

Abstract
Radiolabeled Pittsburgh compound B (PIB) is a benzothiazole imaging agent that usually binds with high affinity, specificity, and stoichiometry to cerebral β-amyloid (Aβ) in patients with Alzheimer’s disease. Among a cohort of ten AD subjects examined postmortem, we describe a case of idiopathic, end-stage Alzheimer’s disease with heavy Aβ deposition yet substantially diminished high-affinity binding of 3H-PIB to cortical homogenates and unfixed cryosections. Cortical tissue samples were analyzed by immunohistochemistry, electron microscopy, ELISA, immunoblotting, MALDI-TOF mass spectrometry, in vitro 3H-PIB binding and 3H-PIB autoradiography. The PIB-refractory subject met the histopathological criteria for AD. However, cortical tissue from this case contained more vascular β-amyloidosis, higher levels of insoluble Aβ40 and Aβ42, and a higher ratio of Aβ40:Aβ42 than did tissue from the nine comparison AD cases. Furthermore, cerebral Aβ from the PIB-refractory subject displayed an unusual distribution of low- and high-molecular weight Aβ oligomers, as well as a distinct pattern of N- and C-terminally truncated Aβ peptides in both the soluble and insoluble cortical extracts. Genetically, the patient was apolipoprotein-Eε4 heterozygous, and exhibited no known AD-associated mutations in the genes of the β-amyloid precursor protein, presenilin-1 or presenilin-2. Our findings suggest that PIB may differentially recognize polymorphic forms of multimeric Aβ in humans with Alzheimer’s disease. In addition, while the prevalence of PIB-refractory cases in the general AD population remains to be determined, the paucity of high-affinity binding sites in this AD case cautions that minimal PIB retention in positron-emission tomography scans of demented patients may not always rule out the presence of Alzheimer-type Aβ pathology.
Keywords: Alzheimer, amyloid-beta, cerebral amyloid angiopathy, diagnosis, imaging, PIB, senile plaques
Introduction
Radiolabeled Pittsburgh compound B (PIB; [N-methyl-11C]2-(4′-methylaminophenyl)-6-hydroxybenzothiazole) is a promising in vivo imaging agent that can bind with high affinity, specificity and stoichiometry to parenchymal amyloid-β deposits [13, 23, 30] and cerebral β-amyloid angiopathy (CAA) [2, 11] in patients with Alzheimer’s disease (AD). Positron-emission tomography (PET) imaging studies indicate that 11C-labeled PIB rapidly enters the brain, where it is retained significantly longer in Aβ-rich regions than in brain areas that lack Aβ deposits [13, 23]. Thus, PIB may be a promising tool for the early diagnosis of AD plaque pathology in living patients [21, 24].
Evaluation of post-mortem or biopsied brain samples from patients who exhibited cortical PIB retention in PET scans indicates that PIB binding in vivo correlates with heavy cerebral Aβ deposition [2, 10, 16]. However, some instances of unexpectedly high uptake in nondemented cases [30], or unexpectedly low retention in clinically diagnosed AD cases [5, 13, 18, 22], have been reported, and some Aβ lesions appear to be refractory to PIB binding [10, 31]. Studies in vitro and in animal models furnish additional evidence that, at nanomolar concentrations similar to those achieved in PET imaging, PIB does not bind to all multimeric assemblies of Aβ equally [14, 29, 31]. Specifically, the density of high-affinity PIB binding sites per molecule of Aβ is much lower in β-amyloid precursor protein (APP)-transgenic mice [14] and in normal aged nonhuman primates [29] compared to AD patients, despite profuse cerebral Aβ deposition in the animal models.
These experiments suggest that PIB may recognize a polymorphic molecular site on multimeric Aβ that is enriched in human AD cases but is altered or concealed in nonhuman species [17, 29]. The occasionally weak correlations between PIB binding and Aβ levels [5, 13, 18, 22, 29, 30] and the presence of poorly binding lesions in some brain regions [10, 31] suggest the possible existence of polymorphic Aβ aggregates in humans as well. Here we report a case of clinically and pathologically confirmed AD with heavy Aβ burden yet strikingly reduced high-affinity PIB binding in vitro compared to a series of nine other AD cases. This finding raises the possibility that some cases of clinicopathologically verifiable AD might not show significant retention of radiolabeled PIB in diagnostic PET scans, and further suggests that PIB can discriminate among naturally-occurring, polymorphic forms of multimeric Aβ in humans.
Materials and Methods
Subject
A 72 year-old woman (case AD1) came to autopsy with a diagnosis of probable Alzheimer’s disease. Ten years earlier she had presented with impairments in short-term memory and visuospatial function. Both began insidiously and slowly worsened with time, and were followed approximately four years later by slowing of gait and increasingly frequent urinary incontinence. She experienced a gradual worsening of cognition in the subsequent years, in addition to worsening gait disturbance and incontinence. Nine years after her initial symptoms, she underwent ventriculoperitoneal shunt placement for possible normal pressure hydrocephalus. Her family reported transient improvement in her gait and incontinence, which then regressed within one month of the shunt placement, likely due to shunt malfunction. Subsequent shunt revisions did not improve her symptoms, and she died shortly thereafter. Aside from her mother, who had Parkinson’s disease but normal cognition, she had no known family history of dementia or other neurodegenerative diseases.
At autopsy, the brain weighed 1110 grams and showed no significant atrophy over the convexity. Vessels of the circle of Willis showed mild-to-moderate, patchy atherosclerosis with 50% occlusion of the right middle cerebral artery. A lesion approximately 1 cm in diameter was noted in the right frontal cortex corresponding to the site of entry of the ventricular shunt, and an old infarct approximately 1.5 cm in diameter was identified in the right medial posterior parieto-occipital cortex. The lateral and third ventricles were markedly dilated, and the hippocampus, amygdala and entorhinal cortex showed moderate atrophy. The caudate nucleus was slightly flattened, but the substantia nigra was normally pigmented. Histopathologically, the case was assigned a Braak AD stage of 6 (Table 1); the apolipoprotein E genotype was ApoE3/4.
Table 1.
Case List.
| Case | Group | Age | Sex | PMI (hr) | Braak stage | ApoE type |
|---|---|---|---|---|---|---|
| AD1 | AD | 72 | f | 3 | VI | 3/4 |
| AD2 | AD | 61 | m | 5.5 | V/VI | 3/4 |
| AD3 | AD | 75 | f | 12 | V/VI | 4/4 |
| AD4 | AD | 91 | f | 2.5 | V/VI | 3/4 |
| AD5 | AD | 61 | m | 4 | V or VI | 3/4 |
| AD6 | AD | 64 | f | 4.5 | VI | 3/4 |
| AD7 | AD | 87 | f | 6 | V/VI | 3/4 |
| AD8 | AD | 84 | m | 4.5 | VI | 3/4 |
| AD9 | AD | 81 | f | 2 | V/VI | 3/3 |
| AD10 | AD | 57 | f | 20 | V/VI | 3/4 |
| AD11* | AD | 86 | f | 2.5 | V | 3/3 |
| AD12* | AD | 74 | f | 2.8 | VI | 2/4 |
| AD13* | AD | 84 | f | 3.5 | VI | 3/4 |
| ND1 | Ctrl | 40 | m | 31 | 0 | 3/4 |
| ND2 | Ctrl | 57 | f | 17 | 0 | 3/3 |
| ND3 | Ctrl | 75 | f | 6 | 0 | 3/3 |
| ND4* | Ctrl | 78 | m | 1.6 | 0 | 3/3 |
Only included in MALDI-TOF MS analysis
PMI: Postmortem interval (hours)
ApoE type: Apolipoprotein E genotype
Comparison subjects
Nine additional, clinically and pathologically confirmed AD cases and three age-matched, nondemented subjects were analyzed for comparison with AD1 (Table 1) in the core analysis. These subjects were part of an earlier comparative study of Aβ in humans and nonhuman primates [29]. Tissue samples from three additional clinically and pathologically confirmed AD cases (which were verified to be PIB-sensitive by in vitro 3HPIB binding analysis) and one age-matched control (Table 1) were used as comparison subjects only in the MALDI-TOF mass spectrometry analyses (below).
Tissue preparation
Bilateral tissue slabs from the core subjects were dissected from the superior temporal gyrus (AD, n=10; ND, n=3) and the paracalcarine occipital cortex (AD, n=7; ND, n=3). The samples from one hemisphere were quickly frozen and stored at −80°C. Frozen tissue later was weighed and Dounce-homogenized in 5 volumes of Tris-buffered saline (TBS) homogenization buffer (50mM Tris-HCl and 150mM NaCl containing complete protease inhibitor [Santa Cruz Biochemicals, Santa Cruz, CA, USA]). Aliquots of the homogenates were stored at −80°C until use in the PIB binding analysis, Aβ-ELISAs, immunoblots and mass spectrometry. Additional blocks of frozen tissue were sectioned at 10μm on a cryostat and mounted onto silanized slides (Fisher Scientific, Waltham, MA, USA). Location-matched tissue from the contralateral hemisphere was fixed in phosphate-buffered, 4% paraformaldehyde, embedded in paraffin wax, and processed for histochemical analysis. In two AD cases, fixed, non-embedded samples from the temporal neocortex were used for ultrastructural investigation (below). All tissue samples were obtained from the Emory University Alzheimer’s Disease Research Center Brain Bank and the New York University Alzheimer’s Disease Center Brain Bank, in accordance with federal and institutional guidelines, and the samples were coded to ensure the anonymity of the subjects.
DNA Sequencing for β-Amyloid Precursor Protein (APP), Presenilin 1 (PSEN-1), and Presenilin 2 (PSEN-2)
Coding exons 3–18 with the corresponding 50bp of intronic junctions of APP and all coding exons from both PSEN-1 and PSEN-2, along with at least 25 base pairs of flanking sequence, were sequenced from genomic DNA extracted from AD1 brain tissue, using primers described in the supplementary material (Tables S1, S2 and S3).
Immunohistochemistry
Tissue sections from all AD cases were processed immunohistochemically using monoclonal anti-Aβ antibodies 6E10 to Aβ1–16 (1:25,000), 4G8 to Aβ17–24 (1:5,000; both from Covance, Princeton, NJ, USA), and 8E1 to the pyroglutamized amino terminus of Aβpyr-glu3-x (1:5,000; Immuno-Biological Laboratories, Gunma, Japan); polyclonal antibodies R361 to Aβ32–40 and R398 to Aβ33–42 (both at 1:15,000, courtesy of Pankaj Mehta, Institute for Basic Research on Developmental Disabilities, Staten Island, NY, USA); monoclonal anti-tau antibodies CP13 to phosphorylated tau serine 202 (1:10,000), MC1 to a pathologic conformation of tau (1:5,000; both courtesy of Peter Davies, Albert Einstein College of Medicine, Bronx, NY, USA), and AT8 to phosphorylated tau serine 202/phosphothreonine 205 (1:5,000; Covance). To rule out the presence of synucleinopathy, we employed a polyclonal antibody to α-synuclein (1:300, courtesy of Bernardino Ghetti, Indiana University, Indianapolis, IN, USA) (see Rosen et al., 2008 for antibody details).
Vectastain Elite kits (Vector Laboratories, Burlingame, CA, USA) were used for avidin-biotin complex (ABC)/horseradish peroxidase immunodetection of antigen-antibody complexes, with diaminobenzidine (DAB) or DAB + nickel as chromogens. Appropriate positive control specimens from cases with previously confirmed pathology were included in all immunostaining experiments. In some instances, sections were lightly counterstained with hematoxylin. Non-immune mouse IgG or rabbit serum was used in place of the monoclonal and polyclonal primary antibodies, respectively, as negative controls. Some additional sections were stained with Congo Red, Thioflavin-T, hematoxylin and eosin, or the Campbell-Gallyas AD-silver stain [29].
Electron microscopy
Fixed tissue slabs from the temporal neocortex of two AD cases (the PIB-refractory case [AD1] and a PIB-sensitive case [AD6]) were cut on a vibratome at 70μm thickness, and regions of heavy Aβ load were identified in adjoining sections immunostained with antibody 6E10. Samples from these regions were dissected from the adjacent, non-immunostained section, washed in phosphate buffer (0.1M, pH 7.4) and immersed in osmium tetroxide (1% in phosphate buffer) for 20 minutes. They were then rinsed in phosphate buffer and dehydrated in graded concentrations of ethanol and propylene oxide. Uranyl acetate (1%) was added to the 70% ethanol (35 minute immersion) to improve contrast in the electron microscope. Next, the blocks were embedded in epoxy resin (Durcupan ACM; Fluka, Ft. Washington, PA, USA) and heated for 48 hours at 60°C. Ultrathin sections were cut with a Leica Ultracut T2 (Nussloch, Germany), collected onto single-slot copper grids, and stained with lead citrate. Thin sections were examined with a Zeiss EM10-C electron microscope (Oberkochen, Germany) and digital images were captured with a Dual View camera (Gatan Inc., Pleasanton, CA, USA).
3H-PIB binding assay
To measure high-affinity PIB binding in cortical homogenates, all samples were simultaneously incubated with 1.0nM 3H-PIB (specific activity=82 Ci/mmol, custom synthesis, GE Healthcare, UK) in a microplate radioligand binding assay [29]. At 1.0nM, the concentration of free 3H-PIB used in our in vitro experiments is comparable to that achieved with 11C-PIB in PET scans [30], and is selective for the high-affinity binding sites on Aβ [2, 11, 12, 14]. The same tissue homogenates used in Aβ ELISAs (below) were diluted 1:33.3 in phosphate-buffered saline (PBS) (pH 7.4) to a final concentration of 5mg wet weight tissue/ml. In a 96 well polypropylene plate, 100μg of wet cortical tissue (20μl of diluted homogenate) from each sample were incubated, in duplicate, with 200μl of 1.0 nM 3H-PIB in 5% ethanol/PBS (nonspecific binding was defined as counts detected in the presence of 1.0 μM unlabeled PIB). After a 2.5-hour incubation (at room temperature, without shaking), samples were quickly transferred to a 96 well Multiscreen HTS Hi Flow FB filter plate and filtered with a vacuum manifold (Millipore Corporation, Billerica, MA, USA). Filters were rinsed 4 times with PBS at room temperature. The filters were dried for 30 minutes at 37°C, after which 50μl of MicroScint 20 (PerkinElmer, Waltham, MA, USA) scintillation fluid were added to each well and the plate was incubated at room temperature for 24 hours, shaking at 100 rpm. 3H-PIB binding was quantified in a TopCount scintillation counter (PerkinElmer) and specific binding was calculated by subtracting nonspecific counts per minute (CPM) (+1.0μM unlabeled PIB). Based on a 15% counting efficiency (established empirically), CPM were converted to femtomoles of bound PIB. PIB binding was linear between 25 and 200μg (wet weight) of AD cortical tissue homogenate added per well with 1.0nM 3H-PIB. To characterize the high-affinity binding components in case AD1 and other AD homogenates, samples were incubated in 1.2nM 3H-PIB with concentrations of unlabeled PIB between 0.1 nM and 1.0μM in a competition binding experiment.
3H-PIB Autoradiography
Slide-mounted, unfixed cryosections were brought to room temperature in air-tight containers and then immersion-fixed in 10% ethanol/PBS for 20 minutes. Sections were incubated in 1.0nM 3H-PIB (specific activity=82Ci/mmol) in 5% ethanol/PBS for one hour at room temperature, rinsed twice with 10% ethanol/PBS and 2 times with ddH2O, both on ice, and then air-dried before exposing directly to a BAS-TR2040 tritium imaging plate (Fujifilm, Stamford, CT, USA). After 2 weeks of exposure, the plate was developed in a BAS-5000 Image Plate Scanner and images were captured with MultiGauge software (Fujifilm). Adjacent cryosections were immunostained with Aβ antibody 6E10, as described above.
ELISA Quantification of Aβ40 and Aβ42
Cortical homogenates were spun at 100,000 × g for one hour at 4°C in a TLA-100.4 ultracentrifuge rotor (Beckman Coulter, Fullerton, CA USA). After collecting buffer-soluble supernatants, the pellet was sonicated in 70% formic acid, spun at 16,100 × g for 60 min at 4°C, and the clear supernatant was collected as the buffer-insoluble extract. For both extracts, total Aβx-40 and Aβx-42 levels were measured by enzyme-linked immunosorbent assays (ELISA) using standardized kits (The Genetics Company, Schlieren, Switzerland) as previously described [28, 29]. Aβ content is expressed relative to the wet weight of the tissue samples (Tables 2, 3).
Table 2.
Soluble Aβ40 and Aβ42 levels in temporal and occipital cortices of the core Alzheimer’s disease and nondemented control cases, as measured by ELISA. Aβ levels are presented relative to wet tissue weight (fmol Aβ/100μg wet weight tissue).
| Group | Case | Temporal Cortex | Occipital Cortex | ||||
|---|---|---|---|---|---|---|---|
| Aβ40 | Aβ42 | Total Aβ | Aβ40 | Aβ42 | Total Aβ | ||
| Alzheimer’s | AD1 | 21.63 | 0.13 | 21.76 | 24.56 | 2.42 | 26.98 |
| AD2 | 0.50 | 0.05 | 0.55 | 0.20 | 0.08 | 0.28 | |
| AD3 | 0.08 | 0.04 | 0.12 | 0.02 | 0.06 | 0.08 | |
| AD4 | 0.08 | 0.07 | 0.15 | 1.34 | 0.16 | 1.51 | |
| AD5 | 1.59 | 0.09 | 1.68 | n/a | n/a | n/a | |
| AD6 | 0.07 | 0.12 | 0.19 | n/a | n/a | n/a | |
| AD7 | 0.01 | 0.01 | 0.04 | 2.80 | 0.51 | 3.31 | |
| AD8 | 0.02 | 0.04 | 0.06 | n/a | n/a | n/a | |
| AD9 | 0 | 0.03 | 0.03 | 0 | 0.05 | 0.05 | |
| AD10 | 0 | 0.01 | 0.02 | 0.02 | 0.08 | 0.11 | |
| AD means: | 2.40 | 0.06 | 2.46 | 4.14 | 0.48 | 4.62 | |
| Nondemented | ND1 | 0 | 0.03 | 0.03 | 0.01 | 0.12 | 0.14 |
| ND2 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ND3 | 0 | 0.01 | 0.01 | 0 | 0 | 0 | |
| ND means: | 0.00 | 0.01 | 0.01 | 0.00 | 0.04 | 0.05 | |
Table 3.
Insoluble Aβ40 and Aβ42 levels in temporal and occipital cortices of the core Alzheimer’s disease and nondemented control cases, as measured by ELISA. Aβ levels are presented relative to wet tissue weight (fmol Aβ/100μg wet weight tissue).
| Group | Case | Temporal Cortex | Occipital Cortex | ||||
|---|---|---|---|---|---|---|---|
| Aβ40 | Aβ42 | Total Aβ | Aβ40 | Aβ42 | Total Aβ | ||
| Alzheimer’s | AD1 | 1126.13 | 790.51 | 1916.63 | 2685.25 | 912.84 | 3598.09 |
| AD2 | 39.84 | 174.88 | 214.72 | 15.49 | 297.96 | 313.45 | |
| AD3 | 62.48 | 97.56 | 160.04 | 5.23 | 186.51 | 191.73 | |
| AD4 | 17.75 | 196.38 | 214.13 | 10.29 | 191.29 | 201.58 | |
| AD5 | 275.37 | 352.57 | 627.94 | n/a | n/a | n/a | |
| AD6 | 3.88 | 444.72 | 448.59 | n/a | n/a | n/a | |
| AD7 | 7.65 | 217.09 | 224.74 | 10.97 | 176.23 | 187.21 | |
| AD8 | 4.07 | 155.58 | 159.66 | n/a | n/a | n/a | |
| AD9 | 0.62 | 145.53 | 146.15 | 0.21 | 59.36 | 59.57 | |
| AD10 | 3.37 | 82.91 | 86.28 | 2.18 | 211.23 | 213.41 | |
| AD means: | 154.12 | 265.77 | 419.89 | 389.95 | 290.77 | 680.72 | |
| Nondemented | ND1 | 0 | 0 | 0 | 0.19 | 0.22 | 0.41 |
| ND2 | 0 | 0.98 | 0.98 | 0.23 | 1.27 | 1.50 | |
| ND3 | 0.03 | 0.23 | 0.26 | 0 | 0.62 | 0.62 | |
| ND means: | 0.01 | 0.40 | 0.41 | 0.14 | 0.70 | 0.84 | |
Western blot
Unfixed cortical tissue blocks were Dounce-homogenized in 4 volumes of PBS. The 20% homogenate was vortexed, probe-sonicated 3 × 5 seconds (level 4, Model 100 ultrasonic dismembrator, Fisher Scientific), centrifuged at 3,000 × g for 5 minutes at 4°C, and the supernatant stored at −80°C until use. Total protein was quantified in the sample with a microplate bicinchoninic acid (BCA) assay (Pierce Biotechnology, Rockford, IL, USA). 60μg of protein was boiled for 5 minutes in 2x Tricine sample buffer and 10x Reducing Agent (Invitrogen, Carlsbad, CA, USA), run on a 10–20% Tricine gel (Invitrogen) and blotted onto a nitrocellulose membrane. The membrane was boiled for 15 minutes in 1xPBS, then blocked at room temperature in 2.5% nonfat milk/0.05% Tween-20 (blocking buffer) for 60 minutes, incubated in primary antibody to Aβ (6E10, 1:1000 diluted in blocking buffer) for 48 hours at 4°C, followed by 90 minutes in horseradish peroxidase-conjugated anti-mouse IgG antibody (1:10,000 in blocking buffer, GE Healthcare, Piscataway, NJ, USA). Antibody binding was detected with Super Signal West Pico electrochemiluminescence substrate (Fisher Scientific) and exposed to Kodak MR Biomax film (Kodak, New Haven, CT, USA).
Immunoprecipitation/MALDI-TOF MS
For mass spectrometry, cortical homogenates were ultracentrifuged to isolate buffer-soluble Aβ (see above). Pellets were resuspended in 2% SDS, spun at 100,000 × g for 60 minutes at 10°C to extract detergent-soluble Aβ, and the pellet containing buffer-insoluble proteins was sequentially extracted with 70% and 90% formic acid (above). Immunoprecipitation of Aβ was performed using paramagnetic beads coated with a cocktail of antibodies 4G8 and 6E10, or C-terminal specific antibody 12F4 to Aβx-42, and the precipitates analyzed by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry, as previously described [32]. Briefly, Dynal M-280 Dynabeads (Invitrogen) coated with sheep anti-mouse antibodies were then coated with monoclonal antibodies 4G8 and 6E10, each at a concentration of 60 μg/ml, or monoclonal antibody 12F4 (Covance) at 120 μg/ul. 50μl of coated beads were added to 1.0ml soluble cortical extract, 1.0ml detergent-soluble extract (after detergent removal using SDS-OUT, according to manufacturer’s instructions, Pierce Biotechnology), or a solution of 100μl formic acid extract, neutralized with 1.0M Tris buffer to pH 7.4 and then diluted up to 10ml volume with deionized water. Following immunoprecipitation and washing, beads were completely dried in a SpeedVac, eluted with 20μl of 0.5% formic acid, dried in a SpeedVac and resuspended in 4μl of 0.1% formic acid. 1μl of IP eluate was mixed with 500μl ethanol and 500μl acetonitrile in 0.1% trifluoroacetic acid and then analyzed in a Micromass TofSpec-2E MALDI-TOF instrument. Major peaks within all spectra were identified with ExPASy’s FindPept software (http://us.expasy.org/tools/findpept.html).
Results
APP, PSEN1, and PSEN2 gene sequencing
No mutations were detected in coding exons 3–18 and 50 bp of intronic junctions of the APP gene sequence in AD1 genomic DNA. Sequencing of all PSEN1 and PSEN2 coding exons and at least 25 base pairs of flanking sequence failed to identify any known AD-associated mutations or polymorphisms that would result in amino acid sequence change to PSEN1 or PSEN2. Conservative minor allele single nucleotide polymorphisms were identified in exons 3, 4, and 11 of PSEN2 (listed Table S4).
Histopathology
Immunohistochemically, subject AD1 had copious dense-cored and diffuse parenchymal senile plaques characteristic of advanced AD, as well as significant large vessel and capillary CAA and tau-positive neurofibrillary pathology (Figs. 1, 2). Parenchymal and vascular Aβ lesions were detected throughout the temporal and occipital cortices of case AD1, with slightly more parenchymal deposits in the temporal cortex and a predilection for vascular amyloid deposition in occipital cortical regions (Figs. 1a, 2). The presence of AD-typical lesions was confirmed by multiple antibodies to Aβ and tau, and by the Congo Red, Thioflavin-T and Campbell-Gallyas silver stains (not shown). Antibody R398 to Aβ42 disclosed a large variety of parenchymal and vascular lesions, similar to antibody 6E10, whereas antibody R361 to Aβ40 labeled primarily CAA and plaque cores (Fig. 3). Antibody 8E1 to Aβpyr-glu3-x, like antibodies 6E10 and R398, stained a broad spectrum of vascular and parenchymal Aβ deposits (not shown). Ultrastructurally, the senile plaques consisted of fibrillar material consistent with amyloid (Fig. 1c), and the neurofibrillary pathology included filamentous ribbons typical of AD (Fig. 1d). The antibody to α-synuclein did not show concomitant cortical Lewy bodies or Lewy neurites in this case (not shown).
Fig. 1. Aβ and tau pathology in the superior temporal gyrus of the PIB-refractory Alzheimer’s case AD1.

(a) Antibody 6E10-immunoreactive senile plaques (two denoted by arrows) and large-vessel CAA (black arrowhead). (b) Antibody CP13-immunoreactive, intra-somatal neurofibrillary tangles (two denoted by arrowheads) and abundant neuropil threads. (c) Ultrastructure of amyloid fibrils in the periphery of a senile plaque. (d) Ultrastructure of a bundle of paired helical filaments (marked by two arrowheads). Case AD1 had significant CAA, and the senile plaques sometimes showed diminished immunostaining in the central region (right-hand arrow in a), but otherwise the histopathology did not differ remarkably from that of the other AD cases. Bar = 100μm in a and b and 200nm in c and d).
Fig. 2. Cerebral β-amyloid angiopathy in the occipital cortex of case AD1.
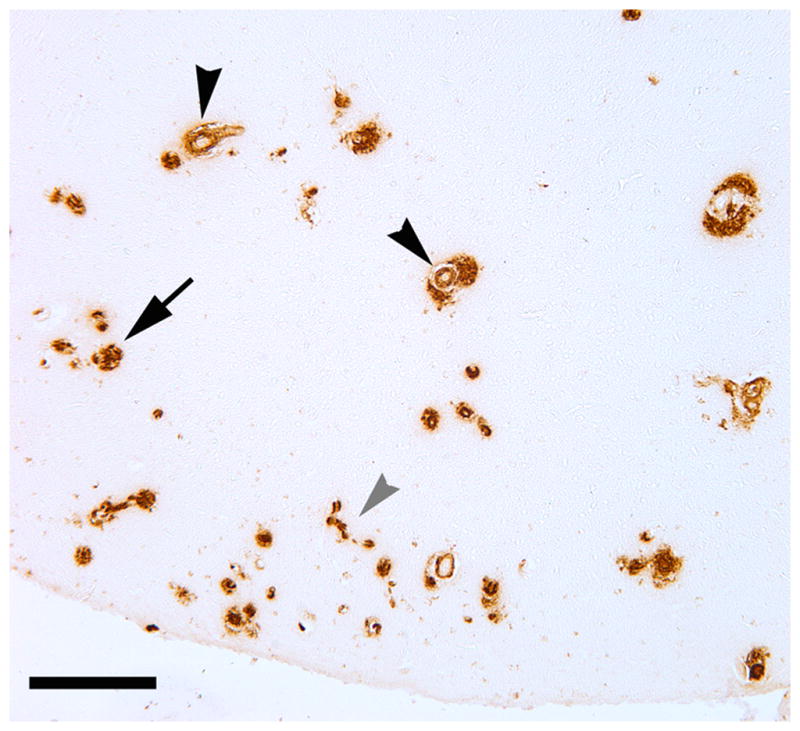
Both large vessels (black arrowheads) and capillaries (grey arrowhead) were affected. CAA of both types was more abundant in the occipital cortex than in the temporal cortex. A plaque-like parenchymal deposit of Aβ is denoted by the arrow. Antibody 4G8; Bar = 200μm.
Fig. 3. Immunostaining for Aβ42 (a) and Aβ40 (b) in nearby temporal cortical sections from case AD1.

The antibody to Aβ42 stained a variety of diffuse and dense parenchymal lesions as well as CAA, but the antibody to Aβ40 stained mainly CAA (the same two immunoreactive blood vessels are marked by arrowheads in both panels) and dense plaque cores (two denoted by arrows in b). Bar = 300μm.
3H-PIB binding and Aβ analysis
PIB binding was markedly reduced in temporal and occipital cortical homogenates of case AD1 compared to 9 other clinically and pathologically confirmed AD subjects (Fig. 4), and was in the range of the non-demented control cases (PIB also bound strongly to homogenates from the AD cases used as controls for the mass spectrometry analysis, below). Despite a paucity of high-affinity PIB binding sites, ELISAs disclosed levels of soluble and insoluble Aβ40 and Aβ42 that greatly exceeded those in both brain regions of all nine comparison AD cases in the core group (Tables 2, 3). The Aβ40:Aβ42 ratios for soluble and insoluble Aβ also were higher in AD1 than in the 9 other AD cases (Fig. 4). A competition curve experiment with 3H-PIB and unlabeled PIB in temporal cortical homogenates from AD1 and a comparison case (AD2) revealed a high-affinity PIB binding site in AD2 (Kd = 3.0nM and Bmax = 290 fmol), comparable to previously published reports [10, 14, 29], but not in AD1 (Fig. 5). Similar results were seen in the occipital cortex (data not shown). Finally, mixing experiments with AD1 and AD2 temporal and occipital cortical homogenates revealed only an additive effect on PIB binding when equal parts of tissue from each case were simultaneously incubated with 1.0nM 3H-PIB (Fig. 6).
Fig. 4. Aβ levels and 3H-PIB binding in temporal and occipital cortical homogenates from AD1, nine comparison AD cases and three nondemented control (ND) cases.

(a, b) By ELISA of soluble and insoluble extracts, AD1 (red arrowheads) exhibited substantially more soluble and insoluble Aβ40 and Aβ42 than did all other AD cases examined in this study. Negligible Aβ was detected in both regions of the 3 ND cases (group means are indicated by the horizontal lines). Levels of soluble Aβ40 and Aβ42 (a) and levels of insoluble Aβ40 and Aβ42 (b) are expressed in fmol Aβ per 100μg wet tissue for each subject. (c) Radioligand binding assays with 3H-PIB reveal high levels of PIB binding to temporal and occipital cortical tissue from all AD cases in this study except AD1, which showed only background levels of PIB binding. (d) The ratio of PIB binding to total Aβ in two cortical regions. When AD1 data points were excluded from the analysis, total levels of insoluble Aβ (Aβ40 and Aβ42) correlate positively with 3H-PIB binding to AD temporal (n=9) and occipital (n=7) cortical homogenates from the comparison AD cases [29].
Fig. 5. Analysis of high-affinity 3H-PIB binding in temporal cortical homogenates from two AD cases.

A homologous competition binding analysis of PIB binding levels in temporal cortical homogenates incubated with 1.2nM 3H-PIB alone and with 1.2nM 3H-PIB co-incubated with concentrations of unlabeled PIB between 1.0 nM and 1.0μM did not reveal a measurable high-affinity PIB binding component in case AD1 (dotted line). In case AD2 (continuous line), a high-affinity PIB binding component was detected with a Kd = 3.0nM and Bmax = 209.28 fmol/100μg wet tissue (AD2: open squares, AD1: filled diamonds). Comparable results were obtained with occipital cortical homogenates (data not shown).
Fig. 6. 3H-PIB binding experiments with mixtures of AD and ND cortical homogenates.
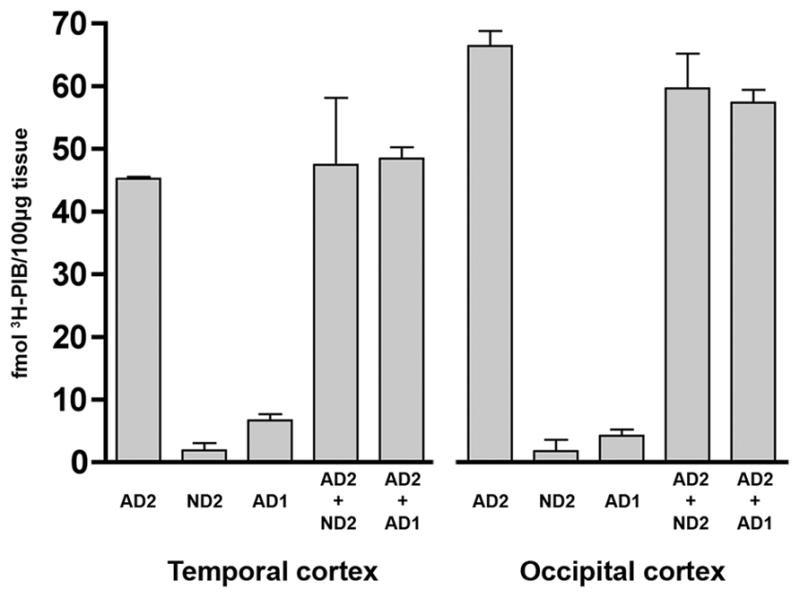
To detect the potential presence of a diffusible element in AD1 cortical tissue that inhibits PIB binding in vitro, we incubated 1.0nM 3H-PIB with 1:1 mixtures of cortical homogenates from a typical PIB-sensitive AD case (AD2) and the PIB-refractory case AD1. We did not identify an inhibitory or synergistic effect on PIB binding in either the temporal or occipital cortical homogenates. Rather, the quantity of PIB binding to mixed homogenates was approximately the sum of total PIB binding to each individual homogenate. Similar results were seen when 1.0nM 3H-PIB was incubated with mixtures of temporal and occipital cortical homogenate from AD2 and a nondemented control case (ND2) that alone exhibited negligible high-affinity PIB binding.
Autoradiography of 1nM 3H-PIB binding in unfixed cryosections revealed some cortical plaques in the superior temporal cortex of case AD1. However, comparison with an adjacent cryosection that was immunostained with antibody 6E10 indicated that only a subset of potential plaques and CAA were labeled (Fig. 7). In the temporal cortex of a comparison AD subject (AD5), 3H-PIB autoradiography and 6E10 immunohistochemistry on adjacent cryosections showed substantial overlap of Aβ-immunoreactive and PIB-positive lesions (Fig. 7).
Fig. 7. 3H-PIB Autoradiography in temporal cortical sections from case AD1 and a PIB-sensitive AD case (AD5).

(a) At a 1.0nM concentration, 3H-PIB binds to few cortical plaques and lightly to some blood vessels in the superior temporal cortex of AD1. (b) In an adjacent cryosection, immunohistochemistry with the 6E10 antibody to Aβ reveals abundant senile plaques as well as CAA in this cortical region. The blue arrowheads denote PIB-positive Aβ plaques and CAA, although the greater part of the Aβ-immunoreactive plaques and vessels did not bind to PIB by autoradiography. (c) In case AD5, 3H-PIB autoradiography and (d) 6E10 immunohistochemistry on adjacent cryosections demonstrate heavy, PIB-positive senile plaque deposition in the superior temporal cortex. Red arrows indicate PIB-positive Aβ lesions identified in adjacent cryosections.
Using antibody 6E10 for immunoblot analysis of temporal cortical extracts from AD1 and seven comparison AD cases, strong bands of 4KDa Aβ monomers were detected in all 8 cases. Light bands corresponding to Aβ dimers (~8KDa) were detectable in several of the AD cases. In the cortical extract from AD1, however, these Aβ dimer bands were of substantially greater intensity relative to the 7 comparison AD cases, and a strong immunoreactive band was detected at ~12 KDa, corresponding to trimeric Aβ. Additionally, numerous higher molecular weight bands migrating between 17 and 44 kDa were detected solely in AD1 temporal cortical tissue. APP levels (~100KDa) were similar in AD1 and the 7 comparison AD cases (Fig. 8). Because our ELISA analyses indicate considerably higher total Aβ levels in AD1 tissue, an immunoblot was conducted with AD1 cortical extract that was diluted to reduce total protein, thereby yielding total Aβ levels that were comparable to the mean Aβ levels in the comparison AD cases. When Aβ levels are normalized as such, the higher molecular weight 6E10-immunoreactive bands, though weaker, are still visible (data not shown).
Fig. 8. Western blot analysis of multimeric Aβ and APP in AD temporal cortex.
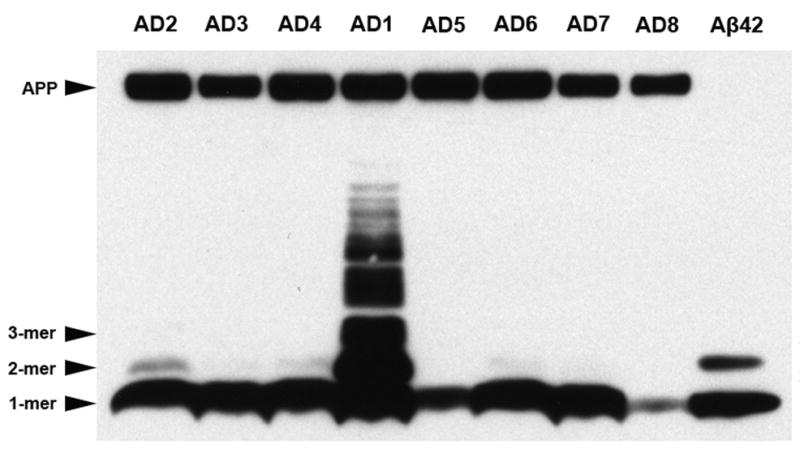
Temporal cortical homogenates containing 60μg of total protein from case AD1 and seven other AD cases were separated by SDS-PAGE and immunoblotted with antibody 6E10 to the N-terminal region of Aβ. A preparation of synthetic Aβ42 (10 ng) is in the far right lane, as a positive control. Strong bands of monomeric Aβ were detected in all AD cases examined. Light bands corresponding to Aβ dimers were detected only in some of the AD lanes under these conditions. In AD1, 6E10 immunoblotting revealed high levels of dimeric and trimeric Aβ, as well as several higher molecular weight, Aβ-immunoreactive bands between 17 KDa and 44KDa. Note that 6E10 also recognizes the Aβ epitope in APP (top band), which was not increased in AD1. When the total protein content in AD1 temporal cortex was diluted to 6.78μg to normalize total Aβ levels to those seen in the comparison AD cases, the higher molecular weight bands, though lighter, were still detected by 6E10 immunoblot analysis, indicating that the presence of these bands cannot be accounted for simply by the overall quantity of Aβ in AD1 temporal cortex (data not shown).
Aβ MALDI-TOF mass spectrometry
MALDI-TOF MS on immunoprecipitated buffer-soluble, SDS-soluble, and formic acid-soluble Aβ from AD1, three comparison (PIB-sensitive) AD cases and one nondemented control case revealed a pattern of N- and C-terminally truncated fragments unique to AD1 temporal cortex (Fig. 9, data from 2 AD cases and the nondemented case not shown). Full-length Aβ40 and Aβ42 were detected in all fractions from all AD cases, although Aβ42 was barely detectable in AD1 buffer-soluble extract, suggesting that the peptide may be preferentially deposited in insoluble lesions and/or cleared from the brain (Fig. 9). Numerous buffer-soluble, C-terminally truncated Aβ fragments were identified in AD1 tissue (Aβ1–38, Aβ1–37, Aβ1–36, Aβ1–34, Aβ1–33, Aβ1–29, Aβ1–28, Aβ1–19, Aβ1–18, Aβ1–16) but not in any of the 3 comparison AD cases. Insoluble Aβx-42 fragments with various N-terminal residues were identified in the AD1 insoluble extract, as were oxidized Aβ40, Aβ42, and Aβx-42 peptides, the majority of which were not seen in the comparison cases.
Fig. 9. Immunoprecipitation/MALDI-TOF MS detection of Aβ peptides in the temporal cortex of case AD1 and a comparison PIB-sensitive AD case (AD12).

By mass spectrometry, we detected substantially more individual Aβ fragments in all cortical fractions of AD1 temporal cortex, as compared to three other AD cases (data from one comparison AD case shown). Numerous C-terminally truncated peptides (Aβ1-x) were seen in the buffer (TBS)-soluble extract, yet Aβ42 was barely detectable. These same truncated peptides also were detected in SDS-soluble extract, along with Aβ40 and Aβ42. N-terminally truncated peptides (Aβx-40 and Aβx-42) and full-length Aβ40 and Aβ42 predominated in formic acid-solubilized fractions (FA 70% and FA 100%) from AD1 cortex. This pattern of terminal-specific truncations in soluble and insoluble cortical fractions was not seen in the comparison AD cases or in a nondemented control case (not shown). The Aβ fragments written in red were particularly abundant, while those in parentheses were just barely detectable. a-42: Immunoprecipitation with monoclonal antibody 12F4, specific to Aβx-42; 4G8/6E10: Immunoprecipitation with two monoclonal antibodies, one to Aβ17–24 (4G8) and one to Aβ1–16 (6E10); TBS: Tris-buffered saline extract; SDS: sodium dodecyl sulfate-soluble extract; FA: formic acid-solubilized extract.
Discussion
Pittsburgh compound B has been shown to bind with high affinity to parenchymal senile plaques [13, 23, 30] and to cerebral β-amyloid angiopathy [2, 11] in Alzheimer’s disease. Previous studies also have reported a significant positive correlation between PIB binding and Aβ levels [2, 10, 16, 31], further supporting the utility of this ligand in the antemortem diagnosis of AD and CAA. In assessing 3H-PIB binding to cortical tissue homogenates in a series of ten AD patients, however, we discovered a case of pathologically confirmed AD (case AD1) with particularly heavy cerebral Aβ deposition, yet negligible high-affinity binding of PIB.
To address potential reasons for the paucity of PIB binding in case AD1, we first considered the ways in which this patient differed from the other AD cases. Histopathologically, AD1 showed numerous diffuse parenchymal Aβ deposits, dense-cored Aβ plaques and neurofibrillary tangles typical of advanced AD, as well as substantial large vessel and capillary CAA (Figs. 1, 2, 3 and 7). However, relative to PIB-sensitive AD cases, autoradiography with 1nM 3H-PIB detected only a fraction of the senile plaques and CAA in AD1 (Fig. 7). Comparison with the adjacent, Aβ-immunostained sections did not indicate preferential binding of PIB to a particular type of Aβ lesion. Although AD1 had substantial CAA, some degree of CAA is usual in AD [1], and vascular amyloid was present (albeit to a lesser degree) in most of the other cases that we examined. Furthermore, PIB has been shown to be a sensitive marker for CAA in vivo [2, 11] and in vitro [19]. Finally, in addition to CAA, abundant dense and diffuse Aβ plaques were present in the cortical samples from AD1 (Fig. 1), yet, despite a heavy burden of Aβ lesions of multiple types, high-affinity PIB binding was unexpectedly meager.
Next, we used ELISA to measure the overall quantity of Aβ and found that the levels of both soluble and insoluble Aβ40 and Aβ42 in AD1 greatly exceeded those in the comparison group of nine AD patients. We then asked whether the relative amounts of Aβ40 and Aβ42 might influence PIB binding. As is usual for AD patients with prominent CAA [15, 27], the Aβ40:Aβ42 ratio was particularly high in AD1 (temporal cortex: Aβ40:Aβ42 = 1.42, vs. 0.22 in the comparison AD group; occipital cortex: Aβ40:Aβ42 = 2.94, vs. 0.04 in the comparison group). However, PIB binding has been shown to correlate strongly with levels of both Aβ40 and Aβ42 in brain tissue [31], and PIB also binds with low stoichiometry but equal affinity to synthetic, aggregated Aβ40 and Aβ42 in vitro [14]. Hence, neither a paucity of Aβ nor the absolute amounts of the two major isoforms of Aβ is a likely explanation for the deficiency of high-affinity PIB binding in this case. However, the influence of the ratio of Aβ isoforms on PIB binding to Aβ generated in vivo deserves further study.
To rule out the presence of a diffusible cortical element in case AD1 that specifically inhibits the high-affinity binding of PIB to Aβ, we then incubated 3H-PIB with mixtures of cortical homogenates from AD1 and a typical PIB-sensitive AD case (AD2). Both in temporal and occipital cortices, PIB binding to AD2 cortical homogenate was neither enhanced nor decreased in the presence of AD1 cortical homogenate, indicating an absence of diffusible factors in the AD1 cortex that inhibit high-affinity PIB binding to Aβ. These data also argue against a diffusible substance in typical AD tissue that stimulates PIB binding to Aβ, as the mixture of AD2 and AD1 cortical homogenates failed to increase PIB binding beyond the sum of the values achieved in the individual homogenates, despite very high amounts of Aβ in the AD1 cortical homogenate.
There is evidence to suggest that PIB binds with high affinity to parenchymal lesions containing N-terminally-truncated and modified Aβpyr-glu3-x peptide isoforms, and that the low stoichiometry of PIB binding to Aβ in the transgenic mouse brain may be attributable to a dearth of such modified Aβ isoforms [8, 20]. In case AD1, however, an N-terminal-specific antibody to Aβpyr-glu3-x labeled a substantial majority of parenchymal and vascular Aβ deposits in the temporal and occipital cortices. By mass spectrometry of temporal cortical samples, we did identify a pattern of N- and C-terminally truncated Aβ peptides unique to AD1: Aβ1-x fragments predominated in the soluble extract, and Aβx-42 peptides were detected mainly in the insoluble extract (Fig. 9). The heterogeneity of C-terminal Aβ fragments in the PIB refractory case may represent the atypical action of carboxyl peptidase(s) in AD1 brain. The possibility of anomalous peptidase activity and/or Aβ clearance mechanisms in this subject, as well as the effect of modified Aβ isoforms on PIB binding affinity, remain to be established. Importantly, these data indicate that deficient high-affinity PIB binding to cortical homogenate from this case is not attributable to an absence of modified or truncated Aβ isoforms that are generally found in mature plaques of the AD brain. Furthermore, in conjunction with the extract-mixing experiments (above), the findings suggest that the presence of abundant Aβ1-x fragments in the soluble extract does not inhibit PIB binding when introduced into cortical extracts from other AD cases.
We then analyzed the distribution of multimeric Aβ by SDS-PAGE/Western blot, and detected Aβ monomers and dimers in both AD1 and most of the comparison AD cases. However, Aβ immunoblots also yielded a dense series of low and higher molecular weight Aβ-immunoreactive bands in AD1 that were not identified, under identical conditions, in any of the comparison AD cases (Fig. 8). Normalization of total Aβ levels indicated that these bands could not be attributed solely to the heavy Aβ load in AD1. Whether these oligomers are indicative of an alternative Aβ folding/aggregation pathway unique to this case, and whether the presence of these oligomers somehow inhibits PIB biding, warrants further investigation. Again, however, the absence of an inhibitory effect of AD1 extract on PIB binding in other AD cases (above) argues against the presence of such an inhibitory factor, at least in a readily diffusible form.
The E4 subtype of apolipoprotein E has been linked to an increased likelihood of developing both AD [4, 25] and CAA [7, 26], and to the overall severity of cerebral β-amyloidosis [9, 33, 34]. However, like AD1, seven of the nine AD cases in the comparison group also were hemizygous for ApoE4, and one was homozygous, yet all of these individuals displayed strong in vitro binding of 3H-PIB, as did the case that was ApoE3/3. Consequently, apolipoprotein E genotype per se does not account for the anomalous PIB binding in AD1. Genetic analysis of the common AD-associated loci in the genes for APP, presenilin1 and presenilin 2 revealed no obvious mutations or functional polymorphisms that might explain the paucity of high-affinity PIB binding in AD1.
Although the PIB-refractory AD case was not imaged with 11C-PIB prior to death, our in vitro findings, using a concentration of free 3H-PIB similar to that achieved in the brain parenchyma of 11C-PIB-scanned patients [30], suggest the possibility that the PET signal would have been diminished or absent in this patient, despite the presence of copious cerebral Aβ. Currently, few cases of suspected AD have been imaged with PIB and then characterized pathologically and biochemically post-mortem; consequently, the frequency of PIB-refractory cases in the AD population is unknown. Although there is compelling evidence that PIB binds only weakly to Aβ deposits in nonhuman species [14, 29, 31], our present results, in conjunction with other findings [5, 13, 18, 22], suggest that PIB imaging may not detect all Aβ lesions in some humans as well [5, 13, 18, 22].
In summary, we describe a case of neuropathologically confirmed AD with heavy cerebral β-amyloidosis yet negligible high-affinity PIB binding in cortical homogenates. On a mechanistic level, the presence of PIB-refractory Aβ in a confirmed case of Alzheimer’s disease implies the existence of molecular strains of Aβ that could be functionally dissimilar with regard to the pathogenesis of AD [17], similar to the heterogeneity of prion strains [3, 6]. In this light, PIB and other binding agents could serve as specific, early diagnostic tools that distinguish polymorphic AD phenotypes, as well as selective in vitro molecular probes for strain-like structural variants of multimeric Aβ [17]. Until the prevalence of PIB-refractory Aβ in senescent humans can be established, our findings indicate that limited 11C-PIB retention in PET images of suspected AD cases should be interpreted with caution.
Supplementary Material
Acknowledgments
This work was supported by RR-00165, PO1AG026423, P50AG025688, the Woodruff Foundation and the Emory University Research Committee.
We thank M. Paul Murphy for helpful discussions, and Jean-Francois Pare, Carolyn Suwyn, Elaine Pranski and Aaron Farberg for excellent technical assistance.
References
- 1.Attems J, Jellinger KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology--a pilot study. Acta Neuropathol. 2004;107:83–90. doi: 10.1007/s00401-003-0796-9. [DOI] [PubMed] [Google Scholar]
- 2.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–434. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- 3.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 4.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 5.Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 6.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 8.Higuchi M. Visualization of brain amyloid and microglial activation in mouse models of Alzheimer’s disease. Curr Alzheimer Res. 2009;6:137–143. doi: 10.2174/156720509787602906. [DOI] [PubMed] [Google Scholar]
- 9.Hyman BT, West HL, Rebeck GW, et al. Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome) Proc Natl Acad Sci U S A. 1995;92:3586–3590. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PIB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- 12.Klunk WE, Engler H, Nordberg A, et al. Imaging the pathology of Alzheimer’s disease: amyloid-imaging with positron emission tomography. Neuroimaging Clin N Am. 2003;13:781–789. ix. doi: 10.1016/s1052-5149(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 13.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 14.Klunk WE, Lopresti BJ, Ikonomovic MD, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar-Singh S. Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008;7(Suppl 1):67–82. doi: 10.1111/j.1601-183X.2007.00380.x. [DOI] [PubMed] [Google Scholar]
- 16.Leinonen V, Alafuzoff I, Aalto S, et al. Assessment of {beta}-Amyloid in a Frontal Cortical Brain Biopsy Specimen and by Positron Emission Tomography With Carbon 11-Labeled Pittsburgh Compound B. Arch Neurol. 2008;65:1304–9. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- 17.LeVine H, III, Walker LC. Molecular polymorphism of Abeta in Alzheimer’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Rinne JO, Mosconi L, et al. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockhart A, Lamb JR, Osredkar T, et al. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 20.Maeda J, Ji B, Irie T, Tomiyama T, et al. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci. 2007;27:10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 22.Nelissen N, Vandenbulcke M, Fannes K, et al. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–2069. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- 23.Nordberg A. Amyloid plaque imaging in vivo: current achievement and future prospects. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S46–50. doi: 10.1007/s00259-007-0700-2. [DOI] [PubMed] [Google Scholar]
- 24.Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 25.Poirier J, Davignon J, Bouthillier D, et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 26.Premkumar DR, Cohen DL, Hedera P, et al. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol. 1996;148:2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 27.Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–30. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosen RF, Farberg AS, Gearing M, et al. Tauopathy with paired helical filaments in an aged chimpanzee. J Comp Neurol. 2008;509:259–270. doi: 10.1002/cne.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosen RF, Walker LC, LeVine H., 3rd PIB binding in aged primate brain: Enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 31.Svedberg MM, Hall H, Hellstrom-Lindahl E, et al. [(11)C]PIB-amyloid binding and levels of Abeta40 and Abeta42 in postmortem brain tissue from Alzheimer patients. Neurochem Int. 2009;54:347–357. doi: 10.1016/j.neuint.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Tomidokoro Y, Lashley T, Rostagno A, et al. Familial Danish dementia: coexistence of Danish and Alzheimer amyloid subunits (ADan AND A{beta}) in the absence of compact plaques. J Biol Chem. 2005;280:36883–36894. doi: 10.1074/jbc.M504038200. [DOI] [PubMed] [Google Scholar]
- 33.Walker LC, Pahnke J, Madauss M, et al. Apolipoprotein E4 promotes the early deposition of Abeta42 and then Abeta40 in the elderly. Acta Neuropathol. 2000;100:36–42. doi: 10.1007/s004010051190. [DOI] [PubMed] [Google Scholar]
- 34.Warzok RW, Kessler C, Apel G, et al. Apolipoprotein E4 promotes incipient Alzheimer pathology in the elderly. Alzheimer Dis Assoc Disord. 1998;12:33–39. doi: 10.1097/00002093-199803000-00005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.