

Summary
Prostatic acid phosphatase (PAP) is a tumor antigen in prostate cancer and the target of several anti-tumor vaccines in earlier clinical trials. Ultimately, the goal of anti-tumor vaccines is to elicit a sustainable immune response, able to eradicate a tumor, or at least restrain its growth. We have investigated plasmid DNA vaccines and have previously conducted a phase 1 trial in which patients with recurrent prostate cancer were vaccinated with a DNA vaccine encoding PAP. In this study, we investigated the immunologic efficacy of subsequent booster immunizations, and conducted more detailed longitudinal immune analysis, to answer several questions aimed at guiding optimal schedules of vaccine administration for future clinical trials. We report that antigen-specific cytolytic T-cell responses were amplified after immunization in 7 of 12 human leukocyte antigen-A2-expressing individuals, and that multiple immunizations seemed necessary to elicit PAP-specific interferon-γ-secreting immune responses detectable by enzyme-linked immunosorbent spot assay. Moreover, among individuals who experienced a ≥200% increase in prostate-specific antigen doubling time, long-term PAP-specific interferon-γ-secreting T-cell responses were detectable in 6 of 8, but in only 1 of 14 individuals without an observed change in prostate-specific antigen doubling time (P=0.001). Finally, we identified that immune responses elicited could be further amplified by subsequent booster immunizations. These results suggest that future trials using this DNA vaccine, and potentially other anti-tumor DNA vaccines, could investigate ongoing schedules of administration with periodic booster immunizations. Moreover, these results suggest that DNA vaccines targeting PAP could potentially be combined in heterologous immunization strategies with other vaccines to further augment PAP-specific T-cell immunity.
Keywords: DNA vaccine, prostate cancer, prostatic acid phosphatase, clinical trial, T-cell immune response monitoring
Anti-tumor vaccines, or active immunotherapies, seek to elicit immune responses within the host that recognize individual or multiple tumor-associated antigens to eliminate tumor cells expressing these antigens. Over the last 2 decades, there has been considerable evaluation of various anti-tumor vaccine approaches in preclinical models and human trials. Prostate cancer is one malignancy in particular where there have been significant efforts, due to several factors: (1) the disease is highly prevalent and the second leading cause of cancer-related death in men in the United States, and therefore a significant health concern for which new therapies are needed; (2) there is typically a long natural history and consequently a potentially long interval of time for the development of a therapeutic anti-tumor immune response; (3) the availability of a serum marker [prostate-specific antigen (PSA)] as a biochemical indicator of minimal residual disease; (4) the prostate is an expendable organ, and consequently autoimmune responses against normal prostate tissue are not a significant concern; and (5) several tissue-specific antigens have already been identified that might serve as targets for vaccines. Within the last decade, several antigen-specific vaccine approaches have been investigated in clinical trials for patients with prostate cancer targeting PSA,1-5 prostatic acid phosphatase (PAP),6,7 prostate-specific membrane antigen,8-10 and multiple carbohydrate antigens.11
Despite the significant effort in developing and evaluating anti-tumor vaccines, there is little consensus as to the “best” antigens to target and the optimal means of targeting these antigens. The prioritization of anti-tumor vaccine antigens, in fact, has been the focus of recent efforts led by the National Cancer Institute.12 Most vaccine approaches targeting individual antigens have focused on eliciting CD8+ T cells, as these are part of the adaptive arm of the immune system with direct cytolytic activity. Consequently, to specifically elicit CD8+ T cells, most approaches have used antigen-loaded antigen-presenting cellular vaccines13-15 or genetic vaccines using viral vaccines or naked DNA plasmids.3-5,8,10 Of these particular approaches, DNA vaccines are generally believed to be a “weaker” immunization strategy given the absence of a concurrent inflammatory antiviral response, and the low level of in-vivo transfection of antigen-presenting cells that occurs after direct administration. However, we have been particularly interested in DNA vaccines as a simpler means of antigen-specific immunization given our experience in rodent models demonstrating that DNA vaccines can elicit antigen-specific CD8+ T-cell responses to an autologous antigen with repetitive immunization and without the need for heterologous immunization strategies.16,17 These findings suggest that repetitive immunization with DNA vaccines might lead to CD8+ anti-tumor T-cell responses as effectively as more cumbersome strategies. Moreover, several anti-tumor DNA vaccines have begun to enter clinical trials and have demonstrated immunologic efficacy and safety.18-20 However, with the continued clinical development of DNA vaccines many questions will need to be answered, such as the number of immunizations necessary to elicit effector CD8+ T cells, the durability of this response, and whether repetitive immunization continues to augment a productive anti-tumor immune response or ultimately leads to tolerant immune responses.
We have recently reported the results of a phase 1 trial in which patients with PSA-recurrent prostate cancer were immunized 6 times at biweekly intervals using a DNA vaccine encoding PAP.21 We demonstrated that after 6 immunizations, PAP-specific T-cell responses were detectable in 10 of 22 individuals. In this report, we have conducted more detailed longitudinal immune analysis designed to answer the following specific questions: (1) Were immune responses elicited early in the course of immunization, or only after multiple immunizations, suggesting that continued immunization might be necessary? (2) Did delayed immune responses occur months after an initial immunization course? (3) Were immune responses, once elicited, durable? and (4) Were the development of immune responses associated with possible clinical responses as determined by changes in PSA doubling time? In addition, 2 patients who previously developed PAP-specific CD8+ T-cell responses were further vaccinated to answer whether PAP-specific T-cell immune responses, once elicited, could be further augmented by subsequent booster immunizations.
MATERIALS AND METHODS
Patient Population, Study Agent, Regulatory Information, and Clinical Trial
We have previously reported the results of a phase 1 trial using a plasmid DNA vaccine, pTVG-HP, in patients with biochemically (serum PSA) recurrent prostate cancer after definitive surgery and/or radiation therapy not receiving androgen deprivation therapy.21 Cryopreserved samples from that trial were used for the analysis described. In addition, that trial was amended to permit the evaluation of booster immunizations using the same pTVG-HP vaccine. Specifically, 2 subjects who had previously been vaccinated, had evidence of a PAP-specific CD8+ T-cell response defined by interferon (IFN)γ enzyme-linked immunosorbent spot (ELISPOT) or T-cell proliferation after immunization, had no significant adverse events during the initial treatment, and who had otherwise stable disease not requiring additional therapy for prostate cancer, were permitted to participate. Subjects were treated at monthly intervals with 100 μg pTVG-HP admixed with 200 μg granulocyte macrophage colony-stimulating factor (GM-CSF; Leukine, sargramostim, Berlex Oncology, Bergenfield, NJ), and coadministered intradermally with a 28-guage needle and syringe as described earlier.21 Patients were evaluated for potential toxicity after every 2 vaccinations by examination and blood tests as reported earlier.21 Serum PSA was evaluated monthly for disease monitoring, and blood was obtained after every 2 booster immunizations for immunologic monitoring. The study protocol was reviewed and approved by all local (including Institutional Review Board of the University of Wisconsin and Biosafety Committee), sponsor (Human Subjects Review Board of the US Army), and federal (Food and Drug Administration, NIH Recombinant DNA Advisory Committee) entities. Both patients gave additional written informed consent for participation.
Peptide-specific T-cell Lines and Evaluation
Peptide-specific T-cell lines were generated by repetitive weekly in-vitro stimulations with peptide-loaded autologous dendritic cells (DC). Briefly, peripheral blood mononuclear cells (PBMC) from human leukocyte antigen (HLA)-A2-expressing individuals were plated at 108 cells/flask in T150 tissue culture flasks for 2 hours at 37°C. Nonadherent cells were then removed, and the adherent cells were cultured for 6 days in the presence of 20 ng/mL rhGM-CSF (R&D Systems, Minneapolis, MN) and 10 ng/mL rhIL-4 (R&D Systems). The resulting DC were then “matured” for 24 to 48 hours by culture in the presence of 150 ng/mL interleukin (IL)-6, 10 ng/mL IL-1β, 10 ng/mL tumor necrosis factor-α, and 1 μg/mL prostaglandin E2. Autologous DC were then pulsed with 10 μg/mL peptide [p18-26, p112-120, or p299-307 derived from PAP, or GILGFVFTL derived from the influenza A matrix protein (pFlu)] for 2 hours, and cultured with autologous T cells selected from PBMC (T-cell negative isolation kit, Dynal, Carlsbad, CA), and obtained before or 2 weeks after immunization, in T-cell medium (Roswell Park Memorial Institute 1640 medium supplemented with l-glutamine, penicillin/streptomycin, β-mercaptoethanol, and 10% human AB serum (Valley Biomedical, Winchester, VA). 20 U/mL rhIL-2 (R&D Systems) and 10 U/mL rhIL-7 (R&D Systems) were added after 24 hours. Cells were restimulated at weekly intervals with irradiated (3000 cGy) peptide-loaded antigen-presenting cells (autologous DC or HLA-A2-expressing B-cell lines) in similar fashion, and T-cell lines were characterized for cytolytic function after 2 to 5 weeks. Cytolytic activity was measured by lactate dehydrogenase release from target cell lines (Cytotox 96 Assay kit, Promega, Madison, WI). Specifically, effector cell lines were plated in 96-well plates at various effector-to-target (E:T) cell ratios. Targets used were T2 cells pulsed with specific peptide or an irrelevant HLA-A2-binding peptide. After 4- to 6-hours at 37°C, plates were centrifuged and 50 μL of culture supernatant was assessed for lactate dehydrogenase concentration spectrophotometrically, according to the manufacturer’s instructions. Controls included wells with effector cells only, wells with media only, target cells only (minimum release), and target cells with 1% Triton X-100 (maximum release). The optical density (OD) signal contributed by the media alone was subtracted from all values. The percent specific lysis was then calculated as: (ODexperimental well – ODeffector only–ODtarget minimum release)/(ODtarget maximum release–ODtarget minimum release). A “positive” cytotoxic T-lymphocyte (CTL) response was defined as the detection of a titratable lysis of peptide-specific T2 cells and significantly higher relative to irrelevant peptide-loaded T2 cells. The first week at which peptide-specific CTL were detected is recorded. Of note, pFlu-specific cultures and CTL analysis were only conducted with pretreatment PBMC specimens from selected patients.
IFNγ ELISPOT
Wells of nitrocellulose 96-well microtiter (ELISPOT) plates were coated with an anti-IFNγ capture monoclonal antibody (Endogen, Rockford, IL), according to the manufacturer’s instructions. Cryopreserved PBMC, obtained at various times before or after vaccination, were then thawed and cultured directly (without in-vitro stimulation) in T-cell medium in these nitrocellulose microtiter (ELISPOT) plates at 2×105 cells/well. Cultures were allowed to proceed for 48- to 72-hours in the presence of media only (no antigen), 2 μg/mL PAP protein (Research Diagnostics Inc, Flanders, NJ), 2 μg/mL PSA protein (Research Diagnostics Inc, negative control), 250 ng/mL tetanus toxoid (Calbiochem, San Diego, CA), or 5 μg/mL phytohemaglutinin (positive mitogenic control, Fisher, Pittsburgh, PA) in 3- to 6-well replicates. ELISPOT plates were then washed and probed for 1.5 hours with a biotinylated anti-IFNγ antibody (Endogen). After incubation, wells were washed, incubated with streptavidin-labeled alkaline phosphatase for 1 hour, and then developed with 5-bromo 4-chloro 3-indoylphosphate/nito blue tetrazolium chloride colorimetric substrate (BioRad, Hercules, CA) for 15 to 30 minutes. The number of spots per well was determined with an automated ELISPOT reader and normalized to 106 starting PBMC. The mean number of spots detected under media-only conditions at each time point was subtracted from the antigen-specific conditions to enumerate antigen-specific IFNγ spot-forming units±SD. Comparison of experimental wells with control no antigen wells was performed using a 2-tailed t test, with P-value less than 0.05 used to define a significant T-cell response. Data from samples for which the phytohemaglutinin-positive control was not significantly positive were not considered viable or adequate for interpretation; however, these data are included for completeness.
Antigen-specific T-cell Proliferation
CD4+ and CD8+ T-cell proliferation in response to antigen stimulation was determined by a 5-day bromodeoxyuridine (BrdU) incorporation assay, using the same methodology and individual lots of test antigens as described earlier.21 For analysis, a proliferation index was determined as the percentage of BrdU+ events for each antigen-stimulated condition divided by the percentage of BrdU+ events with the media-only control. BrdU+ events were assessed at each time point by gating 0.05% events in the no antigen (media only) control group, and applying that gating to all antigen-stimulated conditions.
Pentamer Staining
Cryopreserved PBMC from multiple time points were thawed, resuspended in T-cell medium, and cultured for 1 week with HLA-A2-derived epitopes p112-120 (TLMSAMTNL) and p299-307 (ALDVYNGLL), as well as an HLA-A2-binding PAP-derived nonspecific control peptide p135-143 (ILLWQPIPV). IL-2 was added to 10 IU/mL on the second day of culture. After 1 week, cell cultures were stained for CD3, CD8, and pentamers (ProImmune, Inc, Bradenton, FL) specific for each peptide (versus isotype control) according to the manufacturer’s instructions. The percentage of pentamer-labeled cells at each time point was determined as: [(% CD3+ CD8+ Pentamer+) – (% CD3+ CD8+ Isotype control+)]/(% CD3+ CD8+).
RESULTS
DNA Immunization Augments the Frequency of Antigen-specific Cytolytic T Cells
We have recently reported the results of a phase 1 trial of a DNA vaccine encoding PAP in patients with biochemical recurrence of prostate cancer. In that report, we identified that 10 of 22 patients had evidence of a T-cell response specific for PAP at the conclusion of the 12-week immunization period as evidenced by antigen-specific T-cell proliferation and/or CD8+ IFNγ ELISPOT. To further characterize the resulting immune response, and determine whether cytolytic T cells were specifically amplified after immunization, we analyzed the 12 HLA-A2+ subjects for the presence of PAP epitope-specific T cells22. PBMC were cultured in the presence of 3 HLA-A2-specific peptides derived from the amino-acid sequence of PAP (p18-26, p112-120, and p299-307) and evaluated after weekly in vitro stimulations with peptide for the presence of peptide-specific CTL (Fig. 1). CTL could be detected specific for pFlu after as few as 2 weekly in-vitro stimulations in the majority of patients before immunization, demonstrating the viability of the cells and robustness of the methodology (Fig. 1D). As demonstrated, CTL specific for p18-26 (Fig. 1A), p112-120 (Fig. 1B), and p299-307 (Fig. 1C), previously identified as HLA-A2-specific PAP epitopes, could be detected in multiple individuals after as few as 2 weekly in-vitro stimulations from PBMC obtained after immunization but not from PBMC obtained before immunization. Although these methods are not strictly quantitative, they suggest that the frequency of these cells were augmented after DNA immunization, given that fewer in-vitro stimulations were necessary for detection after immunization.
FIGURE 1.
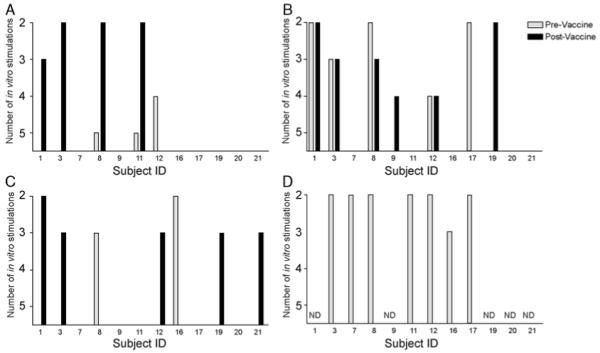
DNA immunization augments the frequency of antigen-specific cytolytic T cells: peripheral blood mononuclear cells from 12 HLA-A2 expressing patients collected before immunization (grey) or after 6 immunizations (black) were cultured in the presence of HLA-A2 epitopes derived from the amino-acid sequence of prostatic acid phosphatase (p18-26, panel A; p112-120, panel B; and p299-307, panel C) or an influenza control 9-mer HLA-A2-specific epitope (GILGFVFTL, panel D). Beginning after 2 weekly in-vitro stimulations, cultures were evaluated for the presence of peptide-specific CTL. Columns indicate the first stimulation during which peptide-specific CTL were detectable. In panel D, CTL analysis was only conducted with pretreatment specimens (grey bars), and with selected patients. CTL indicates cytotoxic T lymphocytes; HLA, human leukocyte antigen; ND, patient samples not assessed.
Several DNA Immunizations Seemed Necessary to Elicit PAP-specific T Cells
In the earlier clinical trial, we observed that approximately half of the patients vaccinated did not have evidence of PAP-specific T cells at the completion of the immunization series. We consequently questioned whether this was potentially because of an inadequate number of immunizations, reasoning that if immune responses were detectable only later in the course of immunization, that potentially 6 immunizations with this DNA vaccine was an insufficient number of immunizations for the majority of patients. PBMC were available pretreatment, and after 2, 4, or 6 immunizations from multiple individuals, and PAP-specific T cells from these time points were assessed by IFNγ ELISPOT without in-vitro stimulation. By this methodology, 4 subjects had evidence of significant PAP-specific IFNγ-secreting T cells after beginning the immunization series and not at baseline (Fig. 2). As demonstrated in Figure 2, responses tended to increase with the number of immunizations, and the majority of significant PAP-specific T cells were detectable only after 6 immunizations.
FIGURE 2.
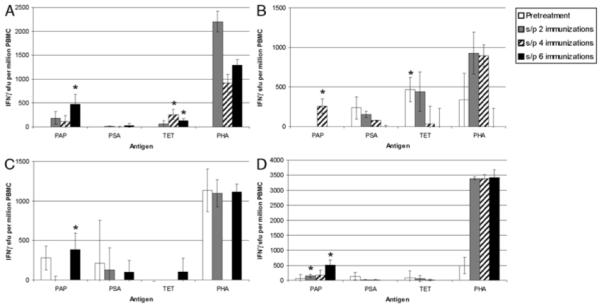
Several DNA immunizations were necessary to elicit PAP-specific T cells: PBMC were collected from 4 patients (panel A, B, C, and D) before immunization, and after 2, 4, or 6 biweekly immunizations. Samples from one of the time points (preimmunization, panel A; and after 4 immunizations, panel C) were not available for 2 of these patients. PBMC were cultured in the presence of PAP protein, prostate-specific antigen (PSA) protein (negative control), tetanus toxoid (TET; immunization control), phytohemaglutinin (PHA; nonspecific mitogenic positive control), or media only for 48 hours, and IFNγ-secreting T cells under each culture condition were enumerated by enzyme-linked immunosorbent spot. Shown are the mean number and SD of antigen-specific IFNγ sfu (number of antigen-specific sfu–number of media-only sfu) per 106 cells at each time point (from quadruplicate assays) for individual patients. Asterisks demonstrate significant responses (P < 0.05, t test) of antigen-specific sfu compared with media only. IFN indicates interferon; PAP, prostatic acid phosphatase; PBMC, peripheral blood mononuclear cells; sfu, spot-forming units.
DNA Immunization Elicited Durable PAP-specific T-cell Immunity in Some Subjects
We next questioned whether immune responses once elicited were durable, or initially detectable, over a year of follow-up. We reasoned that potentially beneficial immune responses, those that might be associated with a better clinical outcome, should be durable. PBMC were available after 3, 6, 9, and/or 12 months from all 22 subjects, and the presence of PAP-specific IFNγ-secreting T cells were assessed by IFNγ ELISPOT as shown above. Of 8 individuals previously reported to have experienced a ≥200% increase in PSA doubling time in the year of follow-up after vaccination (Table 1),21 6 were found to have PAP-specific IFNγ-secreting immune responses at least twice in this year period (Figs. 3A–H). Although some immune responses detectable immediately after immunization seemed durable (Fig. 3B), others seemed to wane over time (Fig. 3I), and others seemed to develop months after the immunization was completed (Figs. 3D, E). Of the 14 individuals who did not experience an increase in PSA doubling time, only 1 was found to have detectable PAP-specific IFNγ-secreting immune responses at least twice in the year of follow-up (Fig. 3J). The association of durable immune responses, defined here as responses detectable at least twice in the 4 quarterly PBMC specimens, was highly associated with a ≥200% increase in PSA doubling time over this same period compared with preimmunization (P=0.001, χ2 test).
TABLE 1.
PSA Doubling Times and Detectable Immune Responses
| PSA Doubling Time in Months (No. Months, No. PSA Values Used for Calculation) |
Immune Response |
||||
|---|---|---|---|---|---|
| Patient No. | Pretreatment | On-treatment | Posttreatment | After Treatment21 | In 12-mo Follow-up |
| 1 | 10.3 (6, 5) | 7.7 (3, 4) | 8.4 (12, 8) | X | |
| 2 | 15.3 (6, 5) | −26 (3, 4) | 13.3 (12, 8) | ||
| 3 (Booster) | 28 (7, 4) | 114 (3, 4) | 57 (12, 6) | X | X |
| 4 | 4.6 (6, 4) | 6.8 (3, 4) | 4.4 (6, 6) | ||
| 5 | 16.2 (7, 4) | −40.3 (3, 4) | 70.7 (12, 6) | X | |
| 6 | 6.9 (6, 4) | 8.2 (3, 4) | 5.5 (12, 7) | X | X |
| 7 | 6.9 (7, 4) | 30.6 (3, 4) | 24.3 (12, 6) | X | |
| 8 | 3.1 (2, 4) | 7.5 (3, 4) | 7.8 (12, 6) | X | |
| 9 | 4.2 (7, 5) | 5.5 (3, 4) | 5.6 (7, 5) | X | |
| 10 | 3.1 (7, 7) | 4.5 (3, 4) | 3 (7, 6) | X | |
| 11 | 30.1 (6, 5) | 28.4 (3, 3) | −198.5 (12, 5) | ||
| 12 | 12.5 (6, 6) | 6 (3, 3) | 20.1 (12, 5) | ||
| 13 (Booster) | 13.9 (10, 4) | 6.1 (3, 3) | 10.5 (6, 3) | X | |
| 14 | 5.8 (6, 5) | 4.1 (3, 4) | −105 (12, 6) | X | |
| 15 | 3.3 (6, 9) | 3.5 (3, 3) | 3.9 (1, 2) | ||
| 16 | 4.2 (5, 5) | 8.6 (3, 4) | 6.4 (7, 5) | X | |
| 17 | 5 (6, 5) | 5.5 (3, 4) | 3.8 (3, 4) | X | |
| 18 | 3 (4, 6) | 11 (3, 4) | 11.4 (12, 7) | X | X |
| 19 | 11.8 (6, 5) | 8.7 (3, 4) | 34 (12, 6) | ||
| 20 | 5.6 (6, 8) | 6.1 (3, 4) | 5.9 (6, 6) | ||
| 21 | 2.5 (6, 4) | 2.5 (3, 4) | 2 (2, 4) | X | |
| 22 | 13 (6, 6) | 23.4 (3, 4) | 9.8 (12, 6) | ||
PSA indicates prostatic acid phosphatase.
PSA doubling times (DT) were calculated for all 22 individuals treated, as previously reported.26 Shown in the table are the PSA DT (in months) for the periods of time pre-treatment, over the 3 months receiving immunization, and post-treatment. All PSA values available were used for the calculation of PSA DT. Values in parentheses include the number of months over which the PSA DT was calculated, and the number of PSA measurements used for the calculation. PSA DT during the on-treatment period or post-treatment period >200% of the pre-treatment period are bolded. PSA DT in the post-treatment period was calculated for 12 months, or until the patient began other therapy. Immune responses detectable immediately after immunization, defined as antigen-specific CD8+ IFNγ responses or T-cell proliferative responses, are shown, as previously reported.26 PAP-specific immune responses detectable by IFNγ ELISPOT at least twice in one-year follow-up (Fig. 3) are shown in the last column.
FIGURE 3.
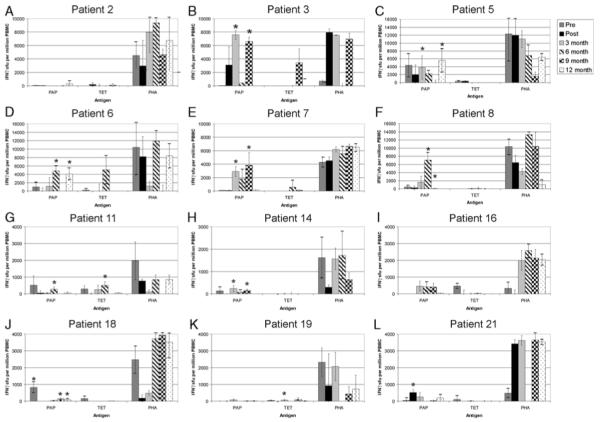
DNA immunization elicited long-term PAP-specific T-cell immunity in multiple subjects: PBMC were collected before immunization, 2 weeks after 6 immunizations (post), and at 3-month intervals thereafter from patients observed to have a ≥200% increase in prostate-specific antigen (PSA) doubling time (panels B, C, E-H, J, K), and representative other patients (panels A, D, I, L). PBMC were cultured in the presence of PAP protein, TET, or PHA as in Figure 2 for 48 to 72 hours and were evaluated for antigen-specific immune responses by IFNγ enzyme-linked immunosorbent spot. Shown are the mean and SD of quadruplicate determinations of IFNγ sfu at each time point. Asterisks denote significant responses (P < 0.05, t test) of antigen-specific (PAP or TET-specific) sfu compared with media only. Data from individual samples for which the PHA-positive control was not demonstrably positive were not considered adequate for interpretation; however, these data are included for completeness. IFN indicates interferon; PAP, prostatic acid phosphatase; PBMC, peripheral blood mononuclear cells; PHA, phytohemaglutinin; sfu, spot-forming units; TET, tetanus toxoid.
DNA Immunization Can Augment PAP-specific T-cell Immunity Previously Elicited
As some immune responses were observed to wane over time, we next questioned whether immune responses, once elicited by DNA immunization, could be augmented with subsequent “booster” immunizations. The clinical protocol, described by McNeel et al,21 was modified to permit subsequent booster immunizations of subjects who had previous evidence of a PAP-specific CD8+ T-cell response after immunization, no evidence of adverse events, and otherwise stable prostate cancer not requiring other therapies. These immunizations were given at a dose of 100 μg plasmid DNA, intradermally, with 200 μg GM-CSF as adjuvant, at monthly intervals for 6 additional immunizations. Blood was collected after every 2 immunizations for immunologic monitoring. Two subjects chose to pursue this, of whom 1 (number 13) began 7 months after completing the initial immunization course. This particular subject, although he had evidence of a CD8+ immune response specific for PAP immediately after the initial immunization course by one assay (CD8+ ELISPOT), had no evidence of an immune response at any other posttreatment time point, and did not demonstrate an increased immune response after the booster immunizations (data not shown). The other subject (number 3), had evidence of PAP-specific T cells after the initial immunizations by multiple methods (Figs. 1A, C, Fig. 2B).21 This subject received monthly booster immunizations beginning 19 months after the initial immunization series. Immediately before receiving this second course, no PAP-specific T cells could be detected by antigen-specific T-cell proliferation (Fig. 4A). By as few as 2 booster immunizations, however, PAP-specific CD4+ and CD8+ T cells were detectably augmented (Fig. 4A). As this particular subject was HLA-A2+, pentamer staining was performed to directly assess for PAP-specific CD8+ T cells. As shown in Figure 4B, PAP-specific CD8+ T cells were clearly augmented shortly after the initial immunization series and the booster immunizations months later. This particular individual remains in long-term follow-up, and to date has required no other therapy for prostate cancer (Fig. 4C). No subject experienced any adverse events greater than grade 1 during the 6-month booster immunization period.
FIGURE 4.
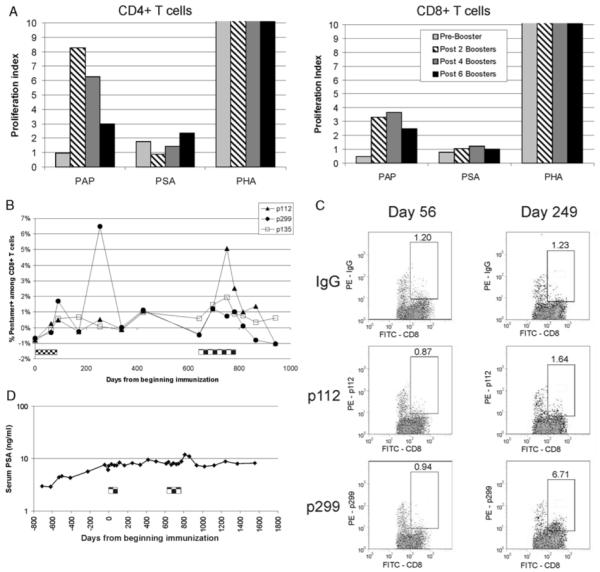
DNA immunization can augment PAP-specific T-cell immunity previously elicited. Panel A: PBMC were collected immediately before, and 2 weeks after 2, 4, or 6 monthly booster immunizations. Cells were cultured in the presence of PAP, PSA, phytohemaglutinin (PHA), or media only, and assayed for proliferation after 5 days by BrdU incorporation. Shown are the fold increases of CD4/8+BrdU+ events (proliferative index) after antigen-specific culture over cells cultured in media only. Panel B: PBMC collected at multiple time points before and after initial immunization and booster immunization (indicated by checkered boxes) were thawed, stimulated in-vitro with peptides for 7 days, and then assayed for peptide-specific CD8+ T cells by pentamer staining. Shown is the percentage of pentamer+ cells among total CD8+ T cells. Panel C: For pentamer staining, samples were gated on CD3+ CD8+ events, with lymphocyte side scatter. Shown are representative staining dot plots from two of the time points indicated; x axis is CD8 staining, nd y axis is pentamer or lgG isotype control. The numbers indicate the percentage of events among CD3+ CD8+ cells. Panel D: Serum PSA values from this same patient. The timing of the initial immunization series and booster immunization series are indicated by the checkered boxes. BrdU indicates bromodeoxyuridine; IFN, interferon; PAP, prostatic acid phosphatase; PBMC, peripheral blood mononuclear cells; PSA, prostate-specific antigen.
DISCUSSION
We have previously reported the results of a clinical trial in which patients with biochemically recurrent prostate cancer were immunized 6 times at 2-week intervals with a DNA vaccine encoding PAP.21 Ultimately, the goal of antitumor vaccines is to elicit a sustainable immune response able to eradicate or restrain a tumor. Hence, most clinical vaccine trials have used schedules of immunization, informed by animal studies, conducted over a limited period of time with the hope of eliciting durable anti-tumor immune responses. This approach is in contrast to most current targeted molecular pharmacologic approaches to the treatment of cancer in which it is generally assumed that chronic therapy is necessary. In addition, as most preclinical immunization studies are performed in inbred rodent strains, estimations of appropriate vaccine schedules may not be uniformly applicable to human trials. Moreover, the means of immunization may greatly affect optimal schedules of administration. To date, despite anti-tumor vaccines having entered phase 3 clinical trials, there has been very little evaluation of whether the number and schedule of immunizations is sufficient to elicit an immune response that is potentially therapeutic and durable. This leaves questions about whether prolonged schedules of administration should be considered, or booster immunizations should be used. In addition, there have been concerns that repetitive immunization might lead to counterproductive tolerant immune responses.23 In an earlier vaccine trial, patients were immunized 6 times at 2-week intervals with a DNA vaccine encoding PAP, a schedule that we had investigated earlier in rats.16 The current analysis was conducted to answer several questions aimed at guiding the schedule of vaccine administration to be used in future clinical trials with this particular DNA vaccine. Although the number of samples evaluable was relatively small, we sought to determine the following:
At what point in the series of 6 vaccinations were immune responses detectable? This was aimed primarily at answering whether 6 vaccinations were sufficient, or whether ongoing immunization should be considered.
Were immune responses, once elicited, durable, and/or were immune responses only detectable months later?
Was the development of a durable immune response associated with beneficial changes in PSA kinetics?
Once immunized, could booster immunizations with the same DNA vaccine subsequently augment an antigen-specific T-cell response.
Using IFNγ ELISPOT as a measure of PAP-specific T cells, we found that in those individuals in whom PAP-specific CD4+ and/or CD8+ T-cell responses were elicited over the course of immunization, the majority of responses were elicited late in the course of DNA immunization. This was similar to what we observed earlier in rats in which although immune responses to the human PAP xenoantigen were elicited after as few as 2 vaccinations, more vaccinations were necessary with a vaccine encoding the rat PAP self-antigen to elicit responses.17 This finding suggests that it could be advantageous to continue immunization beyond 6 initial vaccinations, using DNA immunization alone without a heterologous immunization strategy. Alternatively, it is possible that some individuals were tolerant to PAP at baseline and might not develop detectable PAP-specific T cells. Future studies will explore continued immunization, beyond 6 initial vaccinations, to evaluate these possibilities. However, as suggested earlier, these types of studies must be performed with some caution given that genetic vaccines delivered at frequent intervals over prolonged periods may elicit tolerant responses, and have in fact been investigated by other groups as a means of inducing T-cell tolerance.24
We have identified earlier that rats immunized multiple times with the same DNA vaccine encoding human PAP developed PAP-specific memory CD4+ and CD8+ T cells detectable by antigen-specific T-cell proliferation for at least 1 year after immunization (data not shown). Although in that system the human PAP is a xenoantigen, we similarly identified that several patients had long-lived responses detectable many months after immunization (Fig. 3). Although such findings complicate clinical trial evaluation, as it is difficult to define an appropriate time to measure immune responses, they suggest that immune monitoring at static time points may be insufficient to characterize what is likely a more dynamic process. This may be one reason why many vaccine trials to date have failed to demonstrate an association between the development of an immunologic response and a clinical response. In our previous trial, patients were enrolled with biochemical (serum PSA) recurrence only, and we identified that 8 patients experienced a ≥200% increase in PSA doubling time from pretreatment to the 1-year period of time after treatment.21 We also identified that 10 of 22 patients had a PAP-specific response detectable immediately after immunization, although this was not associated with changes in PSA doubling time.21 Although changes in PSA doubling time have to be interpreted with caution, because this has not been validated prospectively as a clinical trial endpoint, changes in PSA kinetics remain the only marker of possible clinical response in this population of patients with early recurrent disease. In this study, we identified that changes in PSA doubling time were highly associated with the development of late/durable immune responses. Future clinical trials will evaluate whether the durability of PAP-specific T cells, and effector and memory T-cell immune responses in particular, are positively associated with clinical measures such as time to disease progression.
Finally, our studies demonstrate that immune responses to PAP can be subsequently augmented with the same DNA vaccine encoding PAP. In one individual, PAP-specific responses were detected after as few as 2 booster immunizations in terms of proliferative T-cell responses and specific CD8+ T-cell populations. Of note, this particular individual was treated with only 100 μg of plasmid DNA for each immunization. Although the immunization of a single patient is anecdotal, it does provide evidence that comparable doses of plasmid DNA as used in rodent studies, not dosed per weight, can be immunologically effective in human immunization. A similar observation that higher plasmid DNA doses are not necessarily immunologically superior to lower doses was recently reported in a phase 2 DNA vaccine trial for patients with multiple sclerosis.25 Our results suggest that ongoing boosters may have led to decreased antigen-specific proliferative responses, however, this again remains anecdotal. In any case, our findings demonstrate that antigen-specific T-cell immune responses, having been elicited earlier, can be augmented with a DNA vaccine, although optimal schedules of vaccination may need to be evaluated. The specific finding that this DNA vaccine encoding PAP can augment PAP-specific T-cell immunity may be of particular relevance given that a vaccine being evaluated by Dendreon Corporation, sipuleucel-T, is targeting the same PAP antigen and has demonstrated clinical benefit in terms of improving patients’ overall survival.7,26 An “off-the-shelf ” DNA vaccine targeting the PAP antigen could potentially be investigated in combination with that vaccine to maintain or augment long-term PAP-specific T-cell immunity.
Acknowledgments
This study was supported by NIH (K23 RR16489), by the NIH National Gene Vector Laboratory Program, by the NIH NCRR Clinical and Translational Science Award (CTSA) program (1UL1RR025011), and by the US Army Medical Research and Materiel Command Prostate Cancer Research Program (W81XWH-05-1-0404).
Footnotes
All authors have declared that there are no financial conflicts of interest in regards to this study.
REFERENCES
- 1.Meidenbauer N, Harris DT, Spitler LE, et al. Generation of PSA-reactive effector cells after vaccination with a PSA-based vaccine in patients with prostate cancer. Prostate. 2000;43:88–100. doi: 10.1002/(sici)1097-0045(20000501)43:2<88::aid-pros3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 2.Perambakam S, Hallmeyer S, Reddy S, et al. Induction of specific T cell immunity in patients with prostate cancer by vaccination with PSA146-154 peptide. Cancer Immunol Immunother. 2006;55:1033–1042. doi: 10.1007/s00262-005-0090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eder JP, Kantoff PW, Roper K, et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res. 2000;6:1632–1638. [PubMed] [Google Scholar]
- 4.Gulley J, Chen AP, Dahut W, et al. Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate. 2002;53:109–117. doi: 10.1002/pros.10130. [DOI] [PubMed] [Google Scholar]
- 5.Pavlenko M, Roos AK, Lundqvist A, et al. A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer. 2004;91:688–694. doi: 10.1038/sj.bjc.6602019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fong L, Brockstedt D, Benike C, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. 2001;167:7150–7156. doi: 10.4049/jimmunol.167.12.7150. [DOI] [PubMed] [Google Scholar]
- 7.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 8.Mincheff M, Tchakarov S, Zoubak S, et al. Naked DNA and adenoviral immunizations for immunotherapy of prostate cancer: a phase I/II clinical trial. Eur Urol. 2000;38:208–217. doi: 10.1159/000020281. [DOI] [PubMed] [Google Scholar]
- 9.Murphy G, Tjoa B, Ragde H, et al. Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate. 1996;29:371–380. doi: 10.1002/(SICI)1097-0045(199612)29:6<371::AID-PROS5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 10.Wolchok JD, Gregor PD, Nordquist LT, et al. DNA vaccines: an active immunization strategy for prostate cancer. Semin Oncol. 2003;30:659–666. doi: 10.1016/s0093-7754(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 11.Slovin SF, Scher HI. Peptide and carbohydrate vaccines in relapsed prostate cancer: immunogenicity of synthetic vaccines in man–clinical trials at Memorial Sloan-Kettering Cancer Center. Semin Oncol. 1999;26:448–454. [PubMed] [Google Scholar]
- 12.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy GP, Tjoa BA, Simmons SJ, et al. Phase II prostate cancer vaccine trial: report of a study involving 37 patients with disease recurrence following primary treatment. Prostate. 1999;39:54–59. doi: 10.1002/(sici)1097-0045(19990401)39:1<54::aid-pros9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 14.Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–3903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 15.Small EJ, Schellhammer PF, Higano CS, et al. Results of a placebo-controlled phase III trial of immunotherapy with APC8015 for patients with hormone refractory prostate cancer (HRPC) Proc Am Soc Clin Oncol. 2005;23:4500. [Google Scholar]
- 16.Johnson LE, Frye TP, Arnot AR, et al. Safety and immunological efficacy of a prostate cancer plasmid DNA vaccine encoding prostatic acid phosphatase (PAP) Vaccine. 2006;24:293–303. doi: 10.1016/j.vaccine.2005.07.074. [DOI] [PubMed] [Google Scholar]
- 17.Johnson LE, Frye TP, Chinnasamy N, et al. Plasmid DNA vaccine encoding prostatic acid phosphatase is effective in eliciting autologous antigen-specific CD8+ T cells. Cancer Immunol Immunother. 2007;56:885–895. doi: 10.1007/s00262-006-0241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gnjatic S, Altorki NK, Tang DN, et al. NY-ESO-1 DNA vaccine induces T-cell responses that are suppressed by regulatory T cells. Clin Cancer Res. 2009;15:2130–2139. doi: 10.1158/1078-0432.CCR-08-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tagawa ST, Lee P, Snively J, et al. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer. 2003;98:144–154. doi: 10.1002/cncr.11462. [DOI] [PubMed] [Google Scholar]
- 20.Conry RM, Curiel DT, Strong TV, et al. Safety and immunogenicity of a DNA vaccine encoding carcinoembryonic antigen and hepatitis B surface antigen in colorectal carcinoma patients. Clin Cancer Res. 2002;8:2782–2787. [PubMed] [Google Scholar]
- 21.McNeel DG, Dunphy EJ, Davies JG, et al. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27:4047–4054. doi: 10.1200/JCO.2008.19.9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olson BM, Frye TP, Johnson LE, et al. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer Immunol Immunother. 2010;59:943–953. doi: 10.1007/s00262-010-0820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacelle MG, Jensen SM, Fox BA. Partial CD4 depletion reduces regulatory T cells induced by multiple vaccinations and restores therapeutic efficacy. Clin Cancer Res. 2009;15:6881–6890. doi: 10.1158/1078-0432.CCR-09-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrera F, La Cava A, Rizzi M, et al. Gene vaccination for the induction of immune tolerance. Ann N Y Acad Sci. 2007;1110:99–111. doi: 10.1196/annals.1423.012. [DOI] [PubMed] [Google Scholar]
- 25.Garren H, Robinson WH, Krasulova E, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol. 2008;63:611–620. doi: 10.1002/ana.21370. [DOI] [PubMed] [Google Scholar]
- 26.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]