

Abstract
Background
The presence of recurrent high-risk mutations in CDKN2A and CDK4 among melanoma-prone kindreds suggests that a high-throughput, multiplex assay could serve as an effective initial screening tool. Moreover, with the emergence of new melanoma risk single nucleotide polymorphisms (SNPs) through genome-wide association studies, a flexible platform that can easily accommodate these new risk alleles is needed for more accurate genetic risk profiling. To this end, we have developed a novel melanoma-associated mutation detection method using a multiplex bead-based assay. This assay is suitable for high-throughput CDKN2A and CDK4 genotyping and can be eventually adapted to multiple loci across various constituent populations.
Methods
Genomic DNA from a 1603 subjects (1005 in training set, 598 in validation set) were amplified by multiplex PCR using five primer sets followed by multiplex allele-specific primer extension for 39 different known germline variants. The products were then sorted on an xMAP™ (formerly Tag-It™) array and detected by use of the Luminex xMAP™ system. Genotypes were compared to previously-determined sequence data.
Results
In the Toronto training cohort, variants were detected in 145 samples, giving complete concordance between the bead assay and direct sequencing results. Analysis of the 598 samples from the GenoMEL validation set led to identification of 150/155 expected variants (96.77% concordance). Overall, the bead assay correctly genotyped 1540/1603 (96.07%) of all individuals in the study and 1540/1545 (99.68%) of individuals whose mutations were represented in the probe set. Out of a total of 62,512 SNP calls, 62,517 (99.99%) were correctly assigned.
Conclusions
In this initial evaluation, the multiplex bead-based assay for familial melanoma appears to be a highly accurate method for genotyping CDKN2A and CDK4 variants.
Keywords: Melanoma, CDKN2A, CDK4, p14ARF, familial, high-throughput
Introduction
Cutaneous malignant melanoma (CMM) accounts for approximately 2% of new cancer cases diagnosed annually in North America. Of these cases 5–10% of individuals with CMM have an affected family member, indicative of inherited predisposition. The primary locus involved in familial CMM to date is CDKN2A, which encodes the cell cycle regulator protein p16, and when mutated accounts for 25–40% of familial CMM cases(Goldstein, et al., 2006; Goldstein, et al., 2007; Zuo, et al., 1996). The p16 protein is encoded by exons 1α, 2 and 3. An alternative transcript, comprising exons 1β, 2 and 3, encodes a protein, designated p14ARF because the exon 2 sequence is translated in an alternative reading frame relative to that of p16. Germline mutations in exon 1β of p14ARF have been detected in melanoma prone families(Harland, et al., 2005; Hewitt, et al., 2002; Rizos, et al., 2001a; Rizos, et al., 2001b). In addition, rare mutations in the gene encoding cyclin-dependent kinase 4 (CDK4)- the primary enzyme inhibited by p16 binding- have been identified in a small number of CMM kindreds worldwide(Goldstein, et al., 2006; Molven, et al., 2005; Soufir, et al., 1998; Zuo, et al., 1996).
The identification of these genes and their association with familial CMM has highlighted a need for flexible, cost effective, high-throughput genotyping methods, which can eventually be transferred to a diagnostic setting when melanoma risk profiling becomes clinically practical. In this report we describe the development and validation of a novel multiplex bead-based assay to detect CMM associated variants. This assay employs the xTAG™ (formerly Tag-It™) Microsphere-Based Universal Array Genotyping platform (Luminex Molecular Diagnostics, Toronto, Canada) originally described by Bortolin et al.(Bortolin, et al., 2004) in relation to thrombophilia.
The CMM-specific mutation detection assay described here can be readily modified to reflect certain population-specific mutations and to include any novel mutations or low-to-moderate risk alleles that may be shown to be associated with CMM. This study demonstrates the use of a microsphere-based array assay for the detection of 32 common CDKN2A variants, two CDK4 mutations and five p14ARF variants among a large collection of familial melanoma samples from around the world.
Materials and Methods
Patient samples
A total of 1649 individual samples were genotyped. 1018 of these samples were accrued from March 1996 through May 2004 for CDKN2A research-based genetic testing through the Familial Melanoma Clinic at the Toronto-Sunnybrook Regional Cancer Centre (Toronto, Canada) and were from CMM affected patients, their unaffected relatives and spouses. All patients gave informed written consent to participate in this research study allowing a portion of their sample to be utilized for CDKN2A testing and the remainder to be banked for future studies. The remaining 628 samples were from international melanoma pedigrees from the Melanoma Genetics Consortium (GenoMEL www.genomel.org). Ethical committee approval was obtained from all institutions involved. The GenoMEL samples included in the analysis are shown in Table 1. All assay development and initial validation were performed in Toronto. As part of the expanded Boston validation, the device was transferred to the Massachusetts General Hospital and recalibrated. All experiments in Boston were performed to the exact specification of the protocols delineated by Toronto except primer sets were re-synthesized and beads were purchased separately from the manufacturer.
TABLE 1.
| Total | Failed reaction |
Samples scored |
Total variants |
1–8dup8 mutations (not in assay) |
Unprobed variants |
Probed variants |
Correct variants scored |
Missed variants |
Total SNPs interrogated |
SNPs assigned correctly |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| Toronto | 1040 | 22 | 1005 | 158 | 13 | 0 | 145 | 145 | 0 | 39195 | 39195 |
| Barcelona | 46 | 9 | 37 | 25 | 2 | 7a | 16 | 16 | 0 | 1443 | 1443 |
| NCI-US | 62 | 1 | 61 | 43 | 3 | 4b | 36 | 33 | 3h | 2379 | 2376 |
| NCI-Italy | 12 | 1 | 11 | 5 | 0 | 2c | 3 | 3 | 0 | 429 | 429 |
| Genoa | 14 | 2 | 12 | 13 | 0 | 2d | 11 | 11 | 0 | 468 | 468 |
| Leiden | 25 | 1 | 24 | 10 | 0 | 2e | 8 | 8 | 0 | 936 | 936 |
| Queensland | 150 | 10 | 140 | 35 | 3 | 8f | 24 | 24 | 0 | 5460 | 5460 |
| Leeds | 226 | 6 | 220 | 57 | 7 | 0 | 50 | 50 | 0 | 8580 | 8580 |
| Boston | 93 | 0 | 93 | 12 | 2 | 3g | 7 | 5 | 2i | 3627 | 3625 |
| Validation | 628 | 30 | 598 | 200 | 17 | 28 | 155 | 150 | 5 | 23322 | 23317 |
| 0.9677 | 0.9997 | ||||||||||
| Total | 1668 | 52 | 1603 | 358 | 30 | 28 | 300 | 295 | 5 | 62517 | 62512 |
| 0.9833 | 0.9999 |
Affected variants:
p.D84Y, p.L65P, p.R87W (4), p.V59G
p.N71S (2), p.R58X (2)
p.L65P (2)
p.R58X (2)
p.G23R (2)
p.L16P (2), p.L32P (2), p.Q50R (2), p.V51F (2)
p.W15X (2), p.A60T
c.241_254del14 (2), c.IVS2+1G>T
p.A148T(2)
DNA
For the Toronto subjects who were used in development of the assay, genomic DNA was isolated from whole blood with Qiagen QIAamp Blood Kits following the manufacturer’s instructions. DNA samples were stored at −20°C until required. Before analysis, genomic samples were quantified spectrophotometrically by measuring absorbance at 260 nm, diluted to 5 ng/µl, and stored at 4°C. For the GenoMEL validation cases, sample derivation and DNA preparation were separately performed at each site; genetic analysis based on the GenoMEL material has been previously published(Goldstein, et al., 2006; Goldstein, et al., 2007).
DNA amplification
Twenty-five ng of genomic DNA samples were subjected to multiplex PCR amplifications which contained 1µl 10X PCR Buffer (Invitrogen), 0.3 µl 50mM Mg2+, 1 µl dNTPs, 0.6 µl PCR primer mix (0.1 pmol each; IDT), 0.5 µl DMSO, 0.5 µl Platinum® Taq DNA Polymerase (Invitrogen) and ddH2O to a final volume of 10 µl. Samples were amplified for i) CDKN2A exons 1α, 2, 3; and ii) CDK4 exon 2 and p14ARF exon1β under the following cycling conditions: 95°C for 1 min, followed by 40 cycles of 95°C for 30 sec, 54°C for 30 sec, 72°C for 30 sec and then an extension time of 72°C for 7 min. Samples were kept at 4°C until ready for use. A negative PCR control was included in each multiplex reaction. The primers and amplicon sizes for all PCR reactions are shown in Table S1. Multiplex PCR products were analyzed by gel electrophoresis to confirm the presence of discrete bands for each amplicon (data not shown). Since the 1–8dup8 change is a perfect repeat and silent to the allele-specific methodology, it was assayed by a PCR-gel based system and not by the bead analysis.
Amplicons were then subjected to Exonuclease I/Shrimp Alkaline Phosphatase (Exo/SAP; MBI Fermentas) treatment for 1 hour at 37°C to degrade any excess PCR oligonucleotides and inactivate any remaining nucleotides, particularly dCTP, as biotin-dCTP is incorporated during the primer extension reaction. Samples were Exo/SAP inactivated at 80°C for 15 min. The treated PCR products were then diluted 1:2 by the addition of 10 µl of ddH2O.
Allele-Specific Primer Extension reaction
The microsphere-based assay was performed as described by Bortolin et al.(Bortolin, et al., 2004). Treated PCR products were used as the template in an Allele-Specific Primer Extension (ASPE) reaction, using SNP-specific oligonucleotide pairs, one terminating with the wild-type base, the other with the variant base (Table S2).
The ASPE sequence-specific oligos are tagged at the 5’ end with a 24-mer universal tag sequence (Luminex Molecular Diagnostics (formerly TM Bioscience Corporation), Toronto, Canada). The ASPE reaction incorporates Biotin-labeled dCTP, in excess, which is used to quantify reaction products in the Luminex xMAP™ flow cytometer.
ASPE reactions were carried out with 1.25 µl Exo/SAP treated PCR product, 0.5 µl 10X PCR reaction buffer (Tsp; Invitrogen), 0.125 µl 50 mM Mg2+, 0.1 µl 5µM dNTP-C (Amersham), 0.25 µl 5 µM biotin-dCTP (Invitrogen), 0.125 µl ASPE primer mix (1.25 pmol/µl each; IDT), 0.075 µl 5U/µl Tsp and ddH2O to a final volume of 5 µl. The cycling conditions were as follows: 96°C for 2 min, followed by 40 cycles of 94°C for 30 sec, 54°C for 30sec and 74°C for 30sec. Samples were then stored at 4°C until ready for use.
A population of carboxylated microspheres (beads) tagged with complementary (anti-tag) sequences was then hybridized directly to the ASPE products by first denaturing the samples for 2 min at 95°C, and a reporter (streptavidin-conjugated phycoerythrin; SA-PE) was added to detect incorporated biotin.
The hybridized products were then run through the Luminex xMAP™ and classified on the basis of microsphere identification. The presence of each allele was quantified by SA-PE detection as described previously(Bortolin, et al., 2004). Figure 1 gives a schematic outline of the bead-based assay.
Figure 1. The Bead-based Assay.
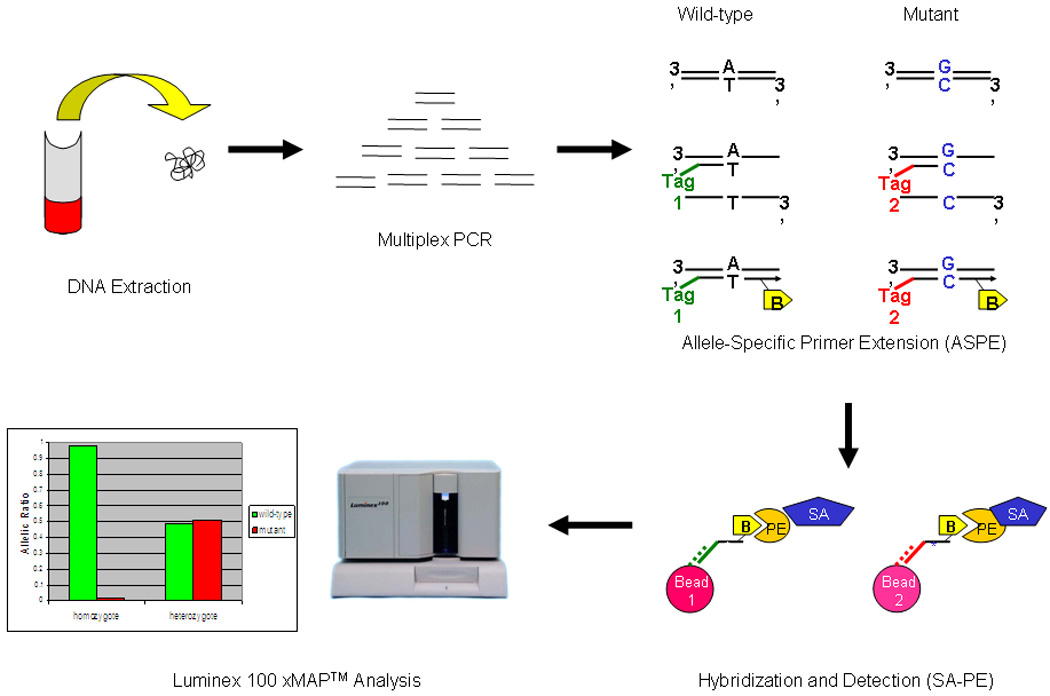
Regions of interest are initially amplified by multiplex PCR (for CDKN2A, the primers are listed in Table S1). Individual probes with the interrogative terminal codon are linked to a unique 24-mer Tag and subjected to ASPE. The ASPE extension step uses biotinylated dCTPs thereby introducing a biotin-labeled moiety into the extended fragment. The 24-mer Tag/biotinylated ASPE fragment is then specifically captured by a color-coded bead adorned with an anti-Tag which fully complements the 24-mer Tag sequence. Streptavidin-conjugated phycoerythrin (SA-PE) is then introduced to detect an intensity signal. In summary, the bead color defines the address (i.e. the individual SNP) while the PE fluorescence intensity defines the amount of the specific SNP fragment.
Statistical Analysis
The raw data, median fluorescent intensity (MFI), was collected for each bead population and base-lined by subtracting the value obtained for a PCR control sample (no DNA) from each sample value (Net MFI). Genotypes were determined from calculating the allelic ratios by using the ratio of Net wild-type or mutant MFI/sum of wild-type and mutant MFI for each SNP. This analysis could then determine wild-type, mutant, and heterozygous SNP genotype
DNA sequencing
Bidirectional DNA sequencing was performed on all 1018 samples using an MJ Base station as described previously(Hogg, et al., 2001) to compare sequences with the bead-based assay genotyping results. The 628 samples supplied by GenoMEL had previously been sequenced at source and at Leeds, as part of studies for other GenoMEL initiatives.
Results
Primary development of the multiplex bead-based assay was carried out on 1018 banked DNA samples from CMM affected individuals, unaffected relatives and spouses referred to the Familial Melanoma Clinic at the Toronto-Sunnybrook Regional Cancer Centre and for which the DNA material was successfully amplified. Since the 1–8dup8 mutation (N=13) is a perfect repeat and undetectable by the ASPE-based strategy, this variant was not incorporated into the bead assay. Thus, for the initial Toronto screen, 1005 samples (i.e. 1018-13) were blindly subjected to analysis for 39 variants (32 CDKN2A variants in one assay, two CDK4 and five p14ARF splice site variants in another, Table S2). In total, 145/1005 (14.42%) samples were shown to harbor an identifiable CDKN2A variant, of which 65 (6.46%) samples carried the p.A148T polymorphism. Figure 2 depicts an example of a heterozygous p.T93P CDKN2A mutation with wild-type sequence at all other SNPs. There was 100% concordance between the bead-based assay and DNA sequencing; we in fact detected a CDK4 p.R24C mutation that was previously undetected and subsequently confirmed by DNA sequencing. None of the p14ARF mutations were identified in the 1005 Toronto samples screened.
Figure 2. Example of a heterozygous p.T93P mutation in an individual.
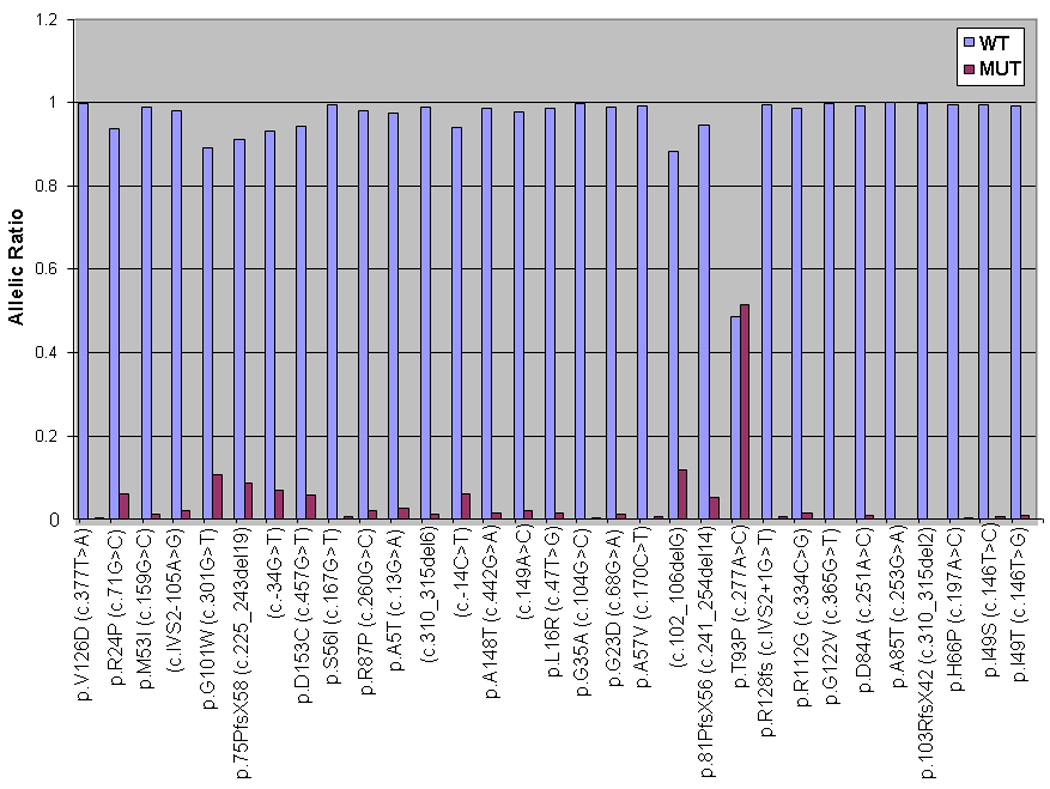
The allelic ratio is approximately 0.5, as expected in a heterozygous state.
Our validation phase was then expanded to include 628 samples from CMM melanoma pedigrees supplied by the international melanoma genetics consortium, GenoMEL. The bead assay was performed blindly on each sample. A total of 30/628 (4.77%) DNA specimens failed to amplify. From the 598 scorable samples, 150/598 (25.08%) were found to harbor a variant using the bead-based assay. When the sequence results were unblinded, 200/598 (33.44%) samples actually carried a variant. There were 45 mutations that could not have been detected based on assay design (seventeen 1–8dup8 mutations and 28 unprobed mutations, Table 1). For the 155 variants that were “eligible” for the assay, 150 were properly identified (150/155; 96.77%).
When the data from the training and validation sets were combined, 295/300 (98.33%) eligible mutations were successfully detected. However, since personal genotyping includes the accurate assignment of wildtype states, on an individual level, 1540/1603 (96.07%) of all available patient samples were correctly genotyped (i.e. variant(s) carrier vs. wildtype). This somewhat overestimates the error rate since 58 individuals had mutations not represented in the probe set. If these individuals were censored, the assay correctly genotyped 1540/1545 (99.68%) of the individuals with genotypes eligible for the assay.
Unlike sequencing, the bead assay performs 39 distinct SNP interrogations from 39 separate probe sets. Thus, it is also reasonable to measure the performance of the assay based on percentage of correct SNP assignments. For ineligible cases, where the mutations were not represented by one of the 39 probes, the assay was agnostic to the actual mutation but still made 39 correct sequence assignments at the individual SNPs. When considering SNP assignments in this study, a total of 62,517 SNPs were analyzed with only 5 incorrect calls. The bead-based assay is therefore 99.98% accurate at SNP scoring.
Four variants which had probe representation but were missed by the bead assay (false negatives) included two c.241_254del14 mutations, one splice site variant (sp3: c.IVS2+1G>T), and two p.A148T polymorphisms. One sample returned a false-positive result by being erroneously called as a p.G23D mutation when in actual fact the sample carried a mutation not included in the SNP panel. This false-positive result cannot be explained, as when we repeated the assay it appeared as wild-type. This was likely due to PCR product quality and/or quantity variations between the original and repeat runs. There were 28 variants among GenoMEL samples that were not included in the original assay (Table 1).
Discussion
A robust “lab-on-a-chip (LOC)” is an appealing notion in the emerging era of point-of-care diagnostics. Inherent to the fabrication of a genetic LOC is the need for a flexible platform that can respond to an enlarging repertoire of risk loci and the specific panel of founder changes embedded within certain populations. In this manuscript, we developed a novel multiplex bead-based assay, which can accurately genotype variants in the two established high-risk melanoma loci (i.e. CDKN2A and CDK4), and validated this assay on a cohort of familial melanoma patients. The fundamental rationale for developing such a system comes from the collective GenoMEL experience. A significant fraction of the 190 CDKN2A and CDK4 mutation-bearing families reported by the consortium harbor recurrent germline mutations(Goldstein, et al., 2006); in fact, the 27 most prevalent variants account for over 80% of the detected mutations among the melanoma families ascertained by GenoMEL. The high frequency of founder changes observed among this cohort makes this an ideal collection to develop this novel mutation-interrogation platform. However, the assay was developed with a single set of testable variants in mind and is not expected to capture all possible CDKN2A mutations latent in the CMM population; thus our assay should be viewed only as a first generation design which can be readily expanded to include additional melanoma risk alleles.
A total of 1603 patient DNA samples were genotyped by both the Tag-It assay and DNA sequencing for exons 1α, 2 and 3 of the CDKN2A gene, exon 2 of the CDK4 gene, and exon1β of p14ARF. With only 5 missed calls, the results between the bead-based assay and DNA sequencing were 98.33% in agreement by genotyping and 99.99% accurate by SNP calls. In terms of per sample cost, direct sequencing (forward and reverse) of the 5 exons (four CDKN2A and one CDK4; 10 separate reactions) costs approximately $20.00 at the MGH sequencing core ($2/sequence reaction) while bead analysis costs approximately $9.35 (25¢ for 70 primer sets + 13¢ per bead color × 70 bead colors). With further refinement, the economic advantage of bead assays could be further enhanced. In this initial proof-of-concept, the 39 variants can be found in 5 exons; however, if the 39 variants were in fact embedded in 39 separate exons, the cost of direct sequencing would be much higher. Although still early in terms of technical development, these results are quite encouraging for this novel bead-based assay.
In terms of assay development, the main technical advantages of this platform are its “plexibility” and flexibility. Both fluorescent-based and MALDI-driven SNP detection offer significantly lower multiplexing capabilities despite their flexibility of design. With newer systems, the bead-based assay can easily accommodate up to 500 SNPs in a single tube; thus, nearly all known variants of CDKN2A, CDK4 and ARF can be incorporated into a single assay along with a host of low-to-moderate risk CMM alleles. Solid array systems (i.e. microarray “chips”) present extraordinary capabilities in terms of parallel SNP analysis (> 1×106 SNPs); however, there is little if any user input into the choice of SNPs or assay design unless a “custom chip” is manufactured. Unlike solid-phase microarrays, however, liquid bead arrays offer several unique advantages: the spherical substrate allows for a greater analytical surface area, the liquid phase mobility encourages more efficient mixing of cognate molecules and the encoded beads can be more readily manipulated. Emerging technologies that capitalize on microfluidics also hold promise for heightened throughput and flexibility(Derveaux, et al., 2008).
A major potential application of the bead-array assay is in SNP-based risk profiling. Over the past few years, a harvest of CMM disease-associated SNPs has been uncovered through large and robust genome-wide association studies(Udayakumar and Tsao, 2009). From a scientific perspective, follow up analysis of putative regions will need to be performed with greater SNP densities. As such, assays need to be rapidly developed for high-throughput analysis without sacrificing multiplexing capabilities. The bead assay is an ideal research platform for saturating these regions with additional SNP markers in hopes of refining the disease-associated intervals. As novel melanoma risk alleles are discovered, the single tube assay, which measures a single allele in a single reaction, will have to be replaced by array-based methods to be capable of analyzing multiple alleles in a single reaction. Thus, the bead assay can be configured to be a truly “personalized” risk predictor allowing SNP panel selection based on historical or phenotypic features.
Bead-based analytics have already been deployed in the clinical setting(Bortolin, et al., 2004) developed the xTAG™ (formerly Tag-It™) platform for the multiplex analysis of a panel of thrombophilia-associated SNPs. They reported 100% concordance between the xTAG™ assay and direct DNA sequencing data for 736 SNP determinations from genomic DNA from 132 patients. They also used the Luminex xMAP™ system and concluded that the xTAG™ platform is a highly accurate, multiplexed, high-throughput SNP-detection assay. Although premature, an extended panel of high, moderate and low-risk alleles could eventually be developed into a useful melanoma risk profiling tool.
There are several limitations to our analysis. Like other allele-specific platforms, variants are interrogated by the bead array rather than discovered. Rare missense mutations, in-frame insertions, large deletions and mutations not included in the assay cannot be successfully recovered. For example, the common 1–8dup8 mutation, in which the first eight amino acids of exon 1α are exactly duplicated, could not be incorporated into the assay as wild-type and mutant sequences cannot be distinguished from each other by oligonucleotide differences. For mutational screening, several studies, including those from GenoMEL, have now reported successes with dHPLC in identifying CDKN2A mutations(Edmunds, et al., 2002; Harland, et al., 2008; Lang, et al., 2005; Marian, et al., 2005; Orlow, et al., 2001). Although direct sequencing and other conformation-based screening strategies, such as dHPLC and high resolution thermal melt (HRM) analysis, unquestionably drive discovery of novel mutations, they are limited in their multiplexing capabilities. Lastly, the maximal utility of this bead array depends on the distribution and frequency of CDKN2A and CDK4 mutations. The GenoMEL consortium enriches for familial cases that have a reasonable founder composition. In some ways, this is an ideal collection to initially validate the assay since a limited panel of variants may in fact capture a significant fraction of cases. However, broader application to the general CMM population may not be appropriate given the current iteration of the assay. In the future, it is possible that other major predisposition genes will be discovered and cost-effective, rapid, high-throughput methods of analyzing populations for founder changes will be developed. The current system reported here can in fact meet this challenge as more genetic information is uncovered.
In conclusion, we have developed and validated the use of a multiplex bead-based assay for the detection of 39 variants associated with familial CMM in the GenoMEL consortium. This multiplex bead-based assay is a fast, sensitive, high sample throughput, cost-effective, and a reliable method for the screening of CDKN2A and CDK4 mutations in a large number of samples. This assay can be easily expanded to include other variants as they are reported and can be readily adapted to specific mutations in specific populations.
Supplementary Material
Acknowledgements
This work was supported by the American Cancer Society (to H. T.), the National Health and Medical Research Council of Australia (to N. K. H.), the NIH (RO1 CA83115) and the EU FP6 Contract nr: LSHC-CT-2006-018702 (to GenoMEL) and the Fondo de Investigaciones Sanitarias, Spain (to S. Puig).
Abbreviations
- CMM
cutaneous malignant melanoma
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- CDK4
cyclin-dependent kinase 4
- ARF
alternative reading frame
- MC1R
melanocortin 1 receptor
- dHPLC
denaturing high-performance liquid chromatography
- SNPs
single-nucleotide polymorphisms
Footnotes
Competing interests: none declared
References
- Bortolin S, Black M, Modi H, Boszko I, Kobler D, Fieldhouse D, Lopes E, Lacroix JM, Grimwood R, Wells P, et al. Analytical validation of the tag-it high-throughput microsphere-based universal array genotyping platform: application to the multiplex detection of a panel of thrombophilia-associated single-nucleotide polymorphisms. Clin Chem. 2004;50(11):2028–2036. doi: 10.1373/clinchem.2004.035071. [DOI] [PubMed] [Google Scholar]
- Derveaux S, Stubbe BG, Braeckmans K, Roelant C, Sato K, Demeester J, De Smedt SC. Synergism between particle-based multiplexing and microfluidics technologies may bring diagnostics closer to the patient. Anal Bioanal Chem. 2008;391(7):2453–2467. doi: 10.1007/s00216-008-2062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds SC, Kelsell DP, Hungerford JL, Cree IA. Mutational analysis of selected genes in the TGFbeta, Wnt, pRb, and p53 pathways in primary uveal melanoma. Invest Ophthalmol Vis Sci. 2002;43(9):2845–2851. [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, Azizi E, Bianchi-Scarra G, Bishop DT, Bressac-de Paillerets B, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66(20):9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, Hayward NK, Demenais F, Bishop DT, Azizi E, Bergman W, Bianchi-Scarra G, Bruno W, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44(2):99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland M, Goldstein AM, Kukalizch K, Taylor C, Hogg D, Puig S, Badenas C, Gruis N, ter Huurne J, Bergman W, et al. A comparison of CDKN2A mutation detection within the Melanoma Genetics Consortium (GenoMEL) Eur J Cancer. 2008;44(9):1269–1274. doi: 10.1016/j.ejca.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland M, Taylor CF, Chambers PA, Kukalizch K, Randerson-Moor JA, Gruis NA, de Snoo FA, ter Huurne JA, Goldstein AM, Tucker MA, et al. A mutation hotspot at the p14ARF splice site. Oncogene. 2005;24(28):4604–4608. doi: 10.1038/sj.onc.1208678. [DOI] [PubMed] [Google Scholar]
- Hewitt C, Lee Wu C, Evans G, Howell A, Elles RG, Jordan R, Sloan P, Read AP, Thakker N. Germline mutation of ARF in a melanoma kindred. Hum Mol Genet. 2002;11(11):1273–1279. doi: 10.1093/hmg/11.11.1273. [DOI] [PubMed] [Google Scholar]
- Hogg D, Liu L, Lassam N. Genetic testing in familial melanoma. In: Nickoloff BJ, editor. Melanoma: Methods and Protocols. Totowa, N.J.: Humana Press Inc.; 2001. [Google Scholar]
- Lang J, Boxer M, MacKie RM. CDKN2A mutations in Scottish families with cutaneous melanoma: results from 32 newly identified families. Br J Dermatol. 2005;153(6):1121–1125. doi: 10.1111/j.1365-2133.2005.06846.x. [DOI] [PubMed] [Google Scholar]
- Marian C, Scope A, Laud K, Friedman E, Pavlotsky F, Yakobson E, Bressac-de Paillerets B, Azizi E. Search for germline alterations in CDKN2A/ARF and CDK4 of 42 Jewish melanoma families with or without neural system tumours. Br J Cancer. 2005;92(12):2278–2285. doi: 10.1038/sj.bjc.6602629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molven A, Grimstvedt MB, Steine SJ, Harland M, Avril MF, Hayward NK, Akslen LA. A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosomes Cancer. 2005;44(1):10–18. doi: 10.1002/gcc.20202. [DOI] [PubMed] [Google Scholar]
- Orlow I, Roy P, Barz A, Canchola R, Song Y, Berwick M. Validation of denaturing high performance liquid chromatography as a rapid detection method for the identification of human INK4A gene mutations. J Mol Diagn. 2001;3(4):158–163. doi: 10.1016/S1525-1578(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Darmanian AP, Holland EA, Mann GJ, Kefford RF. Mutations in the INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J Biol Chem. 2001a;276(44):41424–41434. doi: 10.1074/jbc.M105299200. [DOI] [PubMed] [Google Scholar]
- Rizos H, Puig S, Badenas C, Malvehy J, Darmanian AP, Jimenez L, Mila M, Kefford RF. A melanoma-associated germline mutation in exon 1beta inactivates p14ARF. Oncogene. 2001b;20(39):5543–5547. doi: 10.1038/sj.onc.1204728. [DOI] [PubMed] [Google Scholar]
- Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa-Lyonnet D, Benard J, Bressac-de Paillerets B. Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. Human Molecular Genetics. 1998;7:209–216. doi: 10.1093/hmg/7.2.209. [DOI] [PubMed] [Google Scholar]
- Udayakumar D, Tsao H. Moderate- to low-risk variant alleles of cutaneous malignancies and nevi: lessons from genome-wide association studies. Genome Med. 2009;1(10):95. doi: 10.1186/gm95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, Hayward N, Dracopoli NC. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nature Genetics. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.