

Abstract
Mitochondria are key organelles in eukaryotic cells that not only generate adenosine triphosphate but also perform such critical functions as hosting essential biosynthetic pathways, calcium buffering, and apoptotic signaling. In vivo, mitochondria form dynamic networks that undergo frequent morphologic changes through fission and fusion. In neurons, the imbalance of mitochondrial fission/fusion can influence neuronal physiology, such as synaptic transmission and plasticity, and affect neuronal survival. Core components of the mitochondrial fission/fusion machinery have been identified through genetic studies in model organisms. Mutations in some of these genes in humans have been linked to rare neurodegenerative diseases such as Charcot-Marie-Tooth subtype 2A and autosomal dominant optic atrophy. Recent studies also have implicated aberrant mitochondrial fission/fusion in the pathogenesis of more common neurodegenerative diseases such as Parkinson’s disease. These studies establish mitochondrial dynamics as a new paradigm for neurodegenerative disease research. Compounds that modulate mitochondrial fission/fusion could have therapeutic value in disease intervention.
Introduction
Mitochondria are key organelles in eukaryotic cells thought to originate from ancient bacterial symbionts. Over the billions of years of their symbiosis relationship with their eukaryotic hosts, mitochondria have maintained their DNA, RNA, and protein synthesis systems. These organelles produce chemical energy in a process called oxidative phosphorylation (OXPHOS). Some of the components of the protein complexes involved in OXPHOS are encoded and produced in mitochondria, but most of the subunits of the OXPHOS enzyme complexes are nuclear encoded. Other proteins involved in mitochondrial biogenesis are also nuclear encoded. These include the mitochondrial DNA polymerase γ subunits, the RNA polymerase components, mitochondrial transcription factor A, ribosomal proteins and elongation factors, and metabolic enzymes. The biogenesis and function of mitochondria therefore require coordination and communication between the mitochondrial and nuclear genomes. In addition to housing the electron transport chain on the membrane and essential metabolic pathways in the matrix, mitochondria provide calcium-buffering function and are integrated in a number of signaling processes, such as apoptosis signaling. It is no wonder that mitochondrial dysfunction has been linked to a wide range of human disease conditions [1].
Diseases associated with mitochondrial dysfunction can be loosely grouped into two categories: 1) classic mitochondrial disorders resulting from mutations in mitochondrial DNA or nuclear genes that directly affect mitochondrial gene expression or OXPHOS function; 2) other diseases caused by mitochondrial dysfunction resulting from nuclear gene mutations that primarily disrupt nonrespiratory function. This review focuses on the second category, with particular emphasis on neurodegenerative diseases caused by nuclear gene mutations that affect mitochondrial fission and fusion, evolutionarily conserved mitochondrial membrane remodeling processes that regulate the morphology, and subcellular distribution of mitochondrial units. Mitochondrial morphological dynamics are particularly important for the physiological function of neurons. The importance of mitochondrial dynamics in neuronal function is also illustrated by a growing number of neurodegenerative diseases with prominent defects in mitochondrial morphology, including Parkinson’s disease (PD) and Alzheimer’s disease, which are the most common neurodegenerative diseases.
Genetic Control of Mitochondrial Dynamics
In addition to the static images of kidney bean–shaped structure found in most textbooks, mitochondria often exhibit extensive tubular network morphology in vivo. The dynamics of mitochondrial morphology are determined by the balanced action of fission and fusion processes (Fig. 1). Genetic studies in yeast and Drosophila have been instrumental in defining the core mitochondrial fission/fusion pathway genes [2]. Both the fission and fusion processes involve the remodeling of mitochondrial outer and inner membranes, and different molecules appear to be required for each membrane.
Figure 1.
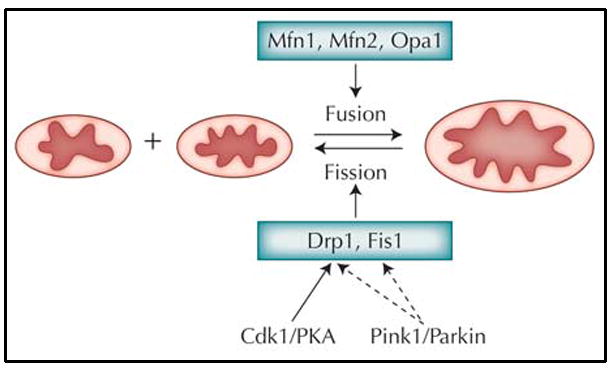
Some of the major players that regulate mitochondrial fission and fusion dynamics. The solid line between Cdk1/PKA and Drp1 indicates direct biochemical interactions between these proteins. The dashed lines between Pink1/Parkin and Drp1 and Fis1 denote hypothetical biochemical interactions for which no experimental evidence is currently available. PKA—adenosine monophosphate–dependent protein kinase.
The fission of mitochondrial outer membrane requires Drp1 and Fis1. Drp1 is a large guanosine triphosphatase (GTPase) related to dynamin that is involved in endocytosis of membrane vesicles. Fis1 is a tetratricopeptide repeat domain–containing protein localized to the outer membrane. Based on work performed in yeast, a model has been proposed in which Fis1 serves as an adaptor protein for recruiting Drp1 from the cytosol to mitochondrial fission sites, where it uses energy generated by GTP hydrolysis to sever mitochondrial tubules [3]. However, the situation might be more complicated in mammalian cells, where Drp1 can be recruited to mitochondria independent of Fis1. From a mechanical point of view, it may appear that the fission of mitochondrial inner and outer membranes can be accomplished simultaneously when Drp1-mediated outer membrane constriction and severance is carried to completion. However, studies in yeast and mammals have found specific proteins localized to the inner mitochondrial membrane, where they are required for the fission process. Examples include the mammalian MTP18 protein [4]. The mechanisms of action of these proteins are unknown. It is possible that they may cooperate with Fis1 from the inside to determine the location of fission sites.
The fusion of mitochondrial outer membrane is controlled by the mitofusins, which also encode large GTPases. In mammals, there are two mitofusins: Mfn1 and Mfn2. In addition to the GTPase domain, mitofusins contain coiled-coil domains that mediate protein–protein interaction. Through the coiled-coil domains, Mfn1 and Mfn2 can form homo-oligomeric and hetero-oligomeric complexes in trans. These homotypic or heterotypic interactions are thought to help bring together the outer membranes of two mitochondria to facilitate fusion [5]. The fusion of mitochondrial inner membrane requires another dynamin family GTPase, Opa1 [6]. Opa1 may also act through homotypic interaction in trans to tether mitochondrial inner membranes. In addition to promoting inner membrane fusion, Opa1 may play a role in controlling inner membrane cristae structure.
Recent studies have revealed important roles for posttranslational modifications in the regulation of mitochondrial fission and fusion machineries. Of all the components of the fission/fusion machinery, Drp1 is most extensively studied. Phosphorylation of Drp1 at Ser585 by the mitotic kinase Cdk1/cyclin B promotes mitochondrial fragmentation observed in cells undergoing mitosis [7•]. Drp1 was also phosphorylated at a conserved serine residue (Ser637) by cyclic adenosine monophosphate (AMP)–dependent protein kinase (PKA) [8•,9•]. In contrast to the effects of phosphorylation at Ser585, phosphorylation at Ser637 is thought to attenuate the GTPase activity of Drp1 and inhibit mitochondrial fission. In addition to affecting mitochondrial morphology, Ser637 site phosphorylation regulates cellular sensitivity to apoptotic stimuli and inner membrane cristae structure. It is thus striking that phosphorylation of DRP1 at nearby residues can have opposite effects on fission activity. The biochemical mechanisms underlying these effects remain to be elucidated.
In addition to phosphorylation, other posttranslational modifications, such as sumoylation and ubiquitination, can regulate Drp1 activity. Modification of Drp1 protein by the small ubiquitin-like modifier (SUMO) protein was found to regulate its fission activity [10]. Sumoylation usually alters the subcellular localization or protects against ubiquitin-mediated destruction of the substrate proteins. This appears to be the case for Drp1. Overexpression of SUMO leads to mitochondrial fragmentation and apoptosis, probably because SUMO1 protects Drp1 from degradation and enhances Drp1 binding to mitochondria. In other settings, sumoylation and ubiquitination often act together to regulate protein function. The mitochondrial E3 ubiquitin ligase MARCH V was identified as regulator of mitochondrial dynamics. Karbowski et al. [11] demonstrated that MARCH V may support fission by facilitating the sub-cellular trafficking and recruitment of Drp1 to sites of mitochondrial division. This occurs without affecting the stability of Drp1 [11]. It remains to be determined whether Drp1 or other regulators of mitochondrial dynamics are directly ubiquitinated by MARCH V.
The activity of Opa1 is regulated by proteolytic processing. Proteolytic cleavage releases OPA1 from the membrane, producing functionally distinct isoforms that are necessary for fusion activity. Several proteases have been identified that may mediate OPA cleavage, including the rhomboid-7 protease of Drosophila and its mammalian counterpart, PARL (presenilin-associated rhomboid-like) [12]. A recent study showed that mammalian PARL is phosphorylated at multiple sites, suggesting that the processing and activity of Opa1 might be tightly regulated in vivo [13]. Together, these studies raised the interesting possibility that cells continually adjust their rates of mitochondrial fission and fusion in response to changing energy demands or environmental stimuli through posttranslational modifications of the fission and fusion proteins.
Regulation of Mitochondrial Dynamics by Physiological or Pathological Stimuli
There appears to be a strong correlation between mitochondrial network morphology and bioenergetics, indicating that the form and function of this organelle are interrelated. For example, mitochondrial bioenergetics in Drp1-depleted cells were profoundly impaired; conversely, inhibition of respiratory chain complex I function alters the organization of the mitochondrial network [14•]. The molecular nature of this link is unclear. In the former case, it is possible that mitochondrial fission/fusion may affect metabolite sharing within the mitochondrial reticulum. Metabolites may become concentrated at the intercisternal space to ensure efficient use by the respiratory chain. Imbalanced fission/fusion may affect this process. In the latter case, it is possible that excessive reactive oxygen species (ROS) production caused by inhibition of electron transport chain may affect mitochondrial morphology. ROSs are produced by mitochondria during normal metabolism. ROS accumulation and the ensuing oxidative stress have been considered a cause of aging. Indeed, aging-related mitochondrial fission/fusion activity changes have been documented [15]. ROS could activate some intracellular signaling pathways that converge on the mitochondrial fission/fusion machinery.
Perturbation of genes involved in other mitochondrial processes also can affect mitochondrial morphology. For example, overexpression of a mouse homologue of human DNA polymerase delta interacting protein or mitochondrial single-stranded DNA binding protein, two proteins involved in mitochondrial DNA replication, increased elongated or fragmented mitochondria in mouse C2C12 myoblast cells, respectively, whereas inactivation of these proteins had the opposite effect [16]. A large-scale RNA interference (RNAi) screen in Caenorhabditis elegans revealed that, after knocking down 719 genes predicted to code for mitochondrial proteins, more than 80% of them caused abnormal mitochondrial morphology, including fragmentation and elongation [17••]. This report strongly suggests that fundamental mitochondrial functions, including metabolism and OXPHOS, are necessary for the maintenance of mitochondrial tubular network morphology. Consistent with this finding, a systematic genetic screen in yeast uncovered 119 essential genes that are required for the maintenance of mitochondrial morphology. Among these, genes were highly enriched for those involved in ergosterol biosynthesis, mitochondrial protein import, actin-dependent transport processes, vesicular trafficking, and ubiquitin proteasome-dependent protein degradation [18]. It thus appears that mitochondrial morphology can be influenced by a wide range of biologic processes and physiological conditions. Cellular signaling pathways underlying these processes may impinge on the fission/fusion machineries to affect mitochondrial morphology.
Mitochondrial Dynamics and Programmed Cell Death
Mitochondria undergo extensive fragmentation during apoptosis before activation of the caspases that lead to cell death. Upon induction of apoptosis, Drp1 translocates from the cytosol to potential sites of mitochondrial division. Inhibition of Drp1 function through the expression of a dominant negative form inhibits mitochondrial fragmentation, loss of membrane potential, and cell death, indicating its important role in apoptosis [19]. However, this conclusion may not apply to all cell death conditions. In a cellular model of Ca2+-induced cell death, Drp1 over-expression fragmented the mitochondrial network, blocked the propagation of intramitochondrial Ca2+ waves, and protected against cell death [20]. Mitochondrial fusion has been frequently shown to exert antiapoptotic activity. However, excessive fusion can lead to cell death, which emphasizes the importance of balanced mitochondrial fission/fusion activities in cellular physiology and cell survival [21]. The finding that Bax and Bak, two proapoptotic regulators, interact with Drp1 and mitofusin further supports the functional link between mitochondrial dynamics and apoptosis. However, there is evidence suggesting that mitochondrial fragmentation per se might not cause apoptosis and that the proapoptotic and profission functions of hFIS1 and Drp1 might be distinct [22]. Further studies are needed to clarify these issues.
Human Neurodegenerative Disorders Caused by Mutations in Mitochondrial Fission/Fusion Genes
Despite the seemingly ubiquitous expression of mitochondrial fission/fusion proteins and the general requirement for mitochondrial dynamics in cellular physiology, mutations in mitochondrial fission/fusion genes in humans have resulted in very specific neurodegenerative diseases. Mutations in Mfn2 cause Charcot-Marie-Tooth subtype 2A (CMT2A), a peripheral neuropathy characterized by muscle weakness and axonal degradation of sensory and motor neurons [23]. Mutations in Opa1 cause the most common form of optic atrophy, autosomal dominant optic atrophy (ADOA) [24]. Patients with ADOA exhibit progressive loss of vision and degeneration of the optic nerve and retinal ganglion cells. A dominant negative mutation in the human DRP1 gene resulted in elongated and tangled mitochondria concentrated at perinuclear region [25]. The patient carrying this mutation died shortly after birth and displayed some symptoms resembling those of ADOA and CMT2A. Moreover, mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1), which appears to be involved in mitochondrial fission, have been associated with CMT neuropathy type 4A [26].
Notably, although some of the human genetic mutations affect mitochondrial morphogenesis, others might cause disease through a fission/fusion-independent mechanism. For example, some Mfn2 mutants retain the ability to promote mitochondrial fusion [27•], suggesting that Mfn2 has other pathogenic roles not related to mitochondrial fusion. One study suggests that Mfn2 might have a role in mitochondrial trafficking [28•], whereas another suggests that Mfn2 can act by tethering mitochondria with the endoplasmic reticulum (ER), thus regulating Ca2+ uptake and signaling [29•]. Disruption of each of these functions could lead to peripheral axon degeneration and neuronal death. To distinguish the contribution of the various functions of the fission/fusion genes in disease pathogenesis, animal models would be instrumental. Knock-out models for Opa1, Mfn1, and Mfn2 have been established in mouse and Drosophila. To more faithfully model the human disease, transgenic animals expressing the exact pathogenic point mutations might be needed.
Involvement of Mitochondrial Fission/Fusion in Other Neurodegenerative Diseases
Mitochondrial defects have been consistently observed in idiopathic as well as familial PD (FPD) cases. Particularly, defects in respiratory complex I are common in idiopathic PD. Evidence supporting a mitochondrial etiology of PD comes from the observation of selective destruction of dopaminergic (DA) neurons in primates and rodents by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a complex I inhibitor; the detection of complex I defects in PD patients; and the recent identification of FPD genes that encode proteins targeted to or associated with mitochondria and affecting mitochondrial function.
FPD genes cloned so far include Syn, Parkin, DJ-1, Pink1, and LRRK2 [30]. Of particular interest is the phosphatase and tensin homologue (PTEN)–induced kinase 1 (Pink1) gene, which encodes a Ser/Thr kinase containing a mitochondrial targeting signal. Recent studies in Drosophila have provided compelling evidence that Pink1 and another FPD gene, Parkin, which encodes an E3 ubiquitin ligase, act in a linear pathway to control mitochondrial morphology in indirect flight muscle and DA neurons, tissues with high energy demand and requirement for mitochondrial function [31–33]. Loss of Pink1 or Parkin leads to mitochondrial aggregation and swelling phenotypes. Overexpression of Parkin can rescue Pink1 loss-of-function phenotypes, suggesting that Parkin acts downstream of Pink1 in this pathway. Genetic studies strongly suggest that the mitochondrial morphology defects in Pink1 and Parkin mutants are due to decreased fission or increased fusion. Enhancing mitochondrial fission activity by the overexpression of Drp1 or reducing fusion activity by the loss of one copy of Opa1 can rescue Pink1 mutant phenotypes in DA neurons [34••]. The biochemical mechanisms linking Pink1/Parkin to mitochondrial fission/fusion machinery remain unclear. Pink1 has recently been shown to phosphorylate Parkin and regulate its mitochondrial association [35], suggesting the mitochondrial substrate(s) of Parkin is likely to be involved in fission/fusion regulation.
In HeLa cells, RNAi-mediated downregulation of Pink1 results in abnormal mitochondrial morphology and altered membrane potential. The morphologic changes of mitochondria can be rescued by expression of wild-type Pink1 but not pathogenic Pink1 mutants. Primary fibroblast cells derived from patients with Pink1 mutations showed a similar defect in mitochondrial morphology [36••]. As with the fly Pink1 mutant, wild-type human Parkin but not pathogenic Parkin mutants could rescue the mitochondrial pathology in human cells. The mitochondrial morphology defects caused by Pink1 inactivation in mammalian cells appears to be qualitatively different from that observed in fly Pink1 mutant muscle or DA neurons. It is not clear whether this is due to cell type–specific effects or species-specific difference of Pink1 function in mitochondrial morphogenesis.
Mutations in Omi/HtrA2 have also been associated with PD. HtrA2 interacts with Pink1, and both are components of a hypothetical stress-sensing pathway. Pink1-dependent phosphorylation of HtrA2 might modulate its proteolytic activity, thereby contributing to an increased resistance of cells to mitochondrial stress [37•]. In the Drosophila eye, HtrA2 genetically acts downstream of Pink1 but functions independently of Parkin. In the same system, rhomboid-7, a mitochondrial protease implicated in Opa1 processing, acts as an upstream component of this pathway by cleaving the precursor forms of Pink1 and Omi [38•]. These data reveal intramembrane proteolysis as an important regulatory mechanism in the Pink1 pathway.
Further supporting a role for mitochondrial fission/fusion in PD is the finding that 6-hydroxydopamine (6-OHDA), a widely used neurotoxin for studying PD, induced profound mitochondrial fragmentation in SH-SY5Y cells [39]. This mitochondrial fragmentation precedes the collapse of mitochondrial membrane potential and cytochrome c release. Silencing of Drp1 prevented 6-OHDA–induced fragmentation of mitochondria and reduced 6-OHDA-induced cell death. These data demonstrate a role for mitochondrial fragmentation and Drp1 function in 6-OHDA–induced cellular toxicity that is relevant to PD pathogenesis.
Spinocerebellar ataxia-12 (SCA12) is caused by CAG repeat expansion in the noncoding region of the PPP2R2B gene encoding regulatory subunits of the protein phosphatase 2A (PP2A) enzyme. Stress-induced translocation of PP2A/Bβ2 to mitochondria promotes apoptosis, whereas silencing of Bβ2 protects hippocampal neurons against various neurotoxic insults. Bβ2 overexpression induces mitochondrial fragmentation, whereas Bβ2 inhibition results in mitochondrial elongation. Genetic studies suggest that mitochondrial fragmentation occurs upstream of apoptosis and is necessary and sufficient for neuronal death [40•]. These data implicate an imbalance of mitochondrial fission/fusion in the pathogenesis of SCA12.
Mitochondrial morphologic defects are also observed in AD. The presumed disease-causing Aβ peptide is localized to mitochondrial inner membrane where it may affect mitochondrial fission/fusion. In mammalian hippocampal neurons, Aβ peptide caused a reduction of Drp1 and Opa1 protein levels and induced aberrant mitochondrial morphology and distribution. Drp1 and Opa1 overexpression attenuated these mitochondrial abnormalities [41•].
In other neurodegenerative diseases, defects in mitochondrial function and distribution have been observed, although a direct link to mitochondrial fission/fusion has not been established. In Huntington’s disease, CAG expansion in the HTT gene is the primary cause. Mitochondrial morphologic defects have been observed in samples from patients with Huntington’s disease. HTT has been shown to interact with HTT-associated protein (HAP), which in turn interacts with kinesin motor and dynein-dynactin [42]. Because the recruitment of Drp1 to mitochondria is known to be regulated by these motor molecules [43], it is possible that mutant HTT affects Drp1 localization to mitochondria. Abnormal phosphorylation and aggregation of tau is associated with a number of neurodegenerative diseases known as tauopathies. Recent studies found that tau affects mitochondrial distribution in fly models: overexpression of human tau in larval motor neurons caused significant morphologic and functional disruption to the neuromuscular junctions. Motor neurons overexpressing tau had a significant reduction in functional mitochondria in the presynaptic terminal, suggesting that tau causes a synaptic dysfunction by reducing functional mitochondria at synapses [44].
Why Are Neurons Preferentially Affected by Aberrant Mitochondrial Fission/Fusion?
The preferential degeneration of neurons—especially neurons with long axons such as peripheral sensory neurons and motor neurons—when mitochondrial dynamics are compromised, reflects several aspects of neuronal physiology that have particular demand for mitochondrial function. First, synaptic transmission, a key feature of neuronal function, requires plasma-membrane-potential maintenance, synaptic neurotransmitter release and reuptake, and build-up of a reserve pool of vesicles for prolonged or high-frequency firing. All of these processes are energy demanding and require a sufficient number of active mitochondria to be present at synaptic sites. Other aspects of synaptogenesis, such as spine morphogenesis, also require sufficient dendritic mitochondrial content and mobility. For example, in cultured hippocampal neurons, the development and plasticity of dendritic spines requires movement of mitochondria into dendritic protrusions. Drp1 and Opa1 activity in these neurons are important for maintaining mitochondrial content required for spine formation [45]. Synaptic plasticity is thought to be the basis of learning and memory, and localized protein translation at specific synapses is a prevailing mechanism of synaptic plasticity. Protein translation is an energy-demanding process, underscoring the importance of dendritic mitochondria in synaptic plasticity.
The extremely polarized morphology of neurons and the considerable distance separating the nerve terminal from the cell body make it necessary to place the site of adenosine triphosphate production near the site of demand. This is especially true for peripheral sensory and motor neurons, which have disproportionately long axons and are the most polarized cells in the body. For these neurons, mitochondrial transport must act cooperatively with mitochondrial fission/fusion to ensure that sufficient mitochondria reach the synapses. Genetic studies in Drosophila have identified Milton and Miro as key proteins that regulate mitochondria transport in axons. Disruption of Milton or Miro (mitochondrial Rho-GTPase), like that of Drp1, leads to a severe loss of mitochondria in the terminals of peripheral or motor neurons and impairs synaptic transmission. Milton is a large coiled-coil–containing protein that localizes to mitochondria [46]. It binds to kinesin heavy chain to mediate the transport of mitochondria along the microtubule cytoskeleton. Miro is a mitochondrial outer membrane protein with two GTPase domains and two Ca2+-binding EF hand motifs [47]. A functional connection between Drp1 and Milton or dMiro is possible, given the interaction between Milton and microtubule motor proteins and the involvement of these motors in Drp1 transport. Further genetic studies among these genes will test their functional relationships.
Other key functions that mitochondria play in neurons are Ca2+ buffering and energy metabolism. Because calcium is a second messenger important for neuronal plasticity and excessive Ca2+ can lead to excitotoxicity, the capacity of mitochondria to buffer synaptic calcium levels is important to normal neuronal physiology. Unlike other cell types, neurons seem to lack the ability to activate the glycolytic pathway after inhibition of OXPHOS by ROSs such as nitric oxide [48]. This places a stronger dependence of neurons on OXPHOS, a central aspect of mitochondrial function, and may contribute to the particular vulnerability of neurons to mitochondrial dysfunction. Other factors may superimpose on these general features of neuronal physiology to confer cell type–specific vulnerability. For example, it was recently realized that mitochondria size and mass are not the same for all neurons (eg, the substantia nigra compacta DA neurons have relatively low mitochondria mass) [49•]. The low mitochondria mass in substantia nigra compacta DA neurons may contribute to the selective vulnerability of these neurons to external or internal toxic stimuli that affect mitochondrial fission and fusion.
Future Directions
Given the general involvement of mitochondrial fission/ fusion in the pathogenesis of a range of neurodegenerative diseases, further studies on this subject promise to offer new insights on disease mechanisms. Some of the topics worthy of future investigation include the following:
Biochemical relationships between disease-associated genes, such as Pink1 and Parkin, involved in PD and the fission/fusion machineries. Possible relationships could include kinase–substrate, E3 ligase–substrate, and Sumo ligase–substrate.
The molecular basis underlying the intimate relationship between mitochondrial form and function. How does fission/fusion affect electron transport chain activity, and how does the energy status of the cell or the functional state of mitochondria affect fission/fusion activity?
How do age-dependent changes of mitochondrial fission/fusion activity arise? Do aging-related signaling pathways and genes such as PI3K and FOXO affect fission/fusion activity?
What is the relationship between mitophagy and mitochondrial dynamics? Mitochondrial fission followed by selective fusion has been shown to segregate dysfunctional mitochondria and facilitate their removal by autophagy [50••]. How is the dysfunctional part of the mitochondria recognized during the fission process? How are dysfunctional mitochondria selected against during subsequent fusion?
Conclusions
Mitochondrial fission and fusion are evolutionarily conserved membrane remodeling processes that respond to a number of physiologic cues or pathologic stimuli. They likely represent an integration point for divergent cellular signaling pathways. Mitochondrial dynamics play an important role in normal neuronal function and are essential for such neuronal processes as synaptogenesis, Ca2+ buffering, axonal transport, and bioenergetics. As a result, dysfunction of mitochondrial dynamics has been linked to a number of neurodegenerative diseases. These findings have important therapeutic implications. Genes controlling mitochondrial dynamics represent potential drug targets for a number of neurodegenerative diseases, and compound screens targeting the mitochondrial fission/fusion process in cellular or animal models could yield promising new therapeutics.
Acknowledgments
The author apologizes to those whose work we could not cite because of space constraints. This work was supported in part by grants from the McKnight Foundation (Brain Disorders Award) and the National Institutes of Health (R01AR054926 and R21NS056878).
Footnotes
Disclosure
No potential conflict of interest relevant to this article was reported.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 3.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tondera D, Czauderna F, Paulick K, et al. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118(Pt 14):3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- 5.Koshiba T, Detmer SA, Kaiser JT, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 6.Cipolat S, Martins de Brito O, Dal Zilio B, et al. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Taguchi N, Ishihara N, Jofuku A, et al. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. This paper describes the effect of Cdk1-mediated phosphorylation of Drp1 on mitochondrial fragmentation frequently observed during mitosis. [DOI] [PubMed] [Google Scholar]
- 8•.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. Chang and Blackstone [9•] and Cribbs and Strack describe phosphorylation of Drp1 by PKA, the reversible dephosphorylation of this site by calcineurin, and the effect of this phosphorylation event on mitochondrial morphogenesis and cell survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. Cribbs and Strack [8•] and Chang and Blackstone describe phosphorylation of Drp1 by PKA, the reversible dephosphorylation of this site by calcineurin, and the effect of this phosphorylation event on mitochondrial morphogenesis and cell survival. [DOI] [PubMed] [Google Scholar]
- 10.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McQuibban GA, Saurya S, Freeman M. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature. 2003;423:537–541. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- 13.Jeyaraju DV, Xu L, Letellier MC, et al. Phosphorylation and cleavage of presenilin-associated rhomboid-like protein (PARL) promotes changes in mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:18562–18567. doi: 10.1073/pnas.0604983103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Benard G, Bellance N, James D, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120(Pt 5):838–848. doi: 10.1242/jcs.03381. This paper describes a bidirectional relationship between mitochondrial bioenergetics and morphology. [DOI] [PubMed] [Google Scholar]
- 15.Jendrach M, Pohl S, Voth M, et al. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech Ageing Dev. 2005;126:813–821. doi: 10.1016/j.mad.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Arakaki N, Nishihama T, Kohda A, et al. Regulation of mitochondrial morphology and cell survival by mitogenin I and mitochondrial single-stranded DNA binding protein. Biochim Biophys Acta. 2006;1760:1364–1372. doi: 10.1016/j.bbagen.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 17••.Ichishita R, Tanaka K, Sugiura Y, et al. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem. 2008;143:449–454. doi: 10.1093/jb/mvm245. This paper describes a large-scale RNAi-based genetic screen for genes that affect mitochondrial morphology when inhibited. Many genes encoding mitochondrially targeted proteins were shown to influence mitochondrial morphology. [DOI] [PubMed] [Google Scholar]
- 18.Altmann K, Westermann B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5410–5417. doi: 10.1091/mbc.E05-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank S, Gaume B, Bergmann-Leitner ES, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 20.Szabadkai G, Simoni AM, Chami M, et al. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 21.Huang P, Yu T, Yoon Y. Mitochondrial clustering induced by overexpression of the mitochondrial fusion protein Mfn2 causes mitochondrial dysfunction and cell death. Eur J Cell Biol. 2007;86:289–302. doi: 10.1016/j.ejcb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 23.Kijima K, Numakura C, Izumino H, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005;116:23–27. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- 24.Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 25.Waterham HR, Koster J, van Roermund CW, et al. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 26.Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglio-side-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–22. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- 27•.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405–414. doi: 10.1083/jcb.200611080. This paper reveals that some of the genetic mutations in Mfn2 associated with CMT2A do not affect mitochondrial fusion activity, suggesting that certain mitochondrial fusion-independent functions of Mfn2 are affected in these cases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Baloh RH, Schmidt RE, Pestronk A, et al. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–430. doi: 10.1523/JNEUROSCI.4798-06.2007. These authors and de Brito and Scorrano [29•] describe two new functions for Mfn2, one related to mitochondrial transport in axons and another in mitochondria-ER tethering and Ca2+ signaling. Disruption of these functions of Mfn2 could contribute to CMT2A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. These authors and Baloh et al. [28•] describe two new functions for Mfn2, one related to mitochondrial transport in axons and another in mitochondria-ER tethering and Ca2+ signaling. Disruption of these functions of Mfn2 could contribute to CMT2A. [DOI] [PubMed] [Google Scholar]
- 30.Bonifati V. Genetics of parkinsonism. Parkinsonism Relat Disord. 2007;13(Suppl 3):S233–S241. doi: 10.1016/S1353-8020(08)70008-7. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Gehrke S, Imai Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 33.Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 34••.Yang Y, Ouyang Y, Yang L, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. This paper provides strong genetic evidence that the mitochondrial morphology defects in indirect flight muscle and DA neurons observed in Drosophila Pink1 mutant are caused by decreased fission or increased fusion. Genetic manipulations that increase fission or inhibit fusion rescued mutant phenotypes. This study for the first time implicates mitochondrial dynamics in DA neuron dysfunction in PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y, Park J, Kim S, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 36••.Exner N, Treske B, Paquet D, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. This paper reported that inhibition of mammalian Pink1 resulted in abnormal mitochondrial morphology, which can be rescued by Parkin overexpression, suggesting that similar to the situation in Drosophila, Pink1 and Parkin may act in a common pathway to regulate mitochondrial morphogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37•.Plun-Favreau H, Klupsch K, Moisoi N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. These authors and Whitworth et al. [38•] reported an interesting relationship between HtrA2 and Pink1, both associated with FPD. Pink1 was found to regulate HtrA2 phosphorylation, whereas HtrA2 may regulate the proteolytic processing of Pink1. [DOI] [PubMed] [Google Scholar]
- 38•.Whitworth AJ, Lee JR, Ho VM, et al. Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson’s disease factors Pink1 and Parkin. Dis Model Mech. 2008;1:168–174. doi: 10.1242/dmm.000109. These authors and Plun-Favreau et al. [37•] reported an interesting relationship between HtrA2 and Pink1, both associated with FPD. Pink1 was found to regulate HtrA2 phosphorylation, whereas HtrA2 may regulate the proteolytic processing of Pink1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez-Lazaro M, Bonekamp NA, Galindo MF, et al. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic Biol Med. 2008;44:1960–1969. doi: 10.1016/j.freeradbiomed.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 40•.Dagda RK, Merrill RA, Cribbs JT, et al. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bbeta2 antagonizes neuronal survival by promoting mitochondrial fission. J Biol Chem. 2008;283:36241–36248. doi: 10.1074/jbc.M800989200. This paper implicated mitochondrial fission/fusion imbalance in the pathogenesis of SCA12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Wang X, Su B, Siedlak SL, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. This paper reported a role for abnormal mitochondrial morphology and distribution in Aβ-induced neurotoxicity in mammalian hippocampal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGuire JR, Rong J, Li SH, et al. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- 43.Varadi A, Johnson-Cadwell LI, Cirulli V, et al. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci. 2004;117(Pt 19):4389–4400. doi: 10.1242/jcs.01299. [DOI] [PubMed] [Google Scholar]
- 44.Chee FC, Mudher A, Cuttle MF, et al. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol Dis. 2005;20:918–928. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 45.Li Z, Okamoto K, Hayashi Y, et al. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Stowers RS, Megeath LJ, Gorska-Andrzejak J, et al. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- 47.Guo X, Macleod GT, Wellington A, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 48.Almeida A, Almeida J, Bolanos JP, et al. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Liang CL, Wang TT, Luby-Phelps K, et al. Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease. Exp Neurol. 2007;203:370–380. doi: 10.1016/j.expneurol.2006.08.015. This paper reported an interesting observation that mitochondria mass is low in mouse substantia nigra DA neurons. This may help explain the particular vulnerability of these neurons to toxic stimuli that affect mitochondrial function. [DOI] [PubMed] [Google Scholar]
- 50••.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. This paper reported an interesting connection between mitochondrial fission/fusion dynamics and elimination of dysfunctional mitochondria by autophagy. It was shown that after fission, the dysfunctional mitochondria are prevented from fusion with other mitochondria and are selectively removed by autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]