

Abstract
The stereospecific cross-coupling of enantioenriched non-benzylic secondary alkyl boron compounds has been achieved. The high selectivity toward product formation over an undesired β-H elimination pathway is achieved via an intramolecular coordination of an ancillary carbonyl to the metal center in the diorganopalladium intermediate.
The Suzuki-Miyaura cross-coupling reaction has emerged as one of the most versatile transformations available for the generation of C-C bonds.2 Although there are many strategies that exist for the cross-coupling of sp2-hybridized organometallics,3 the protocols for the cross-coupling of secondary and potentially enantiomerically enriched sp3-hybridized organometallics has limited precedent.4 In a previous communication, we described a development toward this goal by identifying catalytic reaction conditions for the cross-coupling of cyclic, symmetrical secondary alkyltrifluoroborates with aryl electrophiles.5 However, when applied to symmetrical acyclic substrates, it became evident that the use of our optimized conditions still led to a β-H elimination/isomerization pathway that resulted in mixtures of the desired cross-coupled products as well as the undesired isomerized primary alkylated products.
More recently, other attempts at secondary cross-coupling using various organoboron derivatives have appeared.6 Crudden and coworkers developed a protocol for the preparation of enantioenriched styrene-derived secondary boronate esters and demonstrated their cross-coupling with a variety of aryl iodides.6 Although the reactions proceed in good yields with retention of configuration,7 this method was limited to the cross-coupling of benzylic boron derivatives.
During the course of this investigation, Suginome reported the cross-coupling of α-(acylamino)benzylboronates with aryl bromides and chlorides, which somewhat surprisingly occurred with overall inversion of configuration.8
Herein we report our most recent efforts toward the ultimate goal of cross-coupling non-benzylic, enantioenriched secondary alkyl organoboron reagents with stereochemical fidelity during the cross-coupling event. Subsequent to our studies on secondary alkyltrifluoroborates, efforts were conducted to extend the study of β-trifluoroborato carbonyls10 to the preparation and cross-coupling of acyclic secondary β-trifluoroboratoamides. Using the borylation strategy outlined by Yun and coworkers, a variety of these trifluoroborates were prepared.10a,11 With the desired substrates in hand, an initial screen of catalytic conditions led to the combination of 10 mol of % Pd(OAc)2 and 20 mol % of XPhos, K2CO3 in a cyclopentyl methyl ether (CPME)/H2O solvent system giving the highest isolated yield of potassium N-cyclohexyl-3-(trifluoroborato)butanamide in the coupling reaction with 2-chloroanisole (Table 1, entry 1).
Table 1.
Cross-Coupling of β-Trifluoroboratoamides with Aryl Halidesa
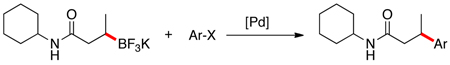
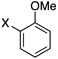
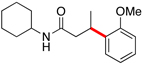
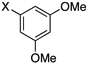
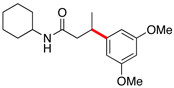
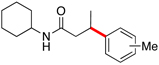
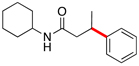
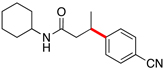
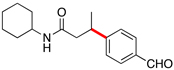
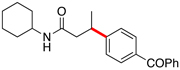
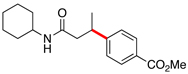
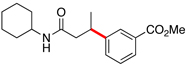
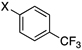
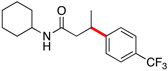
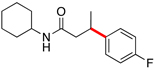
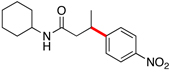
Encouraged by this initial result, further screening revealed that the combination of 10 mol % of XPhos or SPhos with Cs2CO3 (3 equiv) also provided good to excellent yields of the cross-coupled products with both aryl chlorides and - bromides.
Using 10 mol % of Pd(OAc)2 and 20 mol % of XPhos, a variety of electrophilic partners (including those containing ketone-, aldehyde-, ester-, nitrile- and nitro groups) cross-coupled with the model trifluoroborate in good yields. In a number of cases, the use of SPhos as the ligand actually provided higher yields of the cross-coupled product (Table 1, entries 6, 7, 11–13). In all of these examples, <2% of products resulting from β-H elimination or isomerization were isolated.
To investigate the scope of this reaction with respect to the nucleophilic partner, all three sets of suitable catalytic conditions were applied to a variety of amide substrates, in each case generating the cross-coupled products in good yields (Table 2, entries 1–7), again observing little or none of the undesired byproducts.
Table 2.
Cross-Coupling of Various Trifluoroborates with Aryl Halidesa
 | ||||
|---|---|---|---|---|
| entry | RBF3K | ligand/base | % isolated yield | |
| 1 | 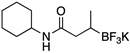 |
XPhos, K2CO3 | 84 | |
| XPhos, Cs2CO3 | 89 | |||
| SPhos, Cs2CO3 | 78 | |||
| 2 | 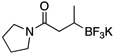 |
XPhos, K2CO3 | 92 | |
| XPhos, Cs2CO3 | 84 | |||
| SPhos, Cs2CO3 | 93 | |||
| 3 | 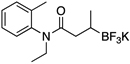 |
XPhos, K2CO3 | 87 | |
| XPhos, Cs2CO3 | 99 | |||
| SPhos, Cs2CO3 | 98 | |||
| 4 | XPhos, K2CO3 | 72 | ||
| XPhos, Cs2CO3 | 78 | |||
| SPhos, Cs2CO3 | 64 | |||
| 5 | 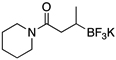 |
XPhos, K2CO3 | 89 | |
| XPhos, Cs2CO3 | 96 | |||
| SPhos, Cs2CO3 | 89 | |||
| 6 | XPhos, K2CO3 | 79 | ||
| XPhos, Cs2CO3 | 91 | |||
| SPhos, Cs2CO3 | 86 | |||
| 7 | 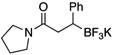 |
XPhos, K2CO3 | 94 | |
| XPhos, Cs2CO3 | 79 | |||
| SPhos, Cs2CO3 | 89 | |||
General Conditions: Pd(OAc)2 (10 mol %), ligand (20 mol %), RBF3K (1 equiv), base (3 equiv) and 6.7:1 CPME/H2O (0.25 M).
With the ultimate goal of developing conditions to generate optically active materials through the use of an appropriate organoboron reagent, we prepared an enantioenriched β-trifluoroboratoamide via an asymmetric β-borylation reaction of the corresponding α,β-unsaturated amide using bis(pinacolato)diboron and (R)-(S)-Josiphos as the chiral ligand (Scheme 1).12
Scheme 1.

Preparation and Cross-Coupling of Enantioenriched β-Trifluoroboratoamide
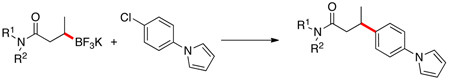
With the enantioenriched secondary organotrifluoroborate in hand, we subjected it to the optimized reaction conditions for the cross-coupling of this family of substrates. Using 10 mol % of Pd(OAc)2, 20 mol % of XPhos, and 3 equiv of K2CO3 in a CPME/H2O solvent system, the cross-coupled product was obtained in an enantiomeric ratio of 95:5 (S:R) in 82% yield for the cross-coupling step. The absolute configurations of the major enantiomers of the borylated starting material and cross-coupled products were determined to be R and S, respectively, by comparison with the authentic S isomers prepared from derivatization of commercially available (S)-3-hydroxybutyric acid and (S)-3-phenylbutyric acid. This complete inversion in stereochemistry during transmetalation for secondary alkyl boron compounds (in substrates that have the potential for β-hydride elimination) represents an important extension to the previously described methods for the cross-coupling of secondary organometallics.
Subsequent cross-couplings with the enantioenriched cyclohexyl amide derivative with aryl chlorides also revealed the same inversion of configuration with no discernable stereochemical erosion detected (eq 1). Interestingly, neither the analogous β–trifluoroboratoketones nor -esters afford the desired coupled products.
 |
(1) |
Although other factors could conceivably be involved, the unique reactivity of β–trifluoroboratoamides supports an hypothesis in which the ancillary carbonyl oxygen plays a role in coordinating with the intermediate diorganopalladium complex. Three beneficial features would derive from this interaction: 1) The coordination could facilitate the transmetalation process, as the conditions optimized for this transformation were not optimal for the cross-coupling of unfunctionalized secondary alkyltrifluoroborates described in our previous communication.5 2) The complexation may also restrict the conformation of the diorganopalladium intermediate, inhibiting a syn-coplanar arrangement of the palladium and the acidic hydrogens alpha to the carbonyl required for β-hydride elimination.13 3) More importantly, the carbonyl interaction with the coordinatively unsaturated palladium could inhibit the metal from interacting agostically with the β-hydrogens, a feature required for β-H elimination (Scheme 2). These characteristics result in the formulation of a new paradigm for successful secondary alkyl cross-coupling with potentially wide implications.
Scheme 2.

Proposed Mechanism for Complete Stereochemical Inversion
As in the Suginome study, the inversion of configuration observed during the cross-coupling reaction with the β-trifluoroboratoamides is attributed to intramolecular coordination of the carbonyl group to the boron. Chiral benzylstannanes,14 silanes15 and α-(acylamino)benzylboronic esters8 have been shown to undergo transmetalation with inversion of configuration, presumably through an SE2 mechanism via an open transition state, a process that is favored in polar solvents. More closely related to the current studies, examples of SE2-type reactions that proceed with inversion of configuration in borate substrates have been reported previously as well.16
In conclusion, the concept of using pendant ligands to serve as hemilabile ligands17 to enhance transmetalation and inhibit the β-hydride elimination pathway in the cross-coupling of secondary organometallic species is highlighted. Additionally, the first cross-coupling of a non-benzylic, enantioenriched secondary alkyl organometallic containing β-hydrogens that proceeds with complete inversion of configuration without any loss of enantioselectivity during the cross-coupling event has been reported.
Supplementary Material
Acknowledgment
G.A.M. thanks the NIH General Medical Sciences (GM035249) and the NSF GOALI program (CHE-0848460) for their generous support of this research. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data. BASF is acknowledged for their generous donation bis(pinacolato)diboron and Solvias for the donation of (R)-(S)-JosiPhos. Petr Valenta, Jason Melvin, and Genette McGrew (University of Pennsylvania) are acknowledged for their assistance in HPLC and SFC analysis.
Footnotes
Supporting Information Available: Experimental details and spectral data of all compounds synthesized. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.These authors contributed equally to this work.
- 2.For reviews on potassium organotrifluoroborates, see: Darses S, Genêt J-P. Eur. J. Org. Chem. 2003:4313. Molander GA, Figueroa R. Aldrichim. Acta. 2005;38:49. Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275. doi: 10.1021/ar050199q. Stefani HA, Cella R, Adriano S. Tetrahedron. 2007;63:3623. Darses S, Genêt J-P. Chem. Rev. 2008;108:288. doi: 10.1021/cr0509758. Molander GA, Canturk B. Angew. Chem. Int. Ed. 2009;48:9240. doi: 10.1002/anie.200904306.
- 3.Kumada coupling: Tamao K, Kiso Y, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:9268. Nakamura N, Matsuo K, Ito S, Nakamura E. J. Am. Chem. Soc. 2004;126:3686. doi: 10.1021/ja049744t. Nagano T, Hayashi T. Org. Lett. 2004;6:1297. doi: 10.1021/ol049779y. Bedford RB, Bruce DW, Frost RM, Goodby JW, Hird M. Chem. Commun. 2004:2822. doi: 10.1039/b413790f. Bedford RB, Bruce DW, Frost RM, Hird M. Chem. Commun. 2005:4161. doi: 10.1039/b507133j. Bedford RB, Betham M, Bruce DW, Danopoulos AA, Frost RM, Hird M. J. Org. Chem. 2006;71:1104. doi: 10.1021/jo052250+. Ohmiya H, Yorimitsu H, Oshima K. J. Am. Chem. Soc. 2006;128:1886. doi: 10.1021/ja057560o. Bica K, Gaertner P. Org. Lett. 2006;8:733. doi: 10.1021/ol052965z. Bedford RB, Betham M, Bruce DW, Davis SA, Frost RM, Hird M. Chem. Commun. 2006:1398. doi: 10.1039/b601014h. Chowdhury RR, Crane AK, Fowler C, Kwong P, Kozak CM. Chem. Commun. 2008:94. doi: 10.1039/b713647a. Suzuki coupling: Brenstrum T, Gerristma DA, Adjabeng GM, Frampton CS, Britten J, Robertson AJ, McNulty J, Capretta A. J. Org. Chem. 2004;69:7635. doi: 10.1021/jo048875+. Gonzalez-Bobes F, Fu GC. J. Am. Chem. Soc. 2006;128:5360. doi: 10.1021/ja0613761. Stille coupling: Powell DA, Maki T, Fu GC. J. Am. Chem. Soc. 2005;127:510. doi: 10.1021/ja0436300. Negishi coupling: Nakamura M, Ito S, Matsuo K, Nakamura E. Synlett. 2005:1794. Hiyama coupling: Strotman NA, Sommer S, Fu GC. Angew. Chem., Int. Ed. 2007;46:3556. doi: 10.1002/anie.200700440.
- 4.(a) Boudier A, Knochel P. Tetrahedron Lett. 1999;40:687. [Google Scholar]; (b) Vyvyan JR, Loitz C, Looper RE, Mattingly CS, Peterson EA, Staben ST. J. Org. Chem. 2004;69:2461. doi: 10.1021/jo035778s. [DOI] [PubMed] [Google Scholar]; (c) Luo X, Zhang H, Duan H, Liu Q, Shu L, Zhang T, Lei A. Org. Lett. 2007;9:4571. doi: 10.1021/ol701995t. [DOI] [PubMed] [Google Scholar]; (d) Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Satoh M, Suzuki A. J. Am. Chem. Soc. 1989;111:314. [Google Scholar]; (e) Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000;122:4020. [Google Scholar]; (f) Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J. Org. Chem. 2002;67:5553. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]; (g) Hayashi T. J. Organomet. Chem. 2002;653:41. [Google Scholar]; (h) Cahiez G, Marquais S. Tetrahedron Lett. 1996;37:1773. [Google Scholar]; (i) Nakao Y, Takeda M, Matsumoto T, Hiyama T. Angew. Chem., Int. Ed. 2010;49:4447. doi: 10.1002/anie.201000816. [DOI] [PubMed] [Google Scholar]
- 5.Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2009;49:4122. [Google Scholar]; (b) Ohmura T, Awano T, Suginome M. Chem. Lett. 2009;38:664. [Google Scholar]; (c) Endo K, Ohkubo T, Hirokami M, Shibata T. J. Am. Chem. Soc. 2010;132:11033. doi: 10.1021/ja105176v. [DOI] [PubMed] [Google Scholar]; (d) Imao D, Glasspoole BW, Laberge VS, Crudden CM. J. Am. Chem. Soc. 2009;131:5024. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 7.(a) Matos K, Soderquist JA. J. Org. Chem. 1998;63:461. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]; (b) Ridgway BH, Woerpel KA. J. Org. Chem. 1998;63:458. doi: 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]
- 8.Ohmura T, Awano T, Suginome M. J. Am. Chem. Soc. 2010;132:13191. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- 9.(a) XPhos (2-dicyclohexylphosphino-2',4',6'-diisopropyl-1,1'-biphenyl). (b) SPhos (2-dicyclohexylphosphino-2’,6’-dimethoxybiphenyl).
- 10.(a) Molander GA, Petrillo DE. Org. Lett. 2008;10:1795. doi: 10.1021/ol800357c. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Jean-Gérard L. J. Org. Chem. 2009;74:1297. doi: 10.1021/jo802453m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Molander GA, Jean-Gérard L. J. Org. Chem. 2009;74:5446. doi: 10.1021/jo900968h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mun S, Lee J-E, Yun J. Org. Lett. 2006;8:4887. doi: 10.1021/ol061955a. See also Bonet A, Gulyás H, Koshevoy IO, Estevan F, Sanaú, Ubeda MA, Fernández E. Chem. Eur. J. 2010;16:6382. doi: 10.1002/chem.200903095. Smith SM, Takacs JM. J. Am. Chem. Soc. 2010;132:1740. doi: 10.1021/ja908257x.
- 12.Chea H, Sim H-S, Yun J. Adv. Synth. Catal. 2009;351:855. [Google Scholar]
- 13.Spessard GO, Miessler GL. Organometallic Chemistry. 1st ed. Upper Saddle River, NJ: Prentice Hall; 1997. [Google Scholar]
- 14.(a) Labadie JW, Stille JK. J. Am. Chem. Soc. 1983;105:6129. [Google Scholar]; (b) Kells KW, Chong JM. J. Am. Chem. Soc. 2004;126:15666. doi: 10.1021/ja044354s. [DOI] [PubMed] [Google Scholar]
- 15.Hatanaka Y, Hiyama T. J. Am. Chem. Soc. 1990;112:7793. [Google Scholar]
- 16.(a) Bergbreiter DE, Rainville DP. J. Organomet. Chem. 1976;121:19. [Google Scholar]; (b) Kabalka GW, Gooch EE., III J. Org. Chem. 1980;45:3578. [Google Scholar]
- 17.(a) Giovannini R, Stüdemann T, Dussin G, Knochel P. Angew. Chem. Int. Ed. 1998;37:2387. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]; (b) Johnson JB, Rovis T. Angew. Chem. Int. Ed. 2008;47:840. doi: 10.1002/anie.200700278. [DOI] [PubMed] [Google Scholar]; (c) Luo X, Zhang H, Duan H, Liu Q, Zhu L, Zhang T, Lei A. Org. Lett. 2007;9:4571. doi: 10.1021/ol701995t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.