

Abstract
This review focuses on mitochondrial abnormalities that occur in the vasculature during aging and explores the link between mitochondrial oxidative stress, chronic low-grade vascular inflammation, increased rate of endothelial apoptosis, and development of vascular diseases in the elderly. Therapeutic strategies targeting the mitochondria for prevention of age-associated vascular dysfunction and disease in old age are considered here based on emerging knowledge of the vasoprotective effects of caloric restriction, caloric restriction mimetics, the GH/IGF-1 axis, and mitochondria-targeted antioxidants.
Keywords: Aging, Mitochondria, Endothelium
Introduction
Mitochondria play important roles in biosynthetic pathways, cellular energetics, cellular redox homeostasis, signaling, calcium buffering, and regulation of programmed cell death. Thus, age-related changes of mitochondria are likely to affect a host of cellular physiological functions simultaneously and contribute to the development of a wide range of age-related diseases. Earlier studies focused on the role of mitochondria in pathological alterations of the brain, heart, and skeletal muscle. However, there is increasing evidence that mitochondria play an important role in vascular pathophysiology as well. Age-specific mortality rates from heart disease and stroke increase exponentially with age throughout the later years of life, accounting for more than 40% of all deaths among people aged over 65. Understanding the role of mitochondria (including alterations in mitochondrial ATP production, regulation of calcium signaling and apoptosis, modulating the activity of sirtuins) in age-related vascular pathophysiological alterations holds promise for developing novel therapeutic approaches to reduce cardiovascular mortality in an aging population. In this review, the effects of aging on mitochondrial ROS production and mitochondrial biogenesis in the vasculature and the signaling role of mitochondria in the aged endothelial and smooth muscle cells are considered in terms of potential mechanisms involved in vascular pathological alterations in aging. The possible benefits of emerging therapeutic strategies that have the potential to promote mitochondrial health in the aged vasculature are also discussed (Fig. 1).
Fig. 1.
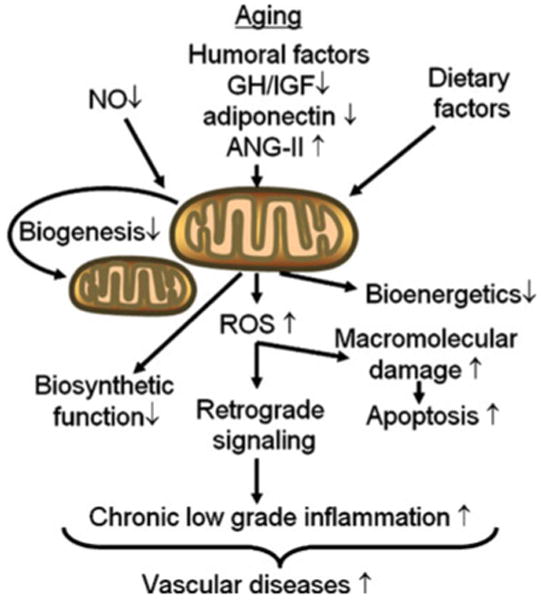
Age-related alterations in mitochondrial function affect endothelial function and phenotype. Aging-induced cell-autonomous mechanisms (e.g., an age-related decline in endothelial NO bioavailability) and age-related changes in the endocrine milieu (e.g., decreases in serum IGF-1 levels) disrupt pathways regulating biogenesis and self-renewal of mitochondria and impair mitochondrial functions. The central importance of aging-induced increases in mitochondrial release of ROS (H2O2, peroxynitrite) in this flowchart is evident, which leads to macromolecular damage in the cells promoting endothelial apoptosis. ROS can react with various proteins and lipids such as LDL, forming highly damaging oxLDL. ROS-mediated activation of retrograde signaling pathways, including NF-κB, lead to chronic low-grade vascular inflammation promoting the development of vascular diseases in the elderly
Mitochondria as sources of reactive oxygen species in aging
Harman first proposed the free radical theory of aging in the 1950s and, in the 1970s, extended the theory to implicate mitochondrial production of reactive oxygen species (ROS, such as O2− and H2O2) as the main cause for age-related macromolecular damage [1]. Since then, a large body of evidence has been published both in support of and against the free radical theory of aging. Although antioxidant supplementation has not been conclusively shown to produce an extension of lifespan in mammals and genetic knockout mice for major cellular antioxidant enzymes show a relatively mild phenotype despite the significant increases in ROS levels, a key role of increased mitochondrial production of ROS in the development of various age-related diseases is supported by the available data. Considerable evidence has been published that with advanced age production of ROS significantly increases in the vasculature, both in endothelial and smooth muscle cells (recently reviewed elsewhere [2]). Recent studies suggest that mitochondrial oxidative stress has an important role in aging-induced vascular dysfunction, which is further exacerbated by an increased activity of cytoplasmic and cell membrane-associated oxidases (including NAD(P)H oxidases) [3, 4]. Our recent understanding of the pathological mechanisms of atherosclerosis suggests that age-related oxidative stress leads to chronic low-grade vascular inflammation [4], which promotes the development of coronary artery disease and stroke in the elderly (see below). It is important to point out that age-related mitochondrial oxidative stress and inflammatory phenotypic changes are present both in endothelial and smooth muscle cells [4, 5]. The role of mitochondrial oxidative stress in cardiac aging leading to mitochondrial protein oxidation and increased mitochondrial DNA mutations has been well documented (for a recent overview, see reference [6]). Further studies are evidently needed to gain more insight in the role of similar aging-induced mitochondrial DNA and protein damage present in vascular endothelial and smooth muscle cells. Another potentially important link between mitochondrial oxidative stress and cardiovascular aging is the induction of programmed cell death [7]. The available evidence suggests that age-related increases in oxidative stress are associated with an increased rate of endothelial apoptosis [17, 21] and leads to microvascular rarefaction [8]. Mitochondrial-derived ROS may also impact vascular relaxation [9]. In that regard, it will be interesting to determine whether pharmacological agents that attenuate mitochondrial oxidative stress are able to prevent apoptotic cell death and/or improve vasomotor function in the aged vasculature. An interesting question that also should be addressed is the possible differences in the responses of endothelial and smooth muscle cells to increased age-related oxidative stress: age-related oxidative stress in the large vessels tends to promote vascular smooth muscle cell proliferation and invasion [10] rather than inducing apoptosis (see below).
The molecular mechanisms responsible for age-related mitochondrial oxidative stress in the vasculature are multifaceted and likely involve cell-autonomous effects, including dysregulation of antioxidant defenses such as peroxynitrite-mediated nitration and inhibition of MnSOD [11], decline in GSH content [12], Nrf2/ARE dysfunction [13], and a dysfunctional electron transport chain [3]. Recent studies showed that the mitochondrial enzyme p66Shc is involved in the regulation of mitochondrial redox status, linking vascular oxidative stress to apoptosis [14]. It is significant that genetic depletion of p66Shc in mice significantly reduces mitochondrial ROS production, which is associated with a 30% increase in lifespan and improved endothelial function [15–17]. In addition to cell-autonomous effects, age-related changes in endocrine regulation are likely to contribute to vascular mitochondrial oxidative stress in aging. Previous studies suggest the existence of a relationship between declining levels of growth hormone (GH) and insulin-like growth factor-1 (IGF-1, the synthesis of which is regulated by GH) and age-related cardiovascular impairment. Importantly, epidemiological studies provide strong evidence that GH and IGF-I deficiency in humans is associated with premature atherosclerosis and elevated cardiovascular disease mortality [18]. There is increasing evidence that IGF-1 confers mitochondrial protection, which likely contributes to its vasoprotective effects in aging. This view is supported by the findings in mice with hypopituitary dwarfism (Ames dwarf), where low plasma IGF-1 levels are associated with increased endothelial ROS generation, mitochondrial oxidative stress, and down-regulation of major anti-oxidant enzymes [19], mimicking the vascular aging phenotype. The available data support the conclusion that supplementation of IGF-1 may exert vasculoprotective effects in aging [20, 21], improving cardiac diastolic function [22], and preventing hippocampal microvascular rarefaction [8, 23, 24]. Recent reports demonstrate that treatment of aged rats with IGF-1 confers mitochondrial protection, including an attenuation of mitochondrial oxidative stress in parenchymal tissues [25]. We hypothesize that GH replacement and/or IGF-1 treatment in aging may also exert mitochondrial protective effects in the aged cardiovascular system as well. The findings that treatment of cultured endothelial cells with IGF-1 in vitro attenuates mitochondrial ROS production strongly support this hypothesis. Further studies are needed to determine the effects of IGF-1 on autophagy of dysfunctional mitochondria and apoptosis in the vasculature. Several traditional cardiovascular risk factors, including dietary factors, can also increase mitochondrial ROS production leading to mitochondrial damage. There is evidence that oxidized low-density lipoprotein, smoking [26], high methionine diet and hyperhomocysteinemia, angiotensin II [27, 28], and hyperglycemia [29] may increase ROS production in mitochondria of endothelial cells. Thus, mitochondrial ROS production likely represents a common pathway through which environmental factors can influence the rate of vascular aging. In that regard, it is significant that, in blood vessels of successfully aging species, metabolic stressors elicit a lower level of mitochondrial oxidative stress than in arteries of shorter-living ones [29], suggesting a possible link between mechanisms regulating increased metabolic stress resistance and slower rate of aging in longer-living species.
Dysregulation of mitochondrial biogenesis in aging
Mitochondria are highly dynamic organelles, and their biogenesis is likely involved in the regulation of cell metabolism and signal transduction. Because mitochondria are particularly susceptive to damage over time, effective control of mitochondrial biogenesis and turnover is critical for the maintenance of energy production, the prevention of accumulation of oxidatively damaged macromolecules, and the promotion of healthy aging [30]. Dysfunction of mitochondrial biogenesis affects the whole organism during aging [30]. Recent advances show that, with age, mitochondrial biogenesis is also impaired in endothelial cells both in conduit arteries [3] and capillaries [31, 32]. The available evidence suggests that mitochondrial biogenesis is also impaired in aged smooth muscle cells [3]. We posit that a decrease in mitochondrial biogenesis can reduce turnover of specific mitochondrial components resulting in the accumulation of oxidized lipids, proteins, and DNA in this cellular compartment, which would lead to a gradual deterioration of various mitochondrial functions, affecting biosynthetic pathways, cellular energetics, cellular redox homeostasis, signaling, calcium buffering, and regulation of apoptosis. It is likely that maintaining a normal turnover of mitochondria during aging would be critical to preventing the deleterious side effects of mitochondrial oxidative stress. Accordingly, maintenance of mitochondrial activity and biogenesis capacity during aging appears to be a key factor in caloric restriction-mediated prevention of age-related diseases in many organs [30, 33]. There is good reason to believe that this is the case in the cardiovascular system as well.
The mechanisms regulating mitochondrial biogenesis and their impairment in the aged vasculature are only partially understood. Mitochondrial biogenesis is governed by a complex interaction of cellular pathways involved in sensing availability of nutrients, stress response, and redox state with endocrine regulatory mechanisms, and it requires the transcriptional regulation of 20% of cellular proteins, coordinated by the interaction of the mitochondrial and nuclear genomes. The view that has emerged during the past decade is that many of the aforementioned endogenous and exogenous factors act through common effector pathways, which involve the peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) and activation of PGC-1α by nitric oxide, CREB, and AMPK. The available evidence suggests that, in the aged vasculature, due to an increased oxidative stress and down-regulation and uncoupling of eNOS [2], bioavailability of nitric oxide is significantly decreased. Lack of NO results in a down-regulation of PGC-1α and other mitochondrial biogenesis factors and consequential dysregulation of constituents of the electron transport chain and other mitochondrial proteins in aged endothelial and smooth muscle cells [3]. Studies on animal models with genetic depletion of eNOS [34] and treated with pharmacological inhibitors of NO synthesis [35] support this model. An aging-associated reduction in AMPK activity may also be an important contributing factor in dysregulation of PGC-1α transcription and impaired mitochondrial biogenesis associated with aging [36]. In addition to the aforementioned intra-cellular regulatory factors, age-related changes in endocrine regulation of cellular energetics (e.g., hypothyroidism, low IGF-1 levels [37]) may also be implicated in impaired mitochondrial biogenesis.
In addition to PGC-1α-regulated mitochondrial biogenesis, another mechanism involved in regulating mitochondrial function during aging is mitochondrial fission/fusion, which permit a constant remodeling of mitochondrial architecture. Recent data suggest that disruption of mitofusin pathways in cells results in complete loss of mitochondrial fusion and severe defects of cellular functions, including poor cellular growth, widespread heterogeneity of mitochondrial membrane potentials, and decreased cellular respiration [38]. Although the role of mitochondrial fission and fusion in vascular aging is poorly understood [39], the available evidence suggest that mitochondrial oxidative stress induced in vitro in endothelial cells results in dysregulation of these processes, whereas lowering the mitochondrial ROS levels can restore the morphological changes in mitochondria and improve mitochondrial function [40].
Mitochondria as signaling organelles in aging
In the past decade, a new era in aging research has emerged that concerns the role of mitochondria in intracellular signaling. Retrograde regulation is the general term for the crosstalk between mitochondria and the nucleus, which encompasses cellular responses to alterations in mitochondrial function. This pathway of communication from mitochondria to the nucleus influences many cellular functions and is thought to play an important role in the aging process. The signaling cascades involved include altered ROS production, Ca2+ dynamics, which activate downstream effectors pathways and transcription factors, the energy-sensing TOR pathway, stress-induced protein kinases, and a mitochondrial stress response pathway similar to the unfolded protein response in the endoplasmic reticulum. The cross-talk between the aforementioned pathways and cellular redox signaling mechanisms in aging is an important area of ongoing research. Mitochondria-initiated apoptosis can be considered as an extreme example of mitochondrial signaling, when signals emanating from the mitochondria drive the programmed breakdown of the entire cell. Although there is increasing evidence suggesting an important role for the aforementioned mechanisms in aging (e.g., the link between mTOR signaling, cardioprotection by caloric restriction [41], and lifespan regulation [42]), their physiological/pathophysiological roles in the cardiovascular system are poorly understood.
The mitochondrial theory of aging [1] postulates that mitochondria-derived ROS contribute to a variety of macromolecular oxidative modifications. Accumulation of such oxidative damage has been proposed to be a primary causal factor in the aging process [1], as well as in development of age-related diseases. However, there is an emerging view that mitochondria-derived ROS, in addition to causing oxidative damage, play important signaling roles as well. Numerous transcription factors, including NF-κB, AP-1, Nrf-2, and P53, are known to be redox-sensitive. Here, we present recent advances on the role of mitochondria-derived ROS in activating NF-κB signaling and inducing a pro-inflammatory vascular phenotype in aging. Our current understanding is that O2−, overproduced in the aged mitochondria in vascular endothelial and smooth muscle cells, is dismutated to H2O2 by Mn-SOD and it is the increased release of H2O2, which is responsible for activating NF-κB in the cytoplasm of aged cells [4]. This concept is supported by the fact that O2− is membrane-impermeable, whereas H2O2 easily penetrates the mitochondrial membranes. Indeed, scavenging of mitochondria-derived H2O2 attenuates NF-κB activation in aged vessels [4], suggesting that the age-related decline in mitochondrial function is, at least in part, responsible for chronic low-grade vascular inflammation in aging [4]. Previous studies have shown that endothelial activation and pro-inflammatory gene expression in aging (e.g., up-regulation of adhesion molecules and iNOS) is promoted, at least in part, by an increased NF-κB activation [4, 43–45]. Chronic activation of NF-κB and endothelial activation are known to predispose arteries to atherosclerosis [46]. Similar to the endothelial cells, age-related oxidative stress in vascular smooth muscle cells also lead to the development of a secretory phenotype promoting proliferation, invasion, matrix fragmentation, and collagenization (recently reviewed elsewhere [10]). Thus, we posit that attenuation of mitochondrial H2O2 release by pharmacological or molecular interventions in vivo should exert vasoprotective effects in aging. Recent studies have shown that, in aging mice, overexpression of human catalase in the mitochondria delays cardiac pathology and attenuates age-related oxidative stress and systemic inflammation [6]. Subsequent studies should determine whether attenuation of mitochondrial ROS production in the endothelial and smooth muscle also prevents age-related low-grade vascular inflammation.
Vascular mitochondria as therapeutic targets in aging
Pathways that attenuate mitochondrial oxidative stress, regulate mitochondrial biogenesis, and/or improve mitochondrial function have recently emerged as potential therapeutic targets for the amelioration of vascular dysfunction and prevention of the development of age-related vascular diseases. Caloric restriction is a dietary regimen, which improves health and slows the aging process in various organisms. There is increasing evidence that caloric restriction confers vasoprotection in aging and in pathological conditions associated with accelerated vascular aging (recently reviewed elsewhere [13]). Although the mechanisms underlying the beneficial vascular effects of caloric restriction are undoubtedly multifaceted, they likely include mitochondrial biogenesis [33] likely due to an increased NO bioavailability [47], an attenuation of mitochondrial ROS production [13, 34], and consequential inhibition of NF-κB activity [48]. The molecular mechanisms responsible for the beneficial cardiovascular effects of caloric restriction are not completely understood and may involve effects mediated by increased levels of adiponectin [49].
Recent studies demonstrated that the polyphenol resveratrol mimics vasoprotective effects of caloric restriction [30, 50, 51], including induction of mitochondrial biogenesis [52] and attenuation of mitochondrial oxidative stress [53, 54] in endothelial cells. The effects of resveratrol are, in part, explained by its ability to activate and/or up-regulate SIRT-1 in these cells, which subsequently deacetylates and activates PGC-1α. In fact, resveratrol is unable to modulate PGC1-α function in SIRT1−/− cells [55]. Resveratrol can also activate the NF-E2-related factor 2 (Nrf2), a transcription factor that regulates expression of numerous ROS detoxifying and antioxidant genes involved in regulation of mitochondrial redox homeostasis [54]. It is significant that resveratrol treatment in aged animals exerts vasoprotective effects and substantially decreases oxidative stress and endothelial apoptosis [50], an effect that likely involves both SIRT1 and Nrf2 mediated pathways. There is increasing evidence suggesting that AMPK regulates both mitochondrial content and mitochondrial antioxidant defenses in endothelial cells [56]. Thus, pharmacological activation of AMPK may also confer vasoprotective effects in aging.
Many of the adverse consequences of mitochondrial oxidative stress are mediated via production of the highly reactive oxidant peroxynitrite, the reaction product of NO and superoxide. Previous studies demonstrated that ONOO- formation is substantially increased in the vasculature of aged animals [11, 57]. The downstream targets of peroxynitrite-induced cytotoxicity are likely multiple (for a comprehensive review see reference [58]) and involve alterations in mitochondrial function. Because there are studies extant suggesting that pharmacological decomposition of peroxynitrite improves vascular dysfunction in aged rodents [59], further studies should determine the effects of peroxynitrite decomposition catalysts on vascular mitochondria in aging.
Several specific mitochondria-targeted antioxidants (e.g., antioxidants conjugated with triphenylphosphonium cation such as mitoquinone, mitovitamin E and mitophenyltertbutyline) are under development and experimental testing; however, their efficacy in attenuating mitochondrial oxidative stress in aging and inhibiting vascular inflammation have not been evaluated yet. These mitochondria-targeted antioxidants achieve concentrations in the mitochondrial matrix several-fold higher than those achieved in the cytosol because of the high negative membrane potential of the inner mitochondrial membrane. Although ongoing human clinical studies with mitochondria-targeted antioxidants involve only non-cardiovascular applications, preclinical studies show promise for prevention of cardiovascular diseases. Recent studies in animal models of hypertension [28] provide proof of concept that mitochondria-targeted antioxidants may confer vasoprotection in aging as well. A recent study reported that oral supplementation with the mitochondrial antioxidants alpha-lipoic acid and coenzyme Q10 reduces apoptosis in the cochlea-aged mice [60], and a similar treatment paradigm was shown to improve endothelial function in the aged aorta [61]. Future studies will undoubtedly exploit the unique biophysical and biochemical properties of mitochondria for the targeted delivery of drugs that affect mitochondrial function and reduce mitochondrial oxidative stress in the aged vasculature.
Perspectives
Although significant progress has been achieved in describing age-related alterations in mitochondrial function and phenotype in the cardiovascular system, there are several questions that have yet to be answered. Importantly, the specific roles for cell-autonomous and non-cell-autonomous mechanisms (e.g., hormonal effects) in age-related mitochondrial alterations need to be elucidated further. A consensus should be reached whether mitochondrial contributions to vascular aging occur primarily via increased mitochondrial ROS or other mechanisms by which mitochondria affect the aging process play an equally important role. For example, future studies should elucidate age-related alterations in mitochondrial autophagy and their contributions to vascular aging. New data on mitochondrial fusion/fission and cellular energetics in vascular aging are also needed. It seems to be likely that age-related changes in nuclear gene expression are different in the major vascular cell types (endothelial and smooth muscle cells, cells of the adventitia). Thus, future studies should consider cell-type specific differences in mitochondrial retrograde signaling in aging as well. There is a reasonable consensus that mitochondrial oxidative stress plays a central role in both induction of apoptotic death and activation of redox-sensitive molecular pathways (e.g., NF-κB-mediated inflammatory processes). These pathways are under intense investigation as therapeutic targets for the prevention of cardiovascular diseases. The concept that evolutionarily conserved pathways (such as SIRT, Nrf2/ARE) control the aging process in mammals, regulating cellular bioenergetics, mitochondrial function, and mitochondrial redox homeostasis lead to the proposal that pharmacological or nutritional modulation of these pathways may be effective in delaying the onset of age-dependent vascular disease.
Acknowledgments
This work was supported by grants from the American Diabetes Association, the American Federation for Aging Research and the NIH (HL077256, AG11370).
Footnotes
Disclosure of potential conflict of interests The authors declare no conflict of interests related to this study.
Contributor Information
Ungvari Zoltan, Email: zoltan-ungvari@ouhsc.edu.
Anna Csiszar, Email: anna-csiszar@ouhsc.edu.
References
- 1.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Csiszar A, Wang M, Lakatta EG, Ungvari ZI. Inflammation and endothelial dysfunction during aging: role of NF-{kappa}B. J Appl Physiol. 2008;105:1333–1341. doi: 10.1152/japplphysiol.90470.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ungvari ZI, Labinskyy N, Gupte SA, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol. 2008;294:H2121–H2128. doi: 10.1152/ajpheart.00012.2008. [DOI] [PubMed] [Google Scholar]
- 4.Ungvari ZI, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith KE, Csiszar A. Increased mitochondrial H2O2 production promotes endothelial NF-kB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 5.Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in rat coronary arteries. FASEB J. 2003;17:1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- 6.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282:R423–R430. doi: 10.1152/ajpregu.00296.2001. [DOI] [PubMed] [Google Scholar]
- 8.Sonntag WE, Lynch CD, Cooney PT, Hutchins PM. Decreases in cerebral microvasculature with age are associated with the decline in growth hormone and insulin-like growth factor 1. Endocrinology. 1997;138:3515–3520. doi: 10.1210/endo.138.8.5330. [DOI] [PubMed] [Google Scholar]
- 9.Gao Q, Zhao X, Ahmad M, Wolin MS. Mitochondrial-derived hydrogen peroxide inhibits relaxation of bovine coronary arterial smooth muscle to hypoxia through stimulation of ERK MAP kinase. Am J Physiol Heart Circ Physiol. 2009;297:H2262–H2269. doi: 10.1152/ajpheart.00817.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19:201–207. doi: 10.1097/MNH.0b013e3283361c0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R, Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009 doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519–528. doi: 10.1161/CIRCRESAHA.107.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napoli C, Martin-Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc Natl Acad Sci USA. 2003;100:2112–2116. doi: 10.1073/pnas.0336359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francia P, delli Gatti C, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110:2889–2895. doi: 10.1161/01.CIR.0000147731.24444.4D. [DOI] [PubMed] [Google Scholar]
- 16.Camici GG, Cosentino F, Tanner FC, Luscher TF. The role of p66Shc deletion in age-associated arterial dysfunction and disease states. J Appl Physiol. 2008;105:1628–1631. doi: 10.1152/japplphysiol.90579.2008. [DOI] [PubMed] [Google Scholar]
- 17.Yamamori T, White AR, Mattagajasingh I, Khanday FA, Haile A, Qi B, Jeon BH, Bugayenko A, Kasuno K, Berkowitz DE, Irani K. P66shc regulates endothelial NO production and endothelium-dependent vasorelaxation: implications for age-associated vascular dysfunction. J Mol Cell Cardiol. 2005;39:992–995. doi: 10.1016/j.yjmcc.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Elhadd TA, Abdu TA, Oxtoby J, Kennedy G, McLaren M, Neary R, Belch JJ, Clayton RN. Biochemical and biophysical markers of endothelial dysfunction in adults with hypopituitarism and severe GH deficiency. J Clin Endocrinol Metab. 2001;86:4223–4232. doi: 10.1210/jcem.86.9.7813. [DOI] [PubMed] [Google Scholar]
- 19.Csiszar A, Labinskyy N, Perez V, Recchia FA, Podlutsky A, Mukhopadhyay P, Losonczy G, Pacher P, Austad SN, Bartke A, Ungvari Z. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008;295:H1882–H1894. doi: 10.1152/ajpheart.412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer's disease. J Neurosci. 2007;27:824–831. doi: 10.1523/JNEUROSCI.4345-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groban L, Pailes NA, Bennett CD, Carter CS, Chappell MC, Kitzman DW, Sonntag WE. Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin II expression in senescent rats. J Gerontol A Biol Sci Med Sci. 2006;61:28–35. doi: 10.1093/gerona/61.1.28. [DOI] [PubMed] [Google Scholar]
- 23.Khan AS, Sane DC, Wannenburg T, Sonntag WE. Growth hormone, insulin-like growth factor-1 and the aging cardiovascular system. Cardiovasc Res. 2002;54:25–35. doi: 10.1016/s0008-6363(01)00533-8. [DOI] [PubMed] [Google Scholar]
- 24.Sonntag WE, Lynch C, Thornton P, Khan A, Bennett S, Ingram R. The effects of growth hormone and IGF-1 deficiency on cerebrovascular and brain ageing. J Anat. 2000;197(Pt 4):575–585. doi: 10.1046/j.1469-7580.2000.19740575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puche JE, Garcia-Fernandez M, Muntane J, Rioja J, Gonzalez-Baron S, Castilla Cortazar I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology. 2008;149:2620–2627. doi: 10.1210/en.2007.1563. [DOI] [PubMed] [Google Scholar]
- 26.Csiszar A, Labinskyy N, Podlutsky A, Kaminski PM, Wolin MS, Zhang C, Mukhopadhyay P, Pacher P, Hu F, de Cabo R, Ballabh P, Ungvari Z. Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am J Physiol Heart Circ Physiol. 2008;294:H2721–H2735. doi: 10.1152/ajpheart.00235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labinskyy N, Mukhopadhyay P, Toth J, Szalai G, Veres M, Losonczy G, Pinto JT, Pacher P, Ballabh P, Podlutsky A, Austad SN, Csiszar A, Ungvari Z. Longevity is associated with increased vascular resistance to high glucose-induced oxidative stress and inflammatory gene expression in Peromyscus leucopus. Am J Physiol Heart Circ Physiol. 2009;296:H946–H956. doi: 10.1152/ajpheart.00693.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008;43:813–819. doi: 10.1016/j.exger.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burns EM, Kruckeberg TW, Comerford LE, Buschmann MT. Thinning of capillary walls and declining numbers of endothelial mitochondria in the cerebral cortex of the aging primate, Macaca nemestrina. J Gerontol. 1979;34:642–650. doi: 10.1093/geronj/34.5.642. [DOI] [PubMed] [Google Scholar]
- 32.Burns EM, Kruckeberg TW, Gaetano PK. Changes with age in cerebral capillary morphology. Neurobiol Aging. 1981;2:283–291. doi: 10.1016/0197-4580(81)90037-3. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci USA. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 35.Addabbo F, Ratliff B, Park HC, Kuo MC, Ungvari Z, Csiszar A, Krasnikov B, Sodhi K, Zhang F, Nasjletti A, Goligorsky MS. The Krebs cycle and mitochondrial mass are early victims of endothelial dysfunction: proteomic approach. Am J Pathol. 2009;174:34–43. doi: 10.2353/ajpath.2009.080650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Echave P, Machado-da-Silva G, Arkell RS, Duchen MR, Jacobson J, Mitter R, Lloyd AC. Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J Cell Sci. 2009;122:4516–4525. doi: 10.1242/jcs.049734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 39.Jendrach M, Pohl S, Voth M, Kowald A, Hammerstein P, Bereiter-Hahn J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech Ageing Dev. 2005;126:813–821. doi: 10.1016/j.mad.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010 doi: 10.1007/s00125-010-1770-4. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linford NJ, Beyer RP, Gollahon K, Krajcik RA, Malloy VL, Demas V, Burmer GC, Rabinovitch PS. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell. 2007;6:673–688. doi: 10.1111/j.1474-9726.2007.00319.x. [DOI] [PubMed] [Google Scholar]
- 42.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Y, Yoon S, Jung KJ, Kim CH, Son TG, Kim MS, Kim YJ, Lee J, Yu BP, Chung HY. Upregulation of aortic adhesion molecules during aging. J Gerontol. 2006;61:232–244. doi: 10.1093/gerona/61.3.232. [DOI] [PubMed] [Google Scholar]
- 44.Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NFkappaB, reduced IkappaBalpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell. 2008;7:805–812. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- 46.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R, Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009;130:518–527. doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation. 2007;116:2809–2817. doi: 10.1161/CIRCULATIONAHA.107.725697. [DOI] [PubMed] [Google Scholar]
- 50.Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA, Zhang Y, Dunn SR, Sharma K, Pleshko N, Woollett LA, Csiszar A, Ikeno Y, Le Couteur D, Elliott PJ, Becker KG, Navas P, Ingram DK, Wolf NS, Ungvari Z, Sinclair DA, de Cabo R. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ungvari ZI, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009 doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G, Pearson K, de Cabo R, Pacher P, Zhang C, Ungvari Z. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H13–H20. doi: 10.1152/ajpheart.00368.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–H1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, de Cabo R, Csiszar A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. 2010 doi: 10.1152/ajpheart.00260.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 56.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 58.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radovits T, Seres L, Gero D, Lin LN, Beller CJ, Chen SH, Zotkina J, Berger I, Groves JT, Szabo C, Szabo G. The peroxynitrite decomposition catalyst FP15 improves ageing-associated cardiac and vascular dysfunction. Mech Ageing Dev. 2007;128:173–181. doi: 10.1016/j.mad.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 60.Someya S, Xu J, Kondo K, Ding D, Salvi RJ, Yamasoba T, Rabinovitch PS, Weindruch R, Leeuwenburgh C, Tanokura M, Prolla TA. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc Natl Acad Sci USA. 2009;106:19432–19437. doi: 10.1073/pnas.0908786106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith AR, Visioli F, Frei B, Hagen TM. Lipoic acid significantly restores, in rats, the age-related decline in vasomotion. Br J Pharmacol. 2008;153:1615–1622. doi: 10.1038/bjp.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]