4 on excitatory synaptic transmission in hippocampus.
Figure 10.
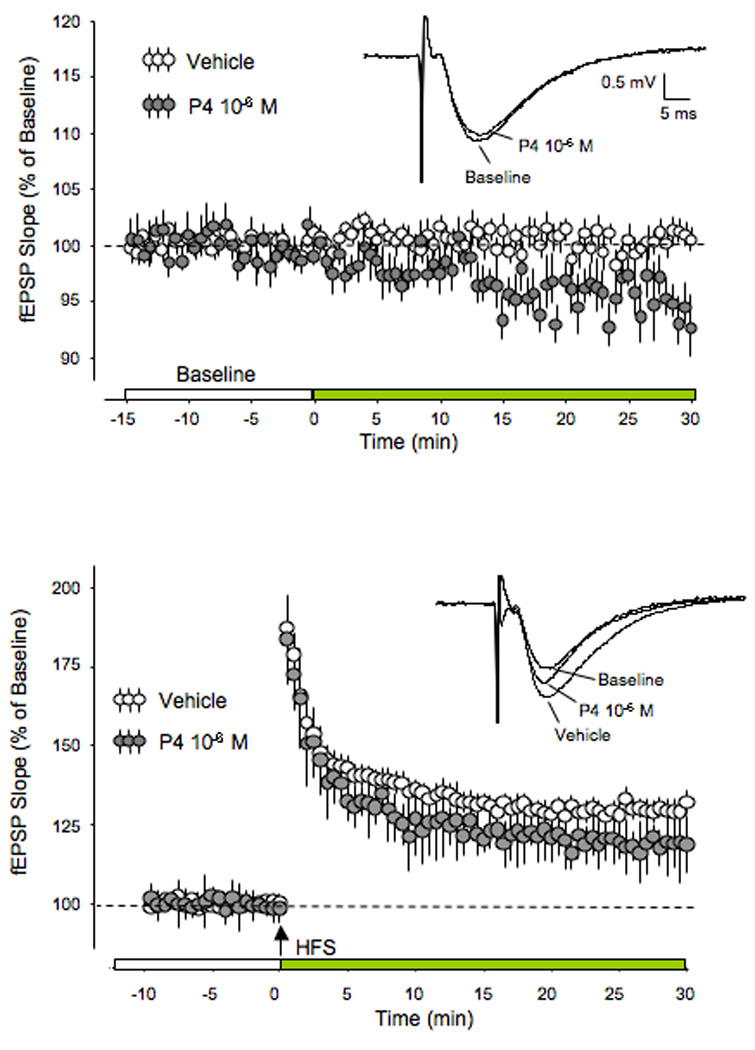
fEPSP Baseline Synaptic Transmission and LTP with Progesterone
(A) fEPSP baseline slope values (vehicle) were measured for 15 min prior to the infusion of P4 (10−6 M). P4 began to elicit a significant decrease in fEPSP during the next 30 min.
(B) fEPSP slope values (vehicle and P4) were measured prior to and following high-frequency stimulation (HFS) designed to induce long-term potentiation (LTP). Following HFS, slices perfused with P4 showed a significant decrease in fEPSP slope values compared to vehicle, resulting in decreased LTP. Reprinted with permission from (Foy et al., 2008a). Copyright 2010 Cold Spring Harbor Laboratory Press.
Figure 11.
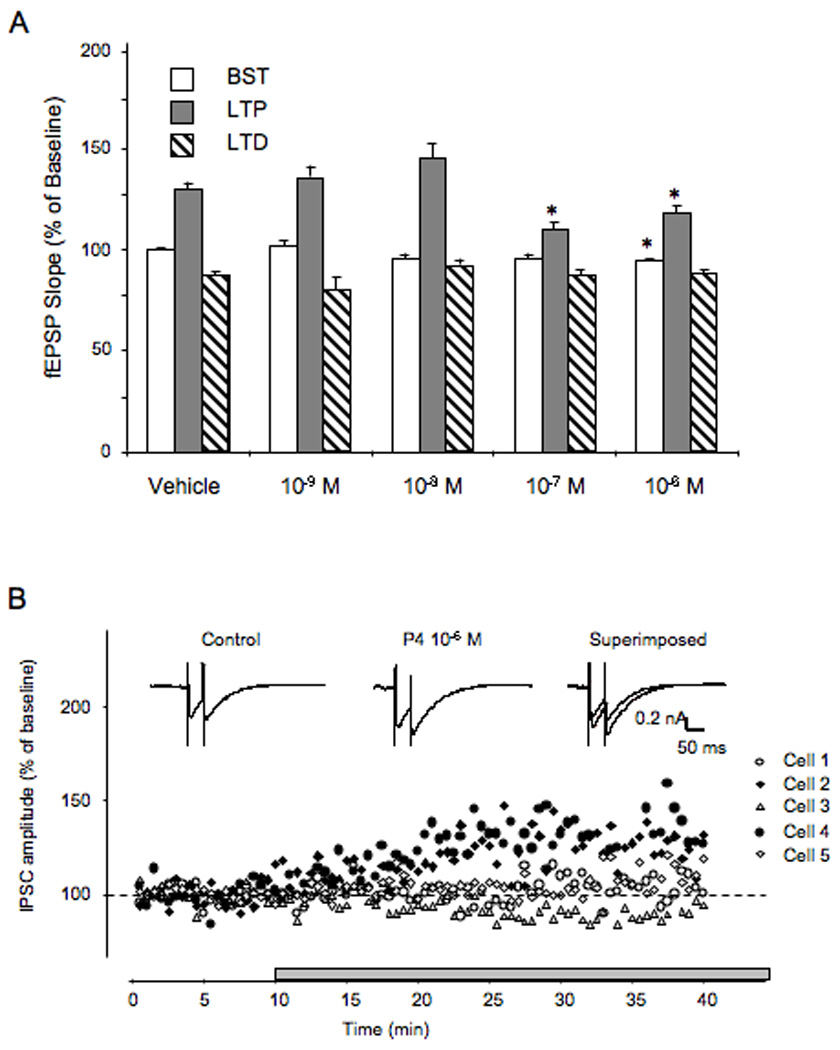
Progesterone Concentration, LTP, LTD and IPSC recordings
(A) Vehicle and P4 at different concentrations (10−9, 10−8, 10−7, and 10−6 M) was compared with fEPSP values during the baseline synaptic transmission (BST), LTP and LTD periods. At the 2 highest concentrations tested, P4 attenuated BST and LTP, but had no effect on LTD.
(B) Using intracellular recordings of inhibitiory post-synaptic currents (IPSCs) from individual CA1 pyramidal neurons, P4 at 10−6 M increased GABAA receptor-mediated current in two of five recorded cells. The currents were recorded with sharp electrodes in discontinuous single electrode volgage clamp mode. Representative current traces recorded before and during P4 application at 10−6 M. Reprinted with permission from (Foy et al., 2008a). Copyright 2010 Cold Spring Harbor Laboratory Press.
The attenuation of baseline synaptic transmission and decreased magnitude of LTP in hippocampal slices resulting from the application of 10−6 M P4 could be due to GABAA receptor activation (Mitchell, Herd, Gunn, Lambert, and Belelli, 2008), either by a direct action of P4, or the action of one of its metabolites. Intracellular recordings were made from individual pyramidal neurons in the presence of NMDA and AMPA receptor antagonists, APV (50 µM) and CNQX (10 µM), respectively (Foy, Akopian, and Thompson, 2008a). The isolated currents were proven to be GABAA receptor-mediated since they were completely blocked by the GABAA receptor antagonist picrotoxin (100 µM). Under these conditions, in two of five cells that were recorded, P4 at 10−6 M increased GABAA receptor-mediated current (Fig. 11B).
X. Conclusions
This review summarized several fundamental characteristics of the effects of the steroid hormones estrogen and progesterone on synaptic plasticity in the mammalian nervous system. Estrogen rapidly enhances both NMDA and AMPA receptor/channel responses elicited by glutamate released from excitatory presynaptic terminals via presumed plasma membrane mechanisms triggering several intracellular signaling cascades. Studies have yet to confirm whether the E2 effects are due to an action directly on the receptors, or indirectly via second messenger processes that in turn influence NMDA and AMPA receptor/channel function.
Estrogen markedly enhances hippocampal LTP. The LTP enhancement after acute E2 application is apparently due to an increase in NMDA and AMPA receptor activation, consistent with biochemical studies that establish the importance of complex signaling intracellular pathways associated with E2 and hippocampal synaptic plasticity. Recent studies have shown that the E2 activation of biochemical cascades leads to cytoskeleton regulation resulting in increased membrane levels of AMPA receptors.
Changes in estrogen levels in cycling female rats have also been correlated with changes in synaptic plasticity, as measured by LTP magnitude. That E2 reverses the effects of age and behavioral stress on hippocampal synaptic plasticity is another interesting finding that has yet to be fully undersood.
Similar to estrogen, progesterone also acts rapidly in hippocampus, but instead of increasing basal synaptic transmission and LTP, P4 decreases hippocampal basal synaptic transmission and LTP. Intracellular recordings so far suggests that this effect is mediated, at least in part, by activation of inhibitory GABAA receptor-mediated activity. This interpretation is consistent with results from other studies linking GABAA with P4 or its metabolites (Herd, Belelli, and Lambert, 2007; Herd, Haythornthwaite, Rosahl, Wafford, Homanics, Lambert, and Belelli, 2008).
The precise function of the limbic system as a steroid hormone-sensitive target in the CNS is not entirely clear, even though limbic structures in general have long been implicated in the feedback control of the hypothalamo-pituitary-gonadal axis. The hippocampus, purported to be involved in a myriad of memory, mood-modulating and attentional functions, is regulated by steroid hormones via mechanisms involving structural and functional plasticity. Perhaps it is through these mechanisms steroid hormones act in concert with hippocampus to regulate behavior.
This chapter has examined studies that suggest the existence of mechanisms by which naturally fluctuating endogenous steroid hormone levels can impact cellular process associated with learning and memory function in mammalian CNS. To the extent that LTP is a mechanism involved in processes of coding and storing information, i.e., in memory formation, E2 appears to enhance these processes, while P4 appears to decrease them. Indeed, the E2-induced enhancement of LTP suggests a possible mechanism by which E2 can exert facilitatory effects on memory processes. Clinical evidence indicates that estrogenic steroids can enhance cognitive function in humans, particularly in postmenopausal women (Henderson, 1997; 2000; Kawas, Resnick, Morrison, Brookmeyer, Corrada, Zonderman, Bacal, Lingle, and Metter, 1997); however, prospective observational studies have yet to find a protective effect of estrogen on either age-related decline in cognition, or the incidence of dementia (Barrett-Conner and Kritz-Silverstein, 1993; Matthews, Cauley, Yaffe, and Zmuda, 1999). A detailed understanding of the mechanisms of action of estrogen and progesterone on hippocampal plasticity, and the roles these steroid hormones play in non-reproductive functions, may have far-reaching implications for hormone therapy to maintain neurological health and function throughout human lifespan.
ACKNOWLEDGEMENTS
This work received institutional support from Loyola Marymount University, and grants from the National Institute of Aging: P01 AG026572 (PI: Roberta Diaz Brinton) and R01 AG023742 (PI: Diana Woodruff-Pak).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta JI, Mayer L, Talboom JS, Zay C, Scheldrup M, Castillo J, Demers LM, Enders CK, Bimonte-Nelson HA. Premarin improves memory, prevents scopolamine-induced amnesia and increases number of basal forebrain choline acetyltransferase positive cells in middle-aged surgically menopausal rats. Horm Behav. 2009;55:454–464. doi: 10.1016/j.yhbeh.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom T. Epileptic seizures in women related to plasma estrogen and progesterone during the menstrual cycle. Acta Neurol Scand. 1976;54:321–347. doi: 10.1111/j.1600-0404.1976.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. Journal of Comparative Physiological Psychology. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends in Neuroscience. 1994;17:13–18. doi: 10.1016/0166-2236(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Foster TC, McNaugton BL. Region-specific age effects on AMPA sensitivity: electrophysiological evidence for loss of synaptic contacts in hippocampal field CA1. Hippocampus. 1992;2:457–468. doi: 10.1002/hipo.450020413. [DOI] [PubMed] [Google Scholar]
- Barrett-Conner E, Kritz-Silverstein D. Estrogen replacement therapy and cognitive function in older women. Journal of the American Medical Association. 1993;269:2637–2641. [PubMed] [Google Scholar]
- Baudry M, Davis JL, Thompson RF. Advances in Synaptic Plasticity. Cambridge, MA: The MIT Press; 2000. [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Berlucchi G, Buchtel HA. Neuronal plasticity: historical roots and evolution of meaning. Exp Brain Res. 2009;192:307–319. doi: 10.1007/s00221-008-1611-6. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci U S A. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi R, Foy MR, Vouimba RM, Thompson RF, Baudry M. Cyclic changes in estradiol regulate synaptic plasticity through the MAP kinase pathway. Proc Natl Acad Sci U S A. 2001;98:13391–13395. doi: 10.1073/pnas.241507698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Proffitt P, Tran J, Luu R. Equilin, a principal component of the estrogen replacement therapy, premarin, increases the growth of cortical neurons via an NMDA receptor dependent mechanism. Experimental Neurology. 1997;147:211–220. doi: 10.1006/exnr.1997.6619. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Thompson RF, Foy MR, Baudry M, Wang J, Finch CE, Morgan TE, Pike CJ, Mack WJ, Stanczyk FZ, Nilsen J. Progesterone receptors: form and function in brain. Front Neuroendocrinol. 2008;29:313–339. doi: 10.1016/j.yfrne.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, Tran J, Proffitt P, Kihil M. 17B-estradiol increases the growth and survival of cultured cortical neurons. Neurochemical Research. 1997;22:1339–1351. doi: 10.1023/a:1022015005508. [DOI] [PubMed] [Google Scholar]
- Camacho-Arroyo I, Guerra-Araiza C, Cerbon MA. Progesterone receptor isoforms are differentially regulated by sex steroids in the rat forebrain. Neuroreport. 1998;9:3993–3996. doi: 10.1097/00001756-199812210-00001. [DOI] [PubMed] [Google Scholar]
- Conneely OM, Maxwell BL, Toft DO, Schrader WT, O'Malley BW. The A and B forms of the chicken progesterone receptor arise by alternate initiation of translation of a unique mRNA. Biochem Biophys Res Commun. 1987;149:493–501. doi: 10.1016/0006-291x(87)90395-0. [DOI] [PubMed] [Google Scholar]
- Cordoba Montoya DA, Carrer HF. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats. Brain Research. 1997;778:430–438. doi: 10.1016/s0006-8993(97)01206-7. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards HE, Epps T, Carlen PL, MacLusky JN. Progestin receptors mediate progesterone suppression of epileptiform activity in tetanized hippocampal slices in vitro. Neuroscience. 2000;101:895–906. doi: 10.1016/s0306-4522(00)00439-5. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- Feng XQ, Dong Y, Fu YM, Zhu YH, Sun JL, Wang Z, Sun FY, Zheng P. Progesterone inhibition of dopamine-induced increase in frequency of spontaneous excitatory postsynaptic currents in rat prelimbic cortical neurons. Neuropharmacology. 2004;46:211–222. doi: 10.1016/j.neuropharm.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Foster TC. Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Research Reviews. 1999;30:236–249. doi: 10.1016/s0165-0173(99)00017-x. [DOI] [PubMed] [Google Scholar]
- Foster TC, Norris CM. Age-associated changes in Ca2+dependent processes: relation to hippocampal synaptic plasticity. Hippocampus. 1997;7:602–612. doi: 10.1002/(SICI)1098-1063(1997)7:6<602::AID-HIPO3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Foy M, Baudry M, Thompson R. Estrogen and hippocampal synaptic plasticity. Neuron Glia Biol. 2004;1:327–338. doi: 10.1017/S1740925X05000165. [DOI] [PubMed] [Google Scholar]
- Foy MR. Neurobiology Program. Kent: Kent State University; 1983. Neuromodulation: Effects of estradiol and THC on brain excitability. [Google Scholar]
- Foy MR. 17beta-estradiol: effect on CA1 hippocampal synaptic plasticity. Neurobiol Learn Mem. 2001;76:239–252. doi: 10.1006/nlme.2001.4018. [DOI] [PubMed] [Google Scholar]
- Foy MR, Akopian G, Thompson RF. Progesterone regulation of synaptic transmission and plasticity in rodent hippocampus. Learn Mem. 2008a;15:820–822. doi: 10.1101/lm.1124708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Baudry M, Foy JG, Thompson RF. 17beta-estradiol modifies stress-induced and age-related changes in hippocampal synaptic plasticity. Behav Neurosci. 2008b;122:301–309. doi: 10.1037/0735-7044.122.2.301. [DOI] [PubMed] [Google Scholar]
- Foy MR, Kim JJ, Shors TJ, Thompson RF. Neurobiological foundations of stress. In: S. M. Yehuda DI, editor. Nutrition, Stress and Medical Disorders. New Jersey: Humana Press; 2005. pp. 37–65. [Google Scholar]
- Foy MR, Stanton ME, Levine S, Thompson RF. Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav Neural Biol. 1987;48:138–149. doi: 10.1016/s0163-1047(87)90664-9. [DOI] [PubMed] [Google Scholar]
- Foy MR, Teyler TJ. 17-alpha-Estradiol and 17-beta-estradiol in hippocampus. Brain Res Bull. 1983;10:735–739. doi: 10.1016/0361-9230(83)90206-x. [DOI] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. Journal of Neuroscience. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y, Detoledo-Morell F, Heller RE. Hippocampal markers of age-related memory dysfunction: behaivoral, electrophysiological and morphological perspectives. Progress in Neurobiology. 1995;45:223–252. doi: 10.1016/0301-0082(94)00047-l. [DOI] [PubMed] [Google Scholar]
- Gellersen B, Fernandes MS, Brosens JJ. Non-genomic progesterone actions in female reproduction. Hum Reprod Update. 2009;15:119–138. doi: 10.1093/humupd/dmn044. [DOI] [PubMed] [Google Scholar]
- Gu Q, Moss RL. 17B-estradiol potentiates kainite-induced currents via activation of the camp cascade. Journal of Neuroscience. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guennoun R, Meffre D, Labombarda F, Gonzalez SL, Deniselle MC, Stein DG, De Nicola AF, Schumacher M. The membrane-associated progesterone-binding protein 25-Dx: expression, cellular localization and up-regulation after brain and spinal cord injuries. Brain Res Rev. 2008;57:493–505. doi: 10.1016/j.brainresrev.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Guerra-Araiza C, Villamar-Cruz O, Gonzalez-Arenas A, Chavira R, Camacho-Arroyo I. Changes in progesterone receptor isoforms content in the rat brain during the oestrous cycle and after oestradiol and progesterone treatments. J Neuroendocrinol. 2003;15:984–990. doi: 10.1046/j.1365-2826.2003.01088.x. [DOI] [PubMed] [Google Scholar]
- Guerriero G. Vertebrate sex steroid receptors: evolution, ligands, and neurodistribution. Ann N Y Acad Sci. 2009;1163:154–168. doi: 10.1111/j.1749-6632.2009.04460.x. [DOI] [PubMed] [Google Scholar]
- Hagihara K, Hirata S, Osada T, Hirai M, Kato J. Distribution of cells containing progesterone receptor mRNA in the female rat di- and telencephalon: an in situ hybridization study. Brain Res Mol Brain Res. 1992;14:239–249. doi: 10.1016/0169-328x(92)90179-f. [DOI] [PubMed] [Google Scholar]
- Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- Henderson VW. The epidemiology of estrogen replacement therapy and Alzheimer's disease. Neurology. 1997;48:S27–S35. doi: 10.1212/wnl.48.5_suppl_7.27s. [DOI] [PubMed] [Google Scholar]
- Henderson VW. Hormone Therapy and the Brain: A Clinical Perspective on the Role of Estrogen. New York: Parthenon Publishing; 2000. [DOI] [PubMed] [Google Scholar]
- Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABA(A) receptors. Pharmacol Ther. 2007;116:20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Herd MB, Haythornthwaite AR, Rosahl TW, Wafford KA, Homanics GE, Lambert JJ, Belelli D. The expression of GABAA beta subunit isoforms in synaptic and extrasynaptic receptor populations of mouse dentate gyrus granule cells. J Physiol. 2008;586:989–1004. doi: 10.1113/jphysiol.2007.146746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Skinkle KL, Hicks TP. Age-dependent, steroid-specific effects of oestrogen on long-term potentiation in rat hippocampal slices. J Physiol. 1999;515(Pt 1):209–220. doi: 10.1111/j.1469-7793.1999.209ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF. Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci U S A. 1996;93:4750–4753. doi: 10.1073/pnas.93.10.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, Lynch G. Cytoskeletal changes underlie estrogen's acute effects on synaptic transmission and plasticity. J Neurosci. 2009;29:12982–12993. doi: 10.1523/JNEUROSCI.3059-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Lynch G. Impaired monosynaptic potentiation in in vitro hippocampal slices from age, memory-deficient rats. Journal of Gerontology. 1977;32:523–533. doi: 10.1093/geronj/32.5.523. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA, Applegate MD. The effects of high Mg2+-Ca2+ ratios on frequency potentiation in hippocampal slices of young and aged rats. Journal of Neurophysiology. 1986;56:797–811. doi: 10.1152/jn.1986.56.3.797. [DOI] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Mendolia-Loffredo S, Shors TJ. High levels of estrogen enhance associative memory formation in ovariectomized females. Psychoneuroendocrinology. 2004;29:883–890. doi: 10.1016/j.psyneuen.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. Long-term potentiation: outstanding questions and attempted synthesis. Philos Trans R Soc Lond B Biol Sci. 2003;358:829–842. doi: 10.1098/rstb.2002.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4:46–56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Matthews K, Cauley J, Yaffe K, Zmuda JM. Estrogen replacement therapy and cognitive decline in older community women. Journal of the American Geriatric Society. 1999;47:518–523. doi: 10.1111/j.1532-5415.1999.tb02563.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocr Rev. 1999;20:279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Gerlach J, Micco D. Putative glucocorticoid receptors in hippocampus and other regions of the rat brain. In: Isaacson R, Pribram K, editors. The Hippocampus, Vol 2: Neurophysiology and behavior. New York: Plenum Press; 1975. [Google Scholar]
- Mitchell EA, Herd MB, Gunn BG, Lambert JJ, Belelli D. Neurosteroid modulation of GABAA receptors: molecular determinants and significance in health and disease. Neurochem Int. 2008;52:588–595. doi: 10.1016/j.neuint.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockage of L-type Ca2+ channels. Journal of Neuroscience. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster TC. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster` Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. Journal of Neuroscience. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. The role of neurosteroids in the pathophysiology and treatment of catamenial epilepsy. Epilepsy Res. 2009;85:1–30. doi: 10.1016/j.eplepsyres.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol. 2003;69:143–179. doi: 10.1016/s0301-0082(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Matzel LD. Long-term potentiation: what's learning got to do with it? Behav Brain Sci. 1997;20:597–614. doi: 10.1017/s0140525x97001593. discussion 614–555. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF. Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science. 1989;244:224–226. doi: 10.1126/science.2704997. [DOI] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS, Waterhouse BD, Woodward DJ. Sex steroid effects on extrahypothalamic CNS. I. Estrogen augments neuronal responsiveness to iontophoretically applied glutamate in the cerebellum. Brain Res. 1987;422:40–51. doi: 10.1016/0006-8993(87)90538-5. [DOI] [PubMed] [Google Scholar]
- Smith SS, Waterhouse BD, Woodward DJ. Locally applied estrogens potentiate glutamate-evoked excitation of cerebellar Purkinje cells. Brain Res. 1988;475:272–282. doi: 10.1016/0006-8993(88)90615-4. [DOI] [PubMed] [Google Scholar]
- Stanton PK. LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus. 1996;6:35–42. doi: 10.1002/(SICI)1098-1063(1996)6:1<35::AID-HIPO7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Terasawa E, Timiras PS. Electrical activity during the estrous cycle of the rat: cyclic changes in limbic structures. Endocrinology. 1968;83:207–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- Teyler TJ. Brain slice preparation: hippocampus. Brain Res Bull. 1980;5:391–403. doi: 10.1016/s0361-9230(80)80009-8. [DOI] [PubMed] [Google Scholar]
- Teyler TJ, Vardaris RM, Lewis D, Rawitch AB. Gonadal steroids: effects on excitability of hippocampal pyramidal cells. Science. 1980;209:1017–1018. doi: 10.1126/science.7190730. [DOI] [PubMed] [Google Scholar]
- Vardaris RM, Teyler TJ. Sex differences in the response of hippocampal CA1 pyramids to gonadal steroids: Effects of testosterone and estradiol on the in vitro slice preparation. Soc Neurosci Abstr. 1980:A153–A159. [Google Scholar]
- Vouimba RM, Foy MR, Thompson RF. 17B-estradiol suppresses expression of long-term depression in aged rats. Brain Research Bulletin. 2000;53:783–787. doi: 10.1016/s0361-9230(00)00377-4. [DOI] [PubMed] [Google Scholar]
- Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–30. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL. Electrophysiological evidence for a rapid membrane action of the gonadal steroid, 17 beta-estradiol, on CA1 pyramidal neurons of the rat hippocampus. Brain Res. 1991;543:148–152. doi: 10.1016/0006-8993(91)91057-8. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Moss RL. Patch-clamp analysis of direct steroidal modulation of glutamate receptor-channels. Journal of Neuroendocrinology. 1994;6:347–355. doi: 10.1111/j.1365-2826.1994.tb00592.x. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. Journal of Neuroscience. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. Journal of Neuroscience. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Berger TW, Barrionuevo G. Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. Journal of Neurophysiology. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, Thompson R, Baudry M. 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A. 2009;106:21936–21941. doi: 10.1073/pnas.0912558106. [DOI] [PMC free article] [PubMed] [Google Scholar]