

Abstract
The gut microbiota enhances the host’s metabolic capacity for processing nutrients and drugs and modulate the activities of multiple pathways in a variety of organ systems. We have probed the systemic metabolic adaptation to gut colonization for 20 days following exposure of axenic mice (n = 35) to a typical environmental microbial background using high-resolution 1H nuclear magnetic resonance (NMR) spectroscopy to analyze urine, plasma, liver, kidney, and colon (5 time points) metabolic profiles. Acquisition of the gut microbiota was associated with rapid increase in body weight (4%) over the first 5 days of colonization with parallel changes in multiple pathways in all compartments analyzed. The colonization process stimulated glycogenesis in the liver prior to triggering increases in hepatic triglyceride synthesis. These changes were associated with modifications of hepatic Cyp8b1 expression and the subsequent alteration of bile acid metabolites, including taurocholate and tauromuricholate, which are essential regulators of lipid absorption. Expression and activity of major drug-metabolizing enzymes (Cyp3a11 and Cyp2c29) were also significantly stimulated. Remarkably, statistical modeling of the interactions between hepatic metabolic profiles and microbial composition analyzed by 16S rRNA gene pyrosequencing revealed strong associations of the Coriobacteriaceae family with both the hepatic triglyceride, glucose, and glycogen levels and the metabolism of xenobiotics. These data demonstrate the importance of microbial activity in metabolic phenotype development, indicating that microbiota manipulation is a useful tool for beneficially modulating xenobiotic metabolism and pharmacokinetics in personalized health care.
IMPORTANCE
Gut bacteria have been associated with various essential biological functions in humans such as energy harvest and regulation of blood pressure. Furthermore, gut microbial colonization occurs after birth in parallel with other critical processes such as immune and cognitive development. Thus, it is essential to understand the bidirectional interaction between the host metabolism and its symbionts. Here, we describe the first evidence of an in vivo association between a family of bacteria and hepatic lipid metabolism. These results provide new insights into the fundamental mechanisms that regulate host-gut microbiota interactions and are thus of wide interest to microbiological, nutrition, metabolic, systems biology, and pharmaceutical research communities. This work will also contribute to developing novel strategies in the alteration of host-gut microbiota relationships which can in turn beneficially modulate the host metabolism.
INTRODUCTION
The gut microbiota (GM) exhibits a relatively low level of diversity compared to those of most soil ecosystems and in humans it is comprised of usually no more than nine phyla of microorganisms, of which only two are dominant: the Firmicutes and the Bacteroidetes (1, 2). This is the result of strong selection pressures inherent in such a specific environment where well-adapted microbes benefit from a regular source of carbon through the digestion of complex carbohydrates, while providing available nutrients to the host as well as potential protection against opportunistic pathogens. This symbiosis is the result of a long coevolution and implies that the host adapts its metabolism to the presence of microbial symbionts.
The issue of modulation of host metabolism, physiology, and homeostasis induced by colonization of germfree (GF) animals by microorganisms was raised as soon as germfree animals were generated (3). Schaedler and coworkers demonstrated in the mid-1960s that the dilated cecum characteristic of germfree mice tended to be reduced back to normal as soon as colonization by microbiota began, in particular when animals were in contact with bacteria of the Bacteroidetes group (4). This study also demonstrated that colonization of a germfree gut was rapid and remarkably stable, establishing within only a week after first exposure. However, a study conducted on germfree rats by Nicholls et al. showed that 3 weeks were necessary to obtain a stabilization and “normalization” of the urinary metabolic phenotype (5), suggesting that beyond the direct effect of microbiota on gut epithelium, other metabolic effects occurred during colonization of adult germfree animals, even when the microbiota was supposed to be established.
In this context, we characterized in a previous study the metabolic phenotypes of germfree and conventional C3H mice to serve as a basis for the following studies on the same germfree mouse model. We demonstrated that the microbiota status affects the systemic metabolism of the host, modulating the metabolic fingerprint of topographically remote organs such as the liver and the kidney (6). Here, we explore the adaptive mechanisms of gut colonization by microbiota using a similar systems biology approach in the same mouse strain. In addition to the nuclear magnetic resonance (NMR)-based metabolic profiling of the animals, as performed in the previous work, we also monitored here the gut microbial establishment by 16S rRNA gene pyrosequencing in order to review the composition of the microbial ecosystem simultaneously with the modifications of the host metabolism induced by the colonization process. In particular, we focused our attention on the evolution of liver metabolism, where important modifications of energy metabolism in conjunction with changes in the expression level of cytochrome P450 (CYP) involved in bile acid and drug detoxification pathways were observed in response to the colonization process. We also highlight a strong correlation between microbial families, such as the Coriobacteriaceae (including the genus Eggerthella), and both the hepatic concentrations of glucose, glycogen, and triglycerides and the activity of Cyp3a11, one of the most active cytochromes in drug metabolism in the mouse.
RESULTS AND DISCUSSION
Gut colonization induces a rapid weight gain associated with stimulation of hepatic glycogenesis and triglyceride synthesis.
This study aimed to improve the understanding of the links between GM and the mechanisms of metabolic adaptation of the host to the presence of its symbiont. In order to understand the impact of a progressive GM acquisition on the host metabolism, 8-week-old female germfree (GF) C3H/Orl mice were exposed for 20 days to the same environment as were their conventionally raised littermates (Conv-R) (n = 35 per group) and body weight was monitored every 5 days. The conventionalization process induced a dramatic increase in total body weight in newly conventionalized (ex-GF) animals in 2 stages (Fig. 1A). Ex-GF animals rapidly acquired 52% of the total weight gain over the first 5 days of colonization (4% of total body weight), followed by a phase of slower weight gain over the further 15 days of observation.
FIG 1 .

Gut microbiota acquisition-induced weight gain is metabolically reflected at the hepatic level. (A) Average body weight along acclimatization in Conv-R (circles) and GF/ex-GF (squares) groups. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (Student’s t test). Numbers of animals are unequal: n = 35 at D0, n = 28 at D5, n = 21 at D10, n = 14 at D15, and n = 7 at D20. (B) O-PLS scores derived from the model calculated from 600-MHz 1H NMR spectra of liver from Conv-R (pink or red symbols) and GF/ex-GF (blue symbols) mice at all time points using time and average weight as Y predictors (two predictive components plus 3 orthogonal components; Q2Y = 0.67, R2Y = 0.85, R2X = 0.46). Time points: D0 (light squares), D5 (circles), D10 (diamonds), D15 (triangles), and D20 (dark squares). (C) Coefficient plot related to the discrimination between GF animals at D0 (bottom) and ex-GF animals at D5 (top). (D) Coefficient plot related to the discrimination between ex-GF animals at D5 (bottom) and ex-GF animals at D20 (top). For panels C and D, metabolites are color coded according to their correlation coefficient, red indicating a very strong positive correlation (r2 > 0.7). The direction of the metabolite indicates the group with which it is positively associated as labeled on the diagram. GPC, glycerophosphocholine; GSH, glutathione.
In order to monitor the link between the physiological response to gut colonization (weight gain) and the systemic modifications of the host metabolism, we constructed several orthogonal projection to latent structure (O-PLS) models (7) derived from the NMR-based metabolic profiles of various biological matrices, as described in Materials and Methods. For example, the O-PLS regression model of hepatic metabolic profiles (1H NMR spectra) against time and mean body weight (used as Y predictors) for both groups is displayed in Fig. 1B. This clearly shows that the 2 phases of weight gain were mirrored at the hepatic metabolic level. Similar effects were also observed in other biological matrices (i.e., kidney, colon, and plasma) as indicated by the O-PLS scores reflecting the systemic metabolic response to weight and time in Fig. S1 in the supplemental material.
In order to understand which metabolic changes were characteristic of these 2 phases, pairwise comparison models were used for each group between GF/ex-GF and Conv-R mice at day 0 (D0), D5, and D20 and within each group between D0 and D5 (phase 1) and between D5 and D20 (phase 2) (Fig. S2). As expected, phase 1 was metabolically characterized in urine by the appearance of gut microbial cometabolites (phenylacetylglycine [PAG] and m-hydroxyphenylpropionic acid [m-HPPA] sulfate) and in colon by an increase of acetate, a well-known fermentation product of anaerobic bacteria, in conjunction with decreased bile acid levels (Fig. S2). Raffinose was more highly expressed in the GF mouse colonic metabotype than in that of Conv-R mice at D0, as has been described previously (6), and decreased during phase 1 of colonization (Fig. S2). Indeed, raffinose is a nondigestible oligosaccharide which requires the microbial enzyme α-galactosidase to initiate its digestion in the gastrointestinal lumen. Similarly to what had been previously observed (6), we therefore expected it to be found in higher concentrations in the GF group than in Conv-R animals and to monitor its degradation as colonization progressed. In the kidney, the GF metabolic profile was characterized by higher levels of osmoprotectants (dimethylamine, betaine, and scylloinositol) than those in Conv-R controls, as previously observed (6), emphasizing the constancy of metabolic perturbations in response to the germfree condition across different studies on the same germfree mouse model. These differences in osmolyte levels were not evident after 5 days of colonization. Remarkably, the liver metabolic profile displayed a large increase in the relative concentrations of glucose associated with high levels of glycogen at D5 (Fig. 1C). This is consistent with previous observations demonstrating that colonization of GF C57BL/6J mice was accompanied by an increase in glucose uptake in the small intestine (8). This pattern is the signature of the activation of the glycogenesis pathway.
The second phase was characterized in the liver metabolic fingerprint by relatively high levels of triglycerides which were associated in urine with higher concentrations of 2-oxoglutarate and 2-oxoisocaproate (Fig. 1D; see also Fig. S2). These latter metabolites are markers of a higher mitochondrial activity and are closely related to insulin secretion in pancreatic islets (9). Taken together, these results emphasize the dramatic changes in energy metabolism triggered by the acclimatization process, characterized by a metabolic switch from glycogenesis as an early response to colonization to triglyceride synthesis as a later adaptive mechanism, and corroborate the role of the GM in hepatic triglyceride metabolism as recently described (10).
Gut colonization alters bile acid metabolite profiles via modulation of hepatic Cyp8b1 expression.
Bile acids are well-known contributors to glucose and lipid metabolism in the liver (11, 12). Besides, the GM is known to alter bile metabolism (13), and consequently, a change in the bile acid profile of the GF mice was expected during the colonization process. The control of the ratio of cholic acid (CA) to muricholic acid (MCA) in the mouse is of particular importance since it determines the overall hydrophobicity of the bile acid pool, which regulates cholesterol absorption and biosynthesis in the liver (14, 15). It was thus hypothesized that bile acids might be involved in the observed modification of energy metabolism, but the dynamics of these changes were not known. The overall hydrophobicity of the bile acid pool is determined by the ratio of the two primary bile acids issued from the classic and the alternative biosynthetic pathways. The former is activated by sterol 12α-hydroxylase (Cyp8b1) to produce the strongly amphipathic CA, while the latter does not require this activation step and, in the mouse, leads to the production of the highly hydrophilic MCA (11). These primary bile acids are then both conjugated to glycine (minor form) or taurine (major form) before their secretion in bile. To test the above-mentioned hypothesis, we thus examined the ratio of taurocholic acid (TCA) to tauromuricholic acid (TMCA) in the evolution of the hepatic bile acid composition. The results were similar to what has been reported in rats (13): the TCA/TMCA ratio was 2:1 in Conv-R mice and 1:1 in GF mice (Fig. 2A). After only 5 days of colonization, this ratio was indistinguishable from that observed in Conv-R mice, indicating that bile acid metabolism adapts quickly to the presence of bacteria in the gut (Fig. 2A).
FIG 2 .
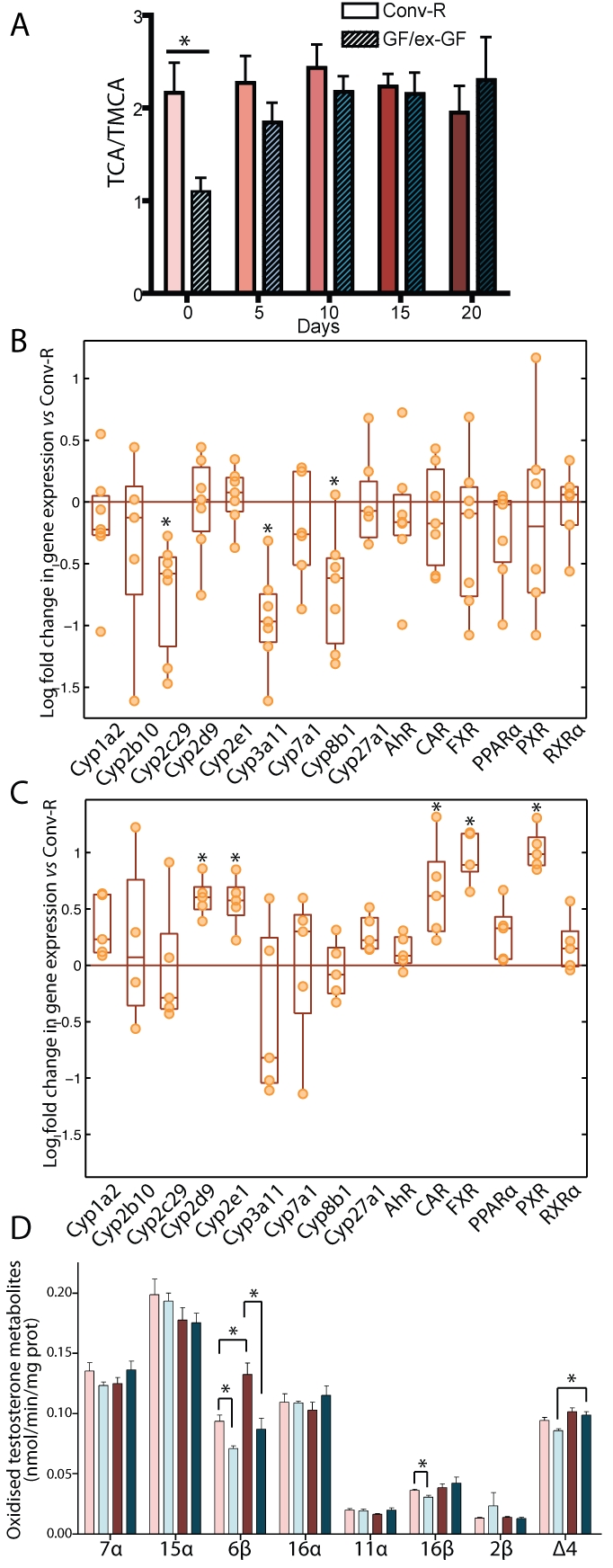
Gut microbiota influences cytochrome P450 expression and activity in liver microsomes. (A) Relative quantification of TCA over TMCA in bile over the acclimatization process. (B) mRNA expression levels of CYPs and nuclear receptors in GF mouse microsomes at D0 expressed relative to those of Conv-R animals (orange line). (C) mRNA expression levels of selected CYPs and nuclear receptors in ex-GF mouse microsomes at D20 expressed relative to those of Conv-R animals (orange line). (D) Profile of oxidized metabolites of testosterone in liver microsomes after 30 min of incubation. Data are means ± standard errors of the means. Symbols: light pink bars, Conv-R animals at D0; light blue bars, GF animals at D0; dark red bars, Conv-R animals at D20; dark blue bars, ex-GF animals at D20. *, P < 0.05 (Student’s t test).
This observation led us to hypothesize that Cyp8b1, the enzyme responsible for the regulation of the TCA/TMCA ratio, was downregulated in GF animals. We then assessed the mRNA expression level by quantitative reverse transcription-PCR (qRT-PCR) of cyp8b1 together with other CYP and nuclear receptors associated with bile acid metabolism and drug detoxification mechanisms potentially affected by the colonization process. As hypothesized, this analysis revealed a significantly lower expression of cyp8b1 in GF animals than in conventional controls (Fig. 2B), and this difference disappeared after 20 days of colonization (Fig. 2C), confirming the involvement of gut microbiota in cyp8b1 induction and the consecutive regulation of the TCA/TMCA ratio. cyp8b1 expression, together with those of cyp7a1 and cyp27a1, is known to be regulated by negative feedback of hydrophobic bile acids such as CA, through the activation of the nuclear receptor FXR (16). However, no downregulation of cyp7a1 and cyp27a1 was observed in GF compared to Conv-R animals. Moreover, despite the significant overexpression of FXR in ex-germfree mice compared to Conv-R animals at D20, neither cyp8b1 nor cyp7a1 and cyp27a1 were downregulated. These results illustrate the complexity of cyp8b1 regulation by GM. These observations are consistent with recently published reviews that discuss the complex regulation of bile acid metabolism (11, 17). Interestingly, suppression of cyp8b1 expression in the mouse liver results in a lower absorption of fatty acids and total cholesterol (11), and therefore, this mechanism of upregulation by GM may participate in the resistance of GF mice to obesity induced by a Western-style diet (18).
Gut colonization strongly influences endogenous xenobiotic detoxification metabolism.
The GM is also known to exert a strong influence on the metabolism of xenobiotics (19). The quantitative RT-PCR analysis revealed that cyp3a11 and cyp2c29 mRNA expression levels were significantly reduced in GF animals (D0) (Fig. 2B), whereas, at D20, these two CYPs were not significantly downregulated anymore.
In order to test the consequence of the downregulation of these CYPs at a more physiological level, the oxidative metabolism of [4-14C]testosterone in liver microsomes was measured as a reflection of the global ability of these enzymes to metabolize sterol compounds. This analysis showed that 6β- and 16β-hydroxylase activities, which are specific activities of Cyp3a11 and Cyp2c29, respectively, were significantly reduced at D0 in GF animals (Fig. 2D). Although mRNA expression levels of cyp3a11 in ex-GF animals were indistinguishable from those of Conv-R mice after 20 days of colonization (Fig. 2C), the specific activity of Cyp3a11 (6β-hydroxylase) was still significantly lower in the ex-GF group at D20, while the 16β-hydroxylase activity (Cyp2c29) was no longer different from conventional levels (Fig. 2C). This indicates that Cyp3a11 exhibited a slower response to the colonization process. In agreement with these results, an overexpression of CAR and PXR, the “xenobiotic sensors,” which are known to induce the expression of cyp3a11 in the mouse (20), was observed at D20 in ex-GF mice (Fig. 2C).
A recently published study assessing the hepatic mRNA levels of some CYPs and transporters in Conv-R and GF IQI mice showed significantly reduced expression levels of cyp1a2 and cyp3a11 in GF animals (21). Although we did not observe any reduction in cyp1a2 expression levels in our investigation, this indicates that the downregulation of cyp3a11 in GF mice is not strain specific and is encountered in at least two different GF strains. This finding is of crucial importance since the 6β-hydroxylase activity, performed in humans by CYP3A4, is responsible for the oxidation of almost 50% of the drugs known to undergo oxidative metabolism in humans (22). Altogether, these results support the core role of the GM in the stimulation of endogenous xenobiotic detoxification pathways.
Chronology of microbiota establishment over the gut colonization process.
All the animals included in this study were housed in the same closed room isolated from any other animal and benefitting from their own filtered ventilation. As a consequence, the germfree mice acquired a consistent flora as shown by the reproducibility of colonization among animals housed in the same or separate cages, as shown on the denaturing gradient gel electrophoresis (DGGE) assays performed at D5 and D20 (Fig. S5).
Microbiota profiling was performed in order to understand the chronology of microbial colonization and the potential influence of microbiota on host metabolism. This was achieved by sequencing the variable regions V1-V2 and V4 of the DNA coding for 16S rRNA extracted from fecal samples at 4 time points (D1, D3, D5, and D20) in both groups. Overall, the gut microbiota ecosystem in ex-GF animals was not identical to that in conventional animals even after 20 days of colonization (Fig. 3). Additionally, ex-GF mice exhibited a lower level of gut microbial complexity than did conventional animals (Fig. 3A).
FIG 3 .
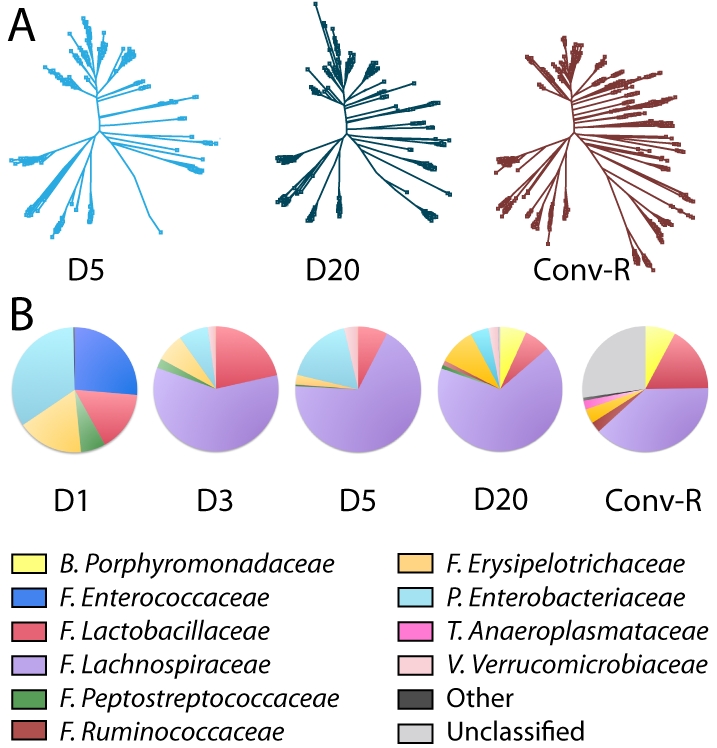
Gut microbiota establishment. (A) Phylogenetic trees illustrating the increasing complexity in gut microbiota in ex-GF animals at D5 and D20 and in Conv-R mice at D20. (B) The family-level composition of the gut microbiota in ex-GF animals at D1, D3, D5, and D20 and in Conv-R mice at D20. This analysis was computed using both V1-V2 and V4 regions. Abbreviations: B, Bacteroidetes; F, Firmicutes; P, Proteobacteria; T, Tenericutes; V, Verrucomicrobia.
The conventional gut microbial ecosystem of these C3H mice sampled at adulthood was composed mainly of Lachnospiraceae (order Clostridiales, phylum Firmicutes), Lactobacillaceae (order Lactobacillales, phylum Firmicutes), and Porphyromonadaceae (order Bacteroidales, phylum Bacteroidetes) and a large proportion of bacteria yet unclassified at the family level. At the phylum level, these observations are consistent with previous studies where Firmicutes and Bacteroidetes dominate the mouse gut microbial ecosystem (23). However, at the family level, our analysis of the C3H mouse microbiota differs from other mouse studies. For instance, compared to a study performed on FvB mice, C3H did not display Clostridiaceae within the order Clostridiales but Lachnospiraceae (24). Similarly, FvB animals exhibited a dominance of the Bacteroidaceae family within the order Bacteroidales, while C3H mice were dominated by the Porphyromonadaceae family (24). This last difference is probably due to a divergence in the technology since Porphyromonadaceae are poorly detected by sequencing of only the V1-V2 region. Here they were detected from the sequencing of the V4 region in complement to the V1-V2 region. This illustrates the difficulty of comparing studies that use different sets of primers or sequences of different regions. Nevertheless, it shows the importance of analyzing the dynamics of the GM at a lower phylogenic level, as the phylum level potentially hides more subtle variations in this ecosystem.
Enterococcaceae, Enterobacteriaceae, Lactobacillaceae, Erysipelotrichaceae, and Peptostreptococcaceae were the first bacterial families to settle in the intestine after exposure to the local environment. These are all facultative anaerobic bacteria (with the exception of Peptostreptococcaceae, which are strict anaerobes) and show a pattern of bacterial colonization (i.e., Gram-positive cocci, lactobacilli, and enterobacteria) similar to the one previously observed in neonates (25). The Enterococcaceae subsequently disappeared rapidly, whereas another family of Firmicutes, the Lachnospiraceae, became the dominant member of this dynamic ecosystem from D3 (Fig. 3B). Verrucomicrobiaceae (phylum Verrucomicrobia) also appeared at D3 and remained relatively stable until D20. At D5, Lachnospiraceae represented a greater proportion than did other families, such as the Lactobacillaceae. Ruminococcaceae represented a small proportion of the total community at D5, and Porphyromonadaceae (phylum Bacteroidetes) were found only at D20 in newly conventionalized animals. Other bacterial families present in conventional animals, such as Anaeroplasmataceae (phylum Tenericutes) (1%), were never established significantly in ex-GF animals over 20 days of colonization. Note that other families and unclassified sequences represented a high percentage of the conventional gut microbiota while these were minor in ex-GF animals. This suggests that bacteria that are highly demanding in terms of culture requirements are underrepresented in databases.
Integration of microbiota establishment and hepatic metabolism discloses a link between Coriobacteriaceae and energy metabolism.
In order to evaluate the associations between the hepatic metabolism and the GM described at taxonomic levels or by the DNA sequences coding for 16S rRNA (V1-V2 and V4) grouped into operational taxonomic units (OTUs), we used an O-PLS-based integration method as described in Materials and Methods. Conventional and ex-GF microbial data obtained at D5 and D20 were pooled and filtered to keep only variables detected in at least 75% of individuals within a group, and these were used as predictors. In addition, a multivariate adjustment was completed to remove the influence of time. Regression models of hepatic metabolic profiles (1H NMR spectra, n = 26) on each taxonomic level (21 models) and OTUs (54 models) were computed. Significant correlations between bacteria and hepatic levels of triglycerides, glycogen, and glucose are presented (Fig. 4; see also Fig. S3 in the supplemental material). The same strategy was repeated using CYP activities as dependent variables instead of NMR spectra (n = 25) (Fig. S4).
FIG 4 .

Correlation heat map between bacteria classified in OTUs (using both V1-V2 and V4 regions) and hepatic energy metabolism. Each OTU is described by the best matching type strain and sequence identity cutoff (see Text S1 in the supplemental material for details). TGs, triglycerides.
Two bacterial phyla (Actinobacteria and Tenericutes) were significantly predicted by the liver metabolic profiles and were both associated with high hepatic levels of triglycerides and low hepatic levels of glycogen and glucose (Fig. S3). The positive correlation of Tenericutes (class Mollicutes) with hepatic triglycerides seems to support previous work which showed that these bacteria bloomed and dominated the gut ecosystem in an obese mouse model fed on a high-fat/high-sugar diet (23). However, the sequences observed at this time by Gordon’s team were further reclassified into the Clostridiales. Therefore, this observation of an association between Tenericutes (class Mollicutes) and hepatic triglycerides constitutes a new finding. In addition, the strongest correlation between bacteria and hepatic triglycerides was observed for Actinobacteria of the family Coriobacteriaceae (Fig. S3). The correlation analysis computed between hepatic metabolic profiles and bacteria classified in OTUs confirmed the connection between the Coriobacteriaceae and hepatic triglycerides, glycogen, and glucose, as 2 of the most strongly correlated OTUs (Eggerthella lenta and Eggerthella hongkongensis, 80% and 90% identity, respectively) belonged to the Coriobacteriaceae family (Fig. 4). Although these bacteria were not detected in ex-GF animals, other correlated bacteria may participate in the metabolic shift described in this group over the colonization process. Recently, a study performed on hamsters reported a strong correlation between unidentified bacteria of the Coriobacteriaceae family and non-high-density-lipoprotein (non-HDL) plasma cholesterol when the metabolism was challenged using grain sorghum lipid extract to improve the HDL/non-HDL ratio (26). In the same study, the authors hypothesized the existence of a link between non-Bifidobacterium members of the Coriobacteriaceae family and cholesterol absorption. Here, we present further evidence of the link between this family of bacteria and the host lipid metabolism, in a different species (i.e., the mouse) and in conventional animals fed a conventional diet, while this link was not observed in conventionally fed hamsters (26), which emphasizes that these two studies are highly complementary. Furthermore, the O-PLS-based integration approach revealed a positive correlation between Eggerthella hongkongensis and the testosterone 6β- and 2β-hydroxylase activity in hepatic microsomes, together with many other bacteria (Fig. S4B). The remarkable correlation pattern of both 6β- and 2β-hydroxy metabolites is the signature of Cyp3a11 activity (27, 28) (Fig. S4B), underlining the involvement of the GM in the stimulation of drug metabolism. Note that the minor metabolite of Cyp3a11, 2β-hydroxytestosterone, was not significantly affected by the colonization process (Fig. 2D), which reflects the fact that only 6β-hydroxylase is a specific activity of Cyp3a11. Altogether, these results suggested that the family Coriobacteriaceae, and particularly the genus Eggerthella, might also be involved in the stimulation of a major hepatic detoxification activity. Interestingly, bacteria of the Coriobacteriaceae family were in the minority in conventional mice, representing approximately 1% of the total number of detected microorganisms. This exemplifies the possibility that bacteria in low proportions in the gut microbial ecosystem might be of extreme importance for the host metabolism and should therefore not be neglected in research. Although we have to be extremely careful in terms of extrapolation to humans, it is noteworthy that non-Bifidobacterium members of the Coriobacteriaceae family are in large proportion in the human gut (29). These findings therefore underline the importance of continuing the research on the link between this family of bacteria and the host lipid metabolism, especially in the context of the ever-growing worldwide obesity epidemic and associated cardiovascular diseases.
Finally, the correlation of OTUs with hepatic metabolism revealed a strong positive association between hepatic triglycerides and a cluster of sequences attributed to Lactobacillus gasseri (98% identity) (Fig. 4). No correlation was found between the presence of Coriobacteriaceae and Lactobacillus gasseri (r2 = 0.05), suggesting that the association between this bacterium and the triglycerides cannot be attributed to the stabilization of one bacterium by the other. This observation is not in accordance with the literature. This bacterium has been reported to be hypoglycemic and hypocholesterolemic in db/db mice and hypercholesterolemic rats (30, 31). It has also been shown that skim milk fermented by L. gasseri reduces mesenteric adipose tissue weight, adipocyte size, and serum leptin concentration in rats (32). It is worth mentioning that a recently published work raised the hypothesis of Lactobacillus involvement in body weight gain in patients treated with vancomycin, an antibiotic that selects lactobacilli (33).
In conclusion, we present here the first attempt at integrating in vivo dynamic gut microbial data over the colonization process with the adaptive mechanisms of the host characterized by its multicompartmental metabolic fingerprint and a range of crucial CYPs involved in drug metabolism. This systems biology approach highlights the essential role of the GM in the energy metabolism of the host through the association of specific clusters of bacteria, such as the Coriobacteriaceae, and the hepatic levels of triglycerides, glucose, and glycogen. These bacteria were also strongly correlated with the specific activity of Cyp3a11, suggesting also an important potential impact on the stimulation of endogenous drug metabolism. These results thus provide new insights into the fundamental mechanisms that regulate host-gut microbiota interactions and provide a basis to further develop novel strategies in the alteration of this relationship in order to beneficially modulate the host metabolism.
MATERIALS AND METHODS
Animal handling and experimental design.
All investigations were conducted according to Swiss ethical legislation on animal experimentation, and the protocol was approved by the Veterinary Office of the canton of Vaud. This parallel experiment involved two groups of 35 C3H/Orl female mice aged 8 weeks (Charles River, France). One group was conventionally raised (pathogen-free Conv-R control), and the other was raised in a germfree environment (maintained in isolators). Throughout the duration of the study, water and food (R03-10) were gamma irradiated and provided ad libitum to both groups.
The day before starting the experiment (D0), seven mice from each group were euthanized by decapitation in order to collect all control samples (urine, plasma, feces, liver, kidney cortex, and proximal colon). To preserve enzymatic activity, liver was rinsed by perfusion of 5 ml of cold NaCl solution through the hepatic portal vein and gall bladder was carefully removed. Intestinal sections were also flushed with cold NaCl solution. All samples were snap-frozen in liquid nitrogen and stored at −40°C for biofluids and −80°C for tissues until analysis.
On the first day of the experiment, all germfree (GF) animals were taken out of isolators and housed in the same environment as Conv-R animals, with bedding that was previously used by Conv-R mice for 3 days in order to expose the germfree mice to the same bacterial ecosystem as the Conv-R group. Then, every 5 days for 20 days (D5, D10, D15, and D20), 7 mice from each group were sacrificed and samples were collected as outlined above.
Sample preparation.
Thirty microliters of urine samples was mixed with 20 µl of phosphate buffer made up in 95% D2O containing 0.1% deuterated 3-(trimethylsilyl)propionic acid (TSP) (pH 7.4) before being placed in 1.7-mm capillary tubes for NMR analysis. The polar phase of extracts of kidney cortex, proximal colon, and plasma samples was extracted as described in Text S1 in the supplemental material.
NMR spectroscopy.
1H NMR spectra of biofluids and tissues were acquired as previously described (6). More details can be found in the supplemental material.
Measurement of relative concentrations of bile acids in bile.
Bile acids were measured using an ultraperformance liquid chromatography (UPLC) system (UPLC Acquity; Waters Ltd., Elstree, United Kingdom) coupled online to an LCT Premier time-of-flight mass spectrometer (Waters MS Technologies, Ltd., Manchester, United Kingdom).
Five microliters of bile samples diluted by a factor of 50 in H2O was injected onto a 2.1- by 100-mm (1.7-µm) HSS T3 Acquity column (Waters Corporation, Milford, MA) and eluted at a flow rate of 500 µl/min using a 20-min gradient of 100% A to 100% B (A = water, 0.1% formic acid; B = methanol, 0.1% formic acid). As bile acids ionize strongly in negative mode, producing a prominent [M-H] ion, samples were analyzed in negative electrospray mode using a scan range of 50 to 1,000 m/z. The capillary voltage was 2.4 kV, the sample cone was 35 V, the desolvation temperature was 350°C, the source temperature was 120°C, and the desolvation gas flow was 900 liters/h. The LCT Premier spectrometer was operated in V optics mode, with a data acquisition rate of 0.1 s and a 0.01-s interscan delay. Leucine enkephalin (m/z, 556.2771) was used as the lock mass; a solution of 200 pg/µl (50:50 acetonitrile [ACN]/H2O) was infused into the instrument at 5 µl/min via an auxiliary sprayer. Data were collected in centroid mode with a scan range of 50 to 1,000 m/z, with lock mass scans collected every 15 s and averaged over 3 scans to perform mass correction. The system was calibrated before data acquisition using a solution of sodium formate.
Assessment of cytochrome P450 global activity on steroid metabolism.
Hepatic microsomal fractions were prepared as described in Text S1 in the supplemental material. The protein concentration of the microsomal fraction was determined according to the method of Lowry et al. (34). The cytochrome P450 amount in the microsomal fractions was assessed in a Kontron dual-wavelength spectrophotometer according to the method described by Omura and Sato (35). Testosterone hydroxylation activities were measured from incubation of liver microsomes using a method derived from that previously described by Waxman (27). Quantification of testosterone hydroxylation activities was achieved by radio-high-pressure-liquid-chromatography (radio-HPLC) detection as described in Text S1 in the supplemental material.
Details regarding quantitative RT-PCR of mRNA extracted from liver samples and microbial profiling are given in the supplemental material.
Multivariate data analysis.
All 1H NMR profiles (from multiple tissues and biofluid samples) were mean centered and scaled to unit variance prior to analysis using Matlab software version R2009a (MathWorks). All O-PLS-derived models were evaluated for goodness of prediction (Q2Y value) using 7-fold cross-validation.
1H NMR profiles of liver, kidney, colon, urine, and plasma metabolites were analyzed by O-PLS methods (7). 1H NMR data were used as independent variables (X matrix) for modeling of time and average total body weight per group (Y predictors) (Fig. 1; see also Fig. S1). Models were calculated using two predictive components and up to 5 orthogonal components.
Effects of time and colonization in 1H NMR data of all investigated biological matrices were then explored by O-PLS discriminant analysis (DA) (pairwise comparisons), where each data matrix was regressed against a dummy matrix indicating the class of samples (Conv-R and GF/ex-GF or time point) (Fig. S2).
Prediction of the presence of bacterial family (using the sum of the data obtained for V1-V2 and V4 regions) by either hepatic 1H NMR-based metabolic profile (Fig. 4; see also Fig. S3) or CYP activities (Fig. S4) for Conv-R and ex-GF animals pooled at D5 and D20 (X matrix) was performed by O-PLS. Microbial data were filtered so that only bacteria detected in at least 75% of animals per group were included in models. Each model uses a single component to predict the intensity of detection of each bacterium (Y predictors). Significance of models (α = 0.05) was tested using 1,000 random permutations, and a P value was calculated by rank determination of the model actual Q2Y value among the Q2Y values calculated for the permutated models. Correlation coefficients extracted from significant models were filtered so that only significant correlations above the threshold defined by Pearson’s critical correlation coefficient (P < 0.05; r2 = 0.33809 when n = 26, and r2 = 0.3882 when n = 25) are displayed. Correlations between taxonomic levels or OTUs and hepatic energy metabolism were computed for triglycerides, glycogen, and glucose at δ 5.31, δ 5.41, and δ 5.22, respectively.
SUPPLEMENTAL MATERIAL
ACKNOWLEDGMENTS
This work was supported by Nestlé as part of the Imperial College London-Nestlé strategic alliance.
We thank Ruth E. Ley and Leslie Dethlefsen for their advice on creating the phylogenetic trees in ARB. We also acknowledge Honglin Dong for delivering the DGGE gels in the supplemental material.
Footnotes
Citation Claus, S. P., S. L. Ellero, B. Berger, L. Krause, A. Bruttin, et al. 2011. Colonization-induced host-gut microbial metabolic interaction. mBio 2(2):e00271-10. doi:10.1128/mBio.00271-10.
REFERENCES
- 1. Dethlefsen L., McFall-Ngai M., Relman D. A. 2007. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ley R. E., Turnbaugh P. J., Klein S., Gordon J. I. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023 [DOI] [PubMed] [Google Scholar]
- 3. Dubos R., Schaedler R. W., Costello R., Hoet P. 1965. Indigenous, normal, and autochthonous flora of the gastrointestinal tract. J. Exp. Med. 122:67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schaedler R. W., Dubos R., Costello R. 1965. Association of germfree mice with bacteria isolated from normal mice. J. Exp. Med. 122:77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicholls A. W., Mortishire-Smith R. J., Nicholson J. K. 2003. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats. Chem. Res. Toxicol. 16:1395–1404 [DOI] [PubMed] [Google Scholar]
- 6. Claus S. P., Tsang T. M., Wang Y., Cloarec O., Skordi E., Martin F. P., Rezzi S., Ross A., Kochhar S., Holmes E., Nicholson J. K. 2008. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol. Syst. Biol. 4:219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trygg J., Wold S. 2002. Orthogonal projections to latent structures (O-PLS). J. Chemom. 16:119–128 [Google Scholar]
- 8. Bäckhed F., Ding H., Wang T., Hooper L. V., Young Koh G., Nagys A., Semenkovich C. F., Gordon J. I. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U. S. A. 102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heissig H., Urban K. A., Hastedt K., Zunkler B. J., Panten U. 2005. Mechanism of the insulin-releasing action of alpha-ketoisocaproate and related alpha-keto acid anions. Mol. Pharmacol. 68:1097–1105 [DOI] [PubMed] [Google Scholar]
- 10. Velagapudi V. R., Hezaveh R., Reigstad C. S., Gopalacharyulu P., Yetukuri L., Islam S., Felin J., Perkins R., Boren J., Oresic M., Backhed F. 2010. The gut microbiota modulates host energy and lipid metabolism in mice. J. Lipid Res. 51:1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mataki C., Magnier B. C., Houten S. M., Annicotte J. S., Argmann C., Thomas C., Overmars H., Kulik W., Metzger D., Auwerx J., Schoonjans K. 2007. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol. Cell. Biol. 27:8330–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., Auwerx J. 2006. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489 [DOI] [PubMed] [Google Scholar]
- 13. Wostmann B. S. 1973. Intestinal bile acids and cholesterol absorption in the germfree rat. J. Nutr. 103:982–990 [DOI] [PubMed] [Google Scholar]
- 14. Duckworth P. F., Vlahcevic Z. R., Studer E. J., Gurley E. C., Heuman D. M., Beg Z. H., Hylemon P. B. 1991. Effect of hydrophobic bile acids on 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity and mRNA levels in the rat. J. Biol. Chem. 266:9413–9418 [PubMed] [Google Scholar]
- 15. Watt S. M., Simmonds W. J. 1984. Effects of four taurine-conjugated bile acids on mucosal uptake and lymphatic absorption of cholesterol in the rat. J. Lipid Res. 25:448–455 [PubMed] [Google Scholar]
- 16. Vlahcevic Z. R., Eggertsen G., Bjorkhem I., Hylemon P. B., Redford K., Pandak W. M. 2000. Regulation of sterol 12alpha-hydroxylase and cholic acid biosynthesis in the rat. Gastroenterology 118:599–607 [DOI] [PubMed] [Google Scholar]
- 17. Chiang J. Y. 2009. Bile acids: regulation of synthesis. J. Lipid Res. 50:11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bäckhed F., Manchester J. K., Semenkovich C. F., Gordon J. I. 2007. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U. S. A. 104:979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nicholson J. K., Holmes E., Wilson I. D. 2005. Gut microorganisms, mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 3:431–438 [DOI] [PubMed] [Google Scholar]
- 20. di Masi A., Marinis E. D., Ascenzi P., Marino M. 2009. Nuclear receptors CAR and PXR: molecular, functional, and biomedical aspects. Mol. Aspects Med. 30:297–343 [DOI] [PubMed] [Google Scholar]
- 21. Toda T., Saito N., Ikarashi N., Ito K., Yamamoto M., Ishige A., Watanabe K., Sugiyama K. 2009. Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica 39:323–334 [DOI] [PubMed] [Google Scholar]
- 22. Williams J. A., Ring B. J., Cantrell V. E., Jones D. R., Eckstein J., Ruterbories K., Hamman M. A., Hall S. D., Wrighton S. A. 2002. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos. 30:883–891 [DOI] [PubMed] [Google Scholar]
- 23. Turnbaugh P. J., Backhed F., Fulton L., Gordon J. I. 2008. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salzman N. H., de Jong H., Paterson Y., Harmsen H. J. M., Welling G. W., Bos N. A. 2002. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology 148:3651–3660 [DOI] [PubMed] [Google Scholar]
- 25. Rotimi V. O., Duerden B. I. 1981. The development of the bacterial flora in normal neonates. J. Med. Microbiol. 14:51–62 [DOI] [PubMed] [Google Scholar]
- 26. Martinez I., Wallace G., Zhang C., Legge R., Benson A. K., Carr T. P., Moriyama E. N., Walter J. 2009. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl. Environ. Microbiol. 75:4175–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waxman D. J. 1991. P450-catalyzed steroid hydroxylation: assay and product identification by thin-layer chromatography. Methods Enzymol. 206:462–476 [DOI] [PubMed] [Google Scholar]
- 28. Waxman D. J., Attisano C., Guengerich F. P., Lapenson D. P. 1988. Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 beta-hydroxylase cytochrome P-450 enzyme. Arch. Biochem. Biophys. 263:424–436 [DOI] [PubMed] [Google Scholar]
- 29. Harmsen H. J., Wildeboer-Veloo A. C., Grijpstra J., Knol J., Degener J. E., Welling G. W. 2000. Development of 16S rRNA-based probes for the Coriobacterium group and the Atopobium cluster and their application for enumeration of Coriobacteriaceae in human feces from volunteers of different age groups. Appl. Environ. Microbiol. 66:4523–4527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Usman, Hosono A. 2000. Effect of administration of Lactobacillus gasseri on serum lipids and fecal steroids in hypercholesterolemic rats. J. Dairy Sci. 83:1705–1711 [DOI] [PubMed] [Google Scholar]
- 31. Usman, Hosono A. 2001. Hypocholesterolemic effect of Lactobacillus gasseri SBT0270 in rats fed a cholesterol-enriched diet. J. Dairy Res. 68:617–624 [DOI] [PubMed] [Google Scholar]
- 32. Hamad E. M., Sato M., Uzu K., Yoshida T., Higashi S., Kawakami H., Kadooka Y., Matsuyama H., Abd El-Gawad I. A., Imaizumi K. 2009. Milk fermented by Lactobacillus gasseri SBT2055 influences adipocyte size via inhibition of dietary fat absorption in Zucker rats. Br. J. Nutr. 101:716–724 [DOI] [PubMed] [Google Scholar]
- 33. Thuny F., Richet H., Casalta J.-P., Angelakis E., Habib G., Raoult D. 2010. Vancomycin treatment of infective endocarditis is linked with recently acquired obesity. PLoS One 5:e9074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 35. Omura T., Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239:2370–2378 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.