

Abstract
Objective
Complement mediated injury of the neuromuscular junction is considered a primary disease mechanism in human myasthenia gravis and animal models of experimentally acquired myasthenia gravis (EAMG). We utilized active and passive models of EAMG to investigate the efficacy of a novel C5 complement inhibitor rEV576, recombinantly produced protein derived from tick saliva, in moderating disease severity.
Methods
Standardized disease severity assessment, serum complement hemolytic activity, serum cytotoxicity, acetylcholine receptor (AChR) antibody concentration, IgG subclassification, and C9 deposition at the neuromuscular junction were used to assess the effect of complement inhibition on EAMG induced by administration of AChR antibody or immunization with purified AChR.
Results
Administration of rEV576 in passive transfer EAMG limited disease severity as evidenced by 100% survival rate and a low disease severity score. In active EAMG, rats with severe and mild EAMG were protected from worsening of disease and had limited weight loss. Serum complement activity (CH50) in severe and mild EAMG was reduced to undetectable levels during treatment, and C9 deposition at the neuromuscular junction was reduced. Treatment with rEV576 resulted in reduction of toxicity of serum from severe and mild EAMG rats. Levels of total AChR IgG, and IgG2a antibodies were similar, but unexpectedly, the concentration of complement fixing IgG1 antibodies was lower in a group of rEV576-treated animals, suggesting an effect of rEV576 on cellular immunity.
Interpretation
Inhibition of complement significantly reduced weakness in two models of EAMG. C5 inhibition could prove to be of significant therapeutic value in human myasthenia gravis.
Myasthenia gravis (MG) is a neuromuscular transmission disease caused primarily by acetylcholine receptor (AChR) autoantibodies,1,2 and several lines of evidence indicate that the fixation of complement at the neuromuscular junction (NMJ) is an important factor in determining disease severity.3 C3 activation fragments and the membrane attack complex are detected at the NMJ of patients with MG, depletion of serum C3 and C4 is appreciated in some patients, and terminal complement components are found in patient sera.4 – 6 In humans, antigenic modulation and direct blockade of the AChR also likely contribute to MG pathology, but their relative significance is not known.7,8
Complement is paramount in driving disease pathology in both models of the experimentally acquired myasthenia gravis (EAMG), whether produced by administration of AChR antibodies or through immunization with purified AChR. With rare exception,9 passive transfer EAMG is induced only by complement-fixing antibodies, and complement depletion by cobra venom factor eliminates NMJ injury and weakness.10 Antibodies directed toward C511 and soluble complement receptor (sCR1)12 are protective in EAMG produced by AChR antibody administration. C5-deficient mice13 or anti-C6 Fab antibody–treated EAMG rats14 are more resistant and develop less severe disease, whereas an absence of cell surface regulators of complement leads to significantly greater disease with EAMG induced by AChR antibodies.15–17
The inhibition of complement as a therapeutic approach is beginning to be applied to human disorders. Pexelizumab, a monoclonal antibody directed against C5, has demonstrated short-term safety and efficacy in humans in myocardial infarction,18 coronary artery bypass graft surgery,19 and lung transplantation.20 A phase 3 trial of eculizumab, a single-chain fragment of anti-C5, in treatment of paroxysmal nocturnal hemoglobinuria21 demonstrated significant clinical benefits with minimal adverse effects. MG would be expected to respond well to complement inhibition especially in acute exacerbations when complement-mediated NMJ injury is expected to be maximal.
rEV576 is a novel complement inhibitor, which appears ideally suited for consideration as a therapeutic agent. It is a 17kDa protein22 identified from the saliva of the tick Ornithodoros moubata and belongs to the lipocalin family of proteins, which inhibit inflammatory mediators.23 Because the agent is derived from ticks that apply themselves to mammalian species for weeks without inducing a reaction, there is an expectation that rEV576 will have limited adverse effects in humans. The compound is not known to cause allergic reactions or to have effects beyond those related to complement inactivation.24 rEV576 specifically targets the C5 activation step and has been shown to inhibit both the classic and the alternative pathways, and act specifically at the C5 step of complement activation.25 The simplest explanation for the activity of rEV576 is direct binding to C5 that prevents interaction with the C5 convertase rather than by blocking the C5a cleavage site.26
In this study, the active and passive models of EAMG were used to investigate the therapeutic activity of rEV576. Rats treated with rEV576 demonstrated dramatic improvements in weakness, recovery from weight loss, and reduced C9 deposition at the NMJ. The findings suggest that this novel complement inhibitor could prove to be an effective treatment in human MG.
Materials and Methods
Animals
Six- to 10-week-old female Lewis rats (Harlan, Indianapolis, IN) were used and maintained in the Case Western Reserve University animal facility. Animals were housed in isolator cages in a pathogen-free environment, and autoclaved rodent chow and water (Clintech Nutrition, Deerfield, IL) were provided ad libitum. All studies were conducted according to approved protocol by the Case Western Reserve University Institutional Animal Care and Use Committee.
Induction of Experimentally Acquired Myasthenia Gravis and Treatment Protocol
EAMG was passively induced using rat anti–mouse muscle AChR McAb-3 antibody (gift from Vanda Lennon, Mayo Clinic, Rochester, MN).27 AChR was purified from the electric organs of Torpedo californica (Pacific Biomarine, Venice, CA) by affinity chromatography.28 The purified AChR was used to induce EAMG and as antigen for in vitro testing. rEV576 was prepared and provided as a gift from Evolutec (Reading, United Kingdom).
In passive EAMG, McAb-3 (1 μg/gm) was administered intraperitoneally (IP) 2 hours after initial treatment with complement inhibitor (3mg/rat IP). After the initial dose, rats were injected with three consecutive doses of rEV576 (1mg every 12 hours). Serum complement activity CH50 was measured on days 2 and 5 of the experiment. In a dose-effectiveness study, 16 animals were divided into 4 groups and received single doses of rEV576 (0.1, 1.0, 10mg, and phosphate-buffered saline [PBS] only) 2 hours before administration of AChR (McAb-3) antibody.
For active EAMG, two independent experiments were performed. In each, 24 rats were immunized subcutaneously at the base of the tail with 40 μg purified AChR emulsified in Complete Freund’s adjuvant (CFA; Sigma, St. Louis, MO) mixed with 10mg of Mycobacterium tuberculosis (10mg in 1 ml CFA, strain H37 RA; Difco, Detroit, MI). Based on behavioral assessments (weight loss, grip strength, disease severity scale), rats were divided at day 32 in groups with severe (S-EAMG) and mild (M-EAMG) disease. Half of the animals in each group were treated IP with 1mg rEV576 for 10 days in 12-hour intervals (days 32–42). As another comparison group, control groups of 12 animals were immunized with CFA only or M. tuberculosis in CFA using the same protocol. Complement activity (CH50) was measured at days 0, 14, and 28, and at the end of treatment.
Animal Assessment
Rats were evaluated in a blinded fashion using weight, disease severity classification, and quantitative strength measurements. For passive EAMG, rats were assessed twice daily at 12-hour intervals. In active EAMG, rats were examined twice weekly and then daily as weakness became evident. A generally accepted disease severity scale was used: 0 = can grip and lift lid of a cage; 1 = can grip but cannot lift the lid of a cage; 2 = unable to grip cage lid; 3 = unable to grip and has hind-limb paralysis; and 4 = moribund.12 A grip strength meter (Columbus Instruments, Columbus, OH) was used to assess forelimb strength. Measurement was performed with digital force gauge (Ametek, Largo, FL), which assesses the peak force by which an animal can grasp a grid pull bar. Before the measurement, each rat was exercised with three to five paw grips; then five grips were registered by the grip strength meter. Mean grip strength in grams for each rat was used for analysis.
CH50: Whole-Complement Hemolytic Assay
Serum complement activity was assessed by CH50 assay according to the manufacturer’s protocol (Sigma).29 One hundred microliters rat serum as a source of complement was diluted 1:200 in gelatin Veronal Buffer (Sigma, St. Louis, MO) and 100 μl of 2 × 108 sensitized sheep erythrocytes was mixed on ice, and the whole reaction was performed at 37°C. At the end of incubation time (60 minutes), sheep red blood cells were removed by centrifugation (700 rpm for 5 minutes) and complement hemolysis was measured at 412nm (Spectra MAX 340; Molecular Devices, Sunnyvale, CA). Serum complement activity was examined at times described in the treatment protocol.
Immunoglobulin G Subclasses
IgG subclasses (IgG, IgG1, IgG2a, IgG2b) antibody levels were determined by enzyme-linked immunosorbent assay (ELISA) in actively induced EAMG. Plates (96-well, Immulon; Fisher Scientific, Pittsburgh, PA) were coated with 100 μl/well goat anti–rat capture antibodies at 4°C overnight, then blocked at 37°C for 1 hour with 200 μl PBS Tween 20 (Sigma). Rat serum samples at dilution 1:200 were added (100 μl/well) and incubated at 37°C for 1 hour. Washed plates were incubated at 37°C for 45 minutes with horseradish peroxidase–conjugated secondary antibodies (goat anti–rat IgG, IgG1, IgG2a, IgG2b; Bethyl Laboratories, Montgomery, TX) diluted at 1:5,000. Colorimetric reaction was developed by incubating 15 to 30 minutes at room temperature with 3,3′5,5′-tetramethylbenzidine (TMB) substrate (Sigma) and stopped with 2M H2SO4. All quantitative standards, samples, or controls were analyzed in triplicate at 450nm (Spectra MAX 340; Molecular Devices). Total IgG, IgG1, IgG2a, and IgG2b concentrations were determined using standard curves, as described in manufacturer’s protocol. The sensitivity of ELISA is in the range of 1.9 to 10,000ng/ml. Samples were diluted to fall within the standard curve range.
Acetylcholine Receptor Antibodies
Sandwich ELISA was used for the detection of AChR antibodies. Serum levels of AChR antibodies were examined 2 and 4 weeks after immunization, and at the end of the experiment. Ninety-six-well plates (Immulon; Fisher Scientific) were coated with 10 μg/ml purified AChR at 4°C overnight and blocked for 2 hours at room temperature with 200 μl PBS Tween 20 (Sigma). Rat serum samples at dilution 1:200 were added (100 μl/well) and incubated at room temperature for 90 minutes. Washed plates were incubated for another 90 minutes with goat anti–rat IgG peroxidase conjugate (Jackson ImmunoResearch, West Grove, PA). The color reaction was developed with peroxidase substrate (Sigma). Reaction was stopped and analyzed. The sensitivity of AChR ELISA was 3 to 80 μg/ml.
Cytotoxicity Assay for Experimentally Acquired Myasthenia Gravis Serum
The Toxilight BioAssay Kit (Cambrex, Rockland, ME) for measuring the release of adenylate kinase from injured cells was used to measure the toxicity of rat serum on the human rhabdomyosarcoma cell line (CCL-136; ATCC, Manassas, VA). RD cells that express AChR30 were cultured in 96-well plates to near confluence, washed twice with Dulbecco’s minimum essential medium (Hyclone, Logan, UT), and incubated in four dilutions (1:2, 1:4, 1:8, and 1:16) for 1 hour at 37°C. Cells were incubated for 15 minutes with 100 μl adenylate detection reagent, and toxicity was measured on Tecan Infinity M200 (TECAN, Salzburg, Austria). Serum samples from each animal were examined in triplicates, and results shown are calculated in relative luminescent unit.
C9 Deposition at the Neuromuscular Junction
For analysis of C9 deposition at the NMJ, 10 μm cryosections of rat diaphragms were mounted on Superfrost Plus treated slides (Fisher Scientific). Diaphragms were collected on the last day of the 10-day treatment phase, at which time experimental and control animals were killed. Sections were washed with PBS and blocked for 45 minutes with 1.5% goat serum (Vector Laboratories, Burlingame, CA), stained for 1 hour with rabbit anti–rat C9 (gift of M. Edward Medof). After washing, a secondary biotinylated anti-rabbit antibody (Vector Laboratories) diluted at 1:400 (30 minutes) was applied. Slides were washed and stained with (5-[4,6-dichlorotriazin-2-ylamino-fluorescein[DTAF] conjugated streptavidin (Jackson ImmunoResearch Laboratories) and Texas Red–labeled bungarotoxin (1:500; Molecular Probes, Eugene, OR) to identify the NMJ. The sections were viewed with Olympus BX50 microscope (Olympus, Center Valley, PA), and digital images were captured with a Spot RT camera (Diagnostic Instruments, Sterling Heights, MI) and analyzed with Image Pro software (Media Cybernetics, Silver Springs, MD). Pixel density measurements were used to quantitate C9 immunoreactivity. A total of 393 NMJs were analyzed in each group. The data were sorted by increasing pixel densities, and total number of NMJs with C9 immunofluorescence intensities was plotted in graph.
Statistical Analysis
The data were analyzed and tested for statistical significance using paired t tests, and analysis of variance was applied where more than two groups were compared. Results were considered significantly different when p < 0.05. C9 deposition and pixel densities were analyzed by analysis of variance and Bonferroni posttest analysis.
Results
rEV576 Is Protective from Passively Induced Experimentally Acquired Myasthenia Gravis in a Dose-Dependent Fashion
In the first set of experiments, we assessed the prophylactic effect of rEV576 in passive EAMG (Fig 1). Treated rats had a 100% survival rate compared with the 66% survival rate of untreated rats (p < 0.003 at day 2). Complement inhibitor–treated rats showed mild weakness with a low disease severity score, whereas all surviving untreated rats developed severe weakness at 24 and 36 hours after EAMG induction (p < 0.0015, except days 0 and 4). After the rEV576 administration, serum complement activity was reduced to undetectable levels and remained so during the first 48 hours of the experiment (p < 0.0005). Serum complement activity was undetectable during treatment and rebounded within 24 hours after the last rEV576 administration (p < 0.10).
Fig 1.
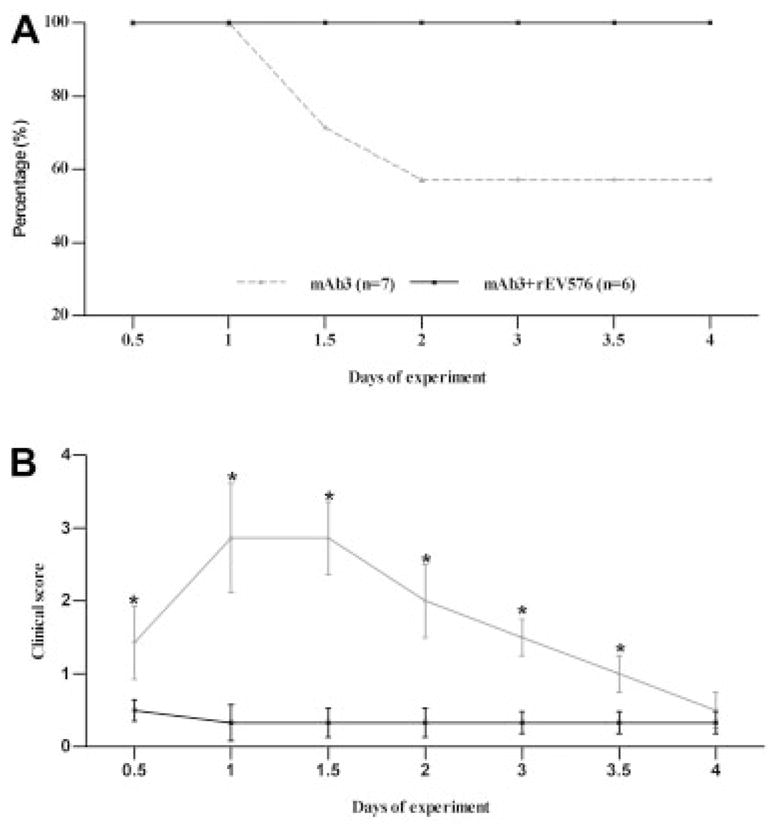
Prophylactic effect of C5 complement inhibitor (rEV576) in passive experimentally acquired myasthenia gravis (EAMG). Survival rate (A) and average clinical score (B) in surviving rats were examined in passively induced EAMG. Female Lewis rats matched for age and weight were injected with McAb3. Untreated animals (n = 7; gray lines) experienced development of severe weakness, and three rats died within 48 hours. EAMG rats treated with rEV576 (n = 6; black lines) were protected from mortality and showed only mild weakness with a low severity score (data expressed as mean score assessed every 12 hours).
We next evaluated the effect of rEV576 dose on weakness and survival (Fig 2) using escalating single doses (0.1, 1.0, and 10mg). Severity of disease evaluated by the severity score and weight loss demonstrated the greatest protective effect with a single 10mg dose of rEV576. However, lower doses of rEV576 were also effective in limiting severity of EAMG (p < 0.01). Disease clinical scores (days 1, 2, and 3) and weight loss (day 2) were significantly different between treated (EAMG + rEV576; p < 0.001) and untreated groups (EAMG only).
Fig 2.

C5 complement inhibitor is protective in a dose-dependent manner. Female Lewis rats were pretreated with single doses of rEV576 (10.0 [black circles], 1.0 [gray squares], 0.1mg [gray triangles]). Incidence and disease severity were significantly lower in rEV576-treated compared with untreated (black triangles) experimentally acquired myasthenia gravis (EAMG) rats (p > 0.01). Severity of disease evaluated by weight loss (A) and clinical score (B) showed statistically significant differences (asterisks) between complement inhibitor–treated (EAMG + rEV576; p > 0.001) and untreated rats (EAMG only). Results are shown as ± standard error of the mean for each experimental group (n = 4).
rEV576 Moderates Severity of Active Experimentally Acquired Myasthenia Gravis
Rats with active EAMG developed weakness by week 4 after AChR immunization. Animals showed signs of either S-EAMG with disease severity score of 2 or greater or M-EAMG with disease severity score of 0 to 1, which allowed assessment of the effect of complement inhibition based on disease severity. Treatment of S-EAMG and M-EAMG rats started within 24 hours of identification of weakness with rEV576 for 10 days. In the first active EAMG experiment, S-EAMG rats all had a disease severity score of 2 and had no difference in grip strength on the day of treatment initiation. However, all the untreated rats required euthanasia within 48 hours of initiation of the treatment portion of the experiment, whereas rEV576 prevented the need for euthanasia (Table). Treatment led to improvement in severity score but not in grip strength from baseline. rEV576 reversed weight loss in the M-EAMG group. All M-EAMG rats had a clinical score of 1 and comparable grip strength (M-EAMG: 489.20 ± 17.35; M-EAMG + rEV576: 494.66 ± 22.76). A second EAMG experiment confirmed the results of the first. None of S-EAMG rats required euthanasia, but the effect of complement inhibition on weight loss and weakness (Fig 3; see the Table) was similar to the first experiment. Untreated S-EAMG rats continued to lose weight (>10%) and grip strength (>25%) by weeks 5 and 6. Rats treated with rEV576 showed significant improvement in disease severity (p < 0.0125). Treatment with M-EAMG was also beneficial. Complement inhibitor–treated rats gained 3.4% of weight, whereas the untreated animals with identical disease severity scores lost an average of 4.6% over the same time, but no significant change occurred in grip strength. In both experiments, the most dramatic differences were found in the S-EAMG group.
Table 1.
Grip Strength and Severity Score in Severe Experimentally Acquired Myasthenia Gravis Rats Treated with Complement Inhibitor rEV576
| Experiments | S-EAMG Group |
S-EAMG + rEV576 Group |
Control Group |
|||
|---|---|---|---|---|---|---|
| Grip Strength | Severity Score | Grip Strength | Severity Score | Grip Strength | Severity Score | |
| Experiment 1 | ||||||
| Day 0 | 380.20 ± 18.31 | 2 | 442.25 ± 10.66 | 2 | 522.40 ± 9.75a | 0 |
| Day 10 | — b — | b | 444.00 ± 9.60 | 1 | 553.06 ± 9.20a | 0 |
| Experiment 2 | ||||||
| Day 0 | 493.28 ± 21.27 | 2 | 486.33 ± 5.76 | 2 | 556.20 ± 10.00a | 0 |
| Day 10 | 479.30 ± 12.42 | 2 | 507.90 ± 11.11c | 1 | 576.33 ± 12.78a | 0 |
Results are shown as ± SEM for each of the groups.
Experimentally acquired myasthenia gravis (EAMG) rats were scored and examined for the grip strength before treatment (day 0) and at the end of treatment with C5 complement inhibitor (day 10). Severity score always improved after treatment with rEV576. n = 4–7 rats in each group.
Significantly greater (p < 0.001) than the corresponding groups immunized with acetylcholine receptor.
All untreated EAMG rats in Experiment 1 were euthanized on days 1 or 2.
Significantly greater (p < 0.0125) than the corresponding group untreated with rEV576 (Experiment 2).
S-EAMG = experimentally acquired myasthenia gravis; SEM = standard error of the mean.
Fig 3.
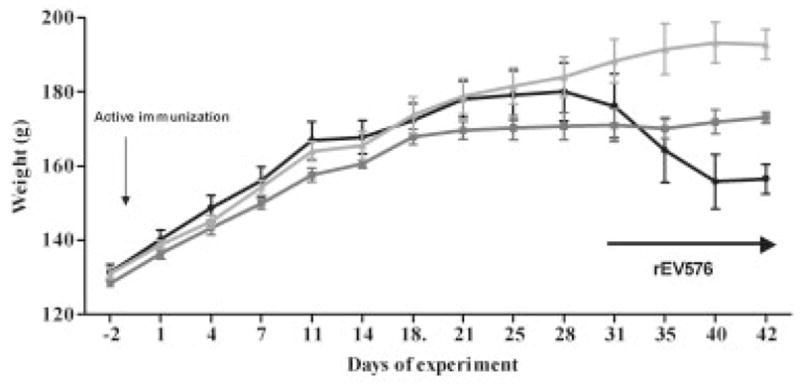
Therapeutic effect of C5 complement inhibitor (rEV576) in actively induced experimentally acquired myasthenia gravis (EAMG). Depending on weight loss and grip strength, we randomly divided rats into two groups. Subsequently, rats were treated intraperitoneally with C5 complement inhibitor or phosphate-buffered saline (see Materials and Methods). Inhibition of complement prevented weight loss in severe EAMG (S-EAMG). Results from one representative experiment are shown as ± standard error of the mean for each of three experimental groups. Black circles represent S-EAMG group (n = 9); dark gray squares represent S-EAMG + rEV576 group (n = 7); light gray triangles represent not immunized group (n = 6).
Serum Complement Activity
Complement activity in active EAMG rats was tested in vitro before, and after, the immunization with AChR. We did not detect significant differences in complement activity among experimental groups at days 0 or 2 and 4 weeks after immunization. Serum complement activity was similar in both S-EAMG and M-EAMG. Administration of complement inhibitor (rEV576) for 10 days at 12-hour intervals resulted in undetectable serum complement activity using the CH50 assay (p < 0.001, data not shown). Similarly, complement activity was undetectable at day 5 of treatment and immediately at the end of treatment (p < 0.001, data not shown).
Acetylcholine Receptor Antibody and Immunoglobulin G Subclass Levels
The levels of specific anti-AChR antibodies in serum were measured before the AChR immunization, on days 14 and 28 after immunization, and at the end of treatment phase (day 42), and there were no differences between S-EAMG and M-EAMG or treated and untreated rats. Ten days of treatment with rEV576 did not affect the AChR antibodies levels in (S-EAMG: 63.504 ± 18.342 μg/ml; S-EAMG + rEV576: 56.758 ± 14.343 μg/ml). The levels of total IgG, IgG2a, and IgG2b (p < 0.057) were comparable among all tested groups. However, M-EAMG–treated rats showed a difference in the IgG1 (p < 0.007) complement-fixing antibody (Fig 4), suggesting an effect of complement inhibition or rEV576 specifically on humoral response. Analysis with Bonferroni posttest correction confirmed a significant decrease (p < 0.001) in the level of IgG1 in complement inhibitor–treated rats.
Fig 4.
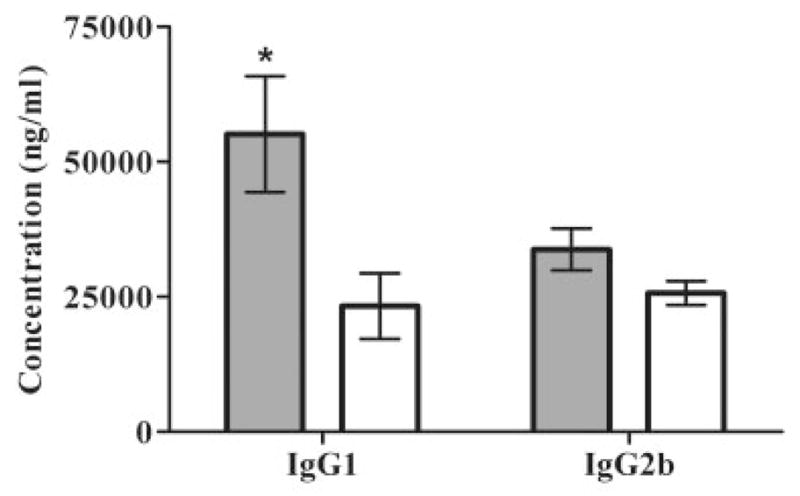
Effect of rEV576 on IgG subclasses in experimentally acquired myasthenia gravis (EAMG). Rat serum from treated and untreated EAMG rats was collected at the end of treatment (6 weeks after immunization). The level of individual IgG subclasses was measured by enzyme-linked immunosorbent assay (ELISA). There were no significant differences in total IgG and IgG2a subclasses in severe (S-EAMG) or mild EAMG (M-EAMG) (not shown). M-EAMG rats treated with rEV576 showed a decreased level of complement-fixing IgG1 (p > 0.001). Results are shown in picograms per milliliter (pg/ml; n = 6 rats per group). Bars indicate ± standard error of the mean of triplicate ELISA determinations with sera from individual rats. Gray bars represent M-EAMG; white bars represent M-EAMG + rEV576.
Treatment with Complement Inhibitor Eliminates Toxicity of Experimentally Acquired Myasthenia Gravis Serum
A bioluminescent assay was used to measure complement-related toxicity of serum on a human rhabdomyosarcoma cell line (CCL-136) expressing AChR. Rat serum from control (not immunized) animals showed only minimal cell damage. The greatest serum toxicity was detected in untreated rats with either S-EAMG or M-EAMG (p < 0.01). Toxicity of sera from rEV576-treated EAMG rats was considerably reduced (Figs 5A, B; p < 0.01), supporting that cell injury is related to complement activation.
Fig 5.

Complement cytotoxicity is attenuated after the treatment with rEV576. Rat serum diluted 1/2, 1/4, 1/8, or 1/16 was incubated for 1 hour with rhabdomyosarcoma cells (5 × 104 cells/well). Treatment with complement inhibitor resulted in a significant reduction of serum toxicity of severe (S-EAMG) (A) and mild experimentally acquired myasthenia gravis (M-EAMG) (B) rats (p < 0.01). Results are expressed as relative luminescent unit (RLU) ± standard error of the mean of six rats per experimental group. (A) Black circles represent S-EAMG group; dark gray squares represent S-EAMG + rEV576 group; light gray triangles represent normal rat serum. (B) Black circles represent M-EAMG group; dark gray squares represent M-EAMG + rEV576 group; light gray triangles represent normal rat serum.
rEV576 Decreases C9 Deposition at Neuromuscular Junction
Bungarotoxin-identified diaphragm end plates were analyzed for C9 deposition by density scan analysis. As shown in Figure 6A, naive, normal rats were negative for C9. The highest C9 deposition was found in untreated EAMG rats. NMJ of EAMG rats showed nearly universal staining for C9. Density scan confirmed significant differences in C9 deposition between rEV576-treated and untreated rats. Figure 6B shows the percentage of diaphragm NMJ with certain fluorescence intensities. Junctions from anti-C5 (rEV576)–treated rats have lower percentage of high-intensity C9 staining (p < 0.001, two-tailed test).
Fig 6.
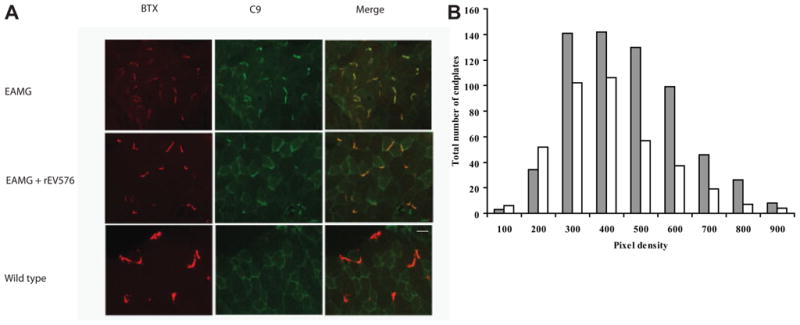
C9 deposition at the neuromuscular junction (NMJ; diaphragm) is significantly reduced in rEV576-treated rats. In comparison with rEV576-treated rats, there is more intense C9 staining in experimentally acquired myasthenia gravis (EAMG) untreated rats. (A) Bungarotoxin (BTX)-stained end plates with reduced C9 deposition in muscle of rEV576-treated rats. Original magnification, 40×. Scale bar = 50 μm). (B) Density scan of fluorescent intensity related to C9 immunoreactivity at the NMJ. Graph shows the number of NMJs with certain C9 fluorescence intensities. Muscle of rEV576-treated rats have a lower percentage of high-intensity staining junctions. The difference between EAMG- (gray bars) and EAMG + rEV576 –treated (white bars) groups is significant (p < 0.0027, two-tailed; n = 6).
Discussion
In EAMG, complement activation by AChR antibodies is an important mechanism for producing NMJ injury and subsequent neuromuscular transmission failure. We have identified that complement inhibition utilizing a novel inhibitor of C5 activation is successful in prevention and reversal of weakness produced in both models of EAMG produced by either AChR antibody administration or AChR immunization. The effect was mediated through complement inactivation as evidenced by dramatically reduced serum complement activity, diminished C9 deposition at the NMJ, and reduced cytotoxicity of serum from treated animals. The sera from rats treated with rEV576 had a reduction of cytotoxicity in an in vitro assay. The assay exploited a human rhabdomyosarcoma cell line, which expresses AChR on cell surfaces and is commonly used as a source of human AChR for assays of AChR antibodies.31,32 Sera from both M-EAMG and S-EAMG produced significant cell injury, but sera of rEV576-treated EAMG rats were no different from control sera in the severity of cytotoxic effects. No difference existed in AChR antibody levels between treated and untreated rats with active EAMG, which limits concern that the groups had inherent differences in AChR antibody production. Assuming the AChR antibody profiles do not differ between treated and untreated rats (see later), the experiment indicates that complement inhibition produced by rEV576 limited cell injury in the assay.
The results lend further support to the well-established hypothesis that the primary mechanism of injury to the NMJ by EAMG is through the action of complement.3 The expected mechanism of rEV576 acting through complement inhibition, in both passive and active EAMG, is demonstrated by the complete abolition of serum complement activity and the reduction of C9 deposition at the NMJ. All groups of rats had similar AChR antibody levels, so that a moderation of EAMG was not mediated through a direct effect on antibody production in active EAMG.
We found that rats with M-EAMG treated with rEV576 produced less IgG1, which in rat activates complement.33 The result is surprising given a lack of precedent for suppression of the C5 step of complement activation to influence humoral immunity; however, there is an emerging literature34 that demonstrates that alterations in complement or complement regulatory proteins may influence antibody profiles. The result suggests that complement activation, or perhaps rEV576 specifically, may have a direct effect on adaptive immunity. Interestingly, IgG1, IgG2a, and IgG2b were no different between the S-EAMG–treated and untreated rats. Perhaps, in more severe disease, complement inhibition effects are masked because of the inherently more active disease process. The alteration of IgG1 subclass does offer the possibility of complement inhibitor treatment also influencing IgG subclasses.
Tick salivary proteins have evolved to be poorly immunogenic as evidenced by a single tick being able to apply itself to a mammal for 2 weeks to finish a blood meal and thousands of ticks infesting a single large mammal without induction of an immune response.35 These observations would support a low likelihood of adverse effects for rEV576 in mammals. In this study and another, more limited, investigation,24 no adverse effects were observed in the experimental animals, although formal toxicity assessments were not preformed.
In human MG and active EAMG, the antibody response is polyclonal, and noncomplement could lead to neuromuscular transmission failure. Decreased AChR half-life by AChR antibody can lead to a reduction of AChR severe enough to compromise neuromuscular transmission,36 and direct effects on AChR channel function could also contribute to transmission failure. Despite these other potential mechanisms, complement-mediated injury of the NMJ is likely a major contributor to the human disease.3,37 The passive model of EAMG used in this investigation does not recapitulate the complexities of the breakdown of self-tolerance in human MG, but does accurately reproduce the final common pathway of antibody-induced, complement-mediated destruction of NMJ. EAMG induced by AChR immunization leads to a loss of self-tolerance and polyclonal AChR antibody production, as occurs in human MG. The data from both models taken together strongly support that rEV576 could prove to be effective for human MG.
Anti-C5,11,17 anti-C6,14 and soluble CR112 have all been shown to limit severity of passive EAMG. Hepburn and colleagues24 also evaluated rEV576 in a passive transfer model of EAMG utilizing a different monoclonal antibody and demonstrated its effectiveness. This study extends the previous study by more precisely characterizing the time course of complement inhibition of rEV576 and extending the study to active EAMG, which more precisely recapitulates the human disease. We conclude that rEV576 would be a dramatically effective treatment for MG patients during acute disease onset, especially in situations of severe weakness. The rapid onset and offset of action could be particularly beneficial in titration of its effects in the intensive care setting.
Acknowledgments
This work was supported by the NIH NEI, National Eye Institute (R24EY014837, H.J.K.). Parasitological Institute of the Slovak Academy of Sciences, Slovak Grant Agency VEGA (2/7186, J.S.) and a Research to Prevent Blindness Challenge departmental grant (L.L.K.).
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol. 2002;2:797–804. doi: 10.1038/nri916. [DOI] [PubMed] [Google Scholar]
- 2.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kusner LL, Kaminski HJ, Soltys J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Expert Rev Clin Immunol. 2008;4:43–52. doi: 10.1586/1744666X.4.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engel AG, Lambert EH, Howard FM. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc. 1977;52:267–280. [PubMed] [Google Scholar]
- 5.Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. 1993;43:1167–1172. doi: 10.1212/wnl.43.6.1167. [DOI] [PubMed] [Google Scholar]
- 6.Romi F, Kristoffersen EK, Aarli JA, Gilhus NE. The role of complement in myasthenia gravis: serological evidence of complement consumption in vivo. J Neuroimmunol. 2005;158:191–194. doi: 10.1016/j.jneuroim.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Drachman DB, Angus CW, Adams RN, et al. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298:1116–1122. doi: 10.1056/NEJM197805182982004. [DOI] [PubMed] [Google Scholar]
- 8.Lindstrom J, Einarson B. Antigenic modulation and receptor loss in experimental autoimmune myasthenia gravis. Muscle Nerve. 1979;2:173–179. doi: 10.1002/mus.880020304. [DOI] [PubMed] [Google Scholar]
- 9.Gomez CM, Richman DP. Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci U S A. 1983;80:4089–4093. doi: 10.1073/pnas.80.13.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lennon VA, Seybold ME, Lindstrom JM, et al. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J Exp Med. 1978;147:973–983. doi: 10.1084/jem.147.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Gong B, Lin F, et al. Anti-c5 antibody treatment ameliorates weakness in experimentally acquired myasthenia gravis. J Immunol. 2007;179:8562–8567. doi: 10.4049/jimmunol.179.12.8562. [DOI] [PubMed] [Google Scholar]
- 12.Piddlesden SJ, Jiang S, Levin JL, et al. Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol. 1996;71:173–177. doi: 10.1016/s0165-5728(96)00144-0. [DOI] [PubMed] [Google Scholar]
- 13.Christadoss P. C5 gene influences the development of murine myasthenia gravis. J Immunol. 1988;140:2589–2592. [PubMed] [Google Scholar]
- 14.Biesecker G, Gomez CM. Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J Immunol. 1989;142:2654–2659. [PubMed] [Google Scholar]
- 15.Lin F, Kaminski HJ, Conti-Fine BM, et al. Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest. 2002;110:1269–1274. doi: 10.1172/JCI16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaminski HJ, Kusner LL, Richmonds C, et al. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol. 2006;202:287–293. doi: 10.1016/j.expneurol.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Morgan BP, Chamberlain-Banoub J, Neal JW, et al. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol. 2006;146:294–302. doi: 10.1111/j.1365-2249.2006.03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong PW, Mahaffey KW, Chang W-C, et al. Concerning the mechanism of pexelizumab’s benefit in acute myocardial infarction. Am Heart J. 2006;151:787–790. doi: 10.1016/j.ahj.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Fitch JCK, Rollins S, Matis L, et al. Pharmacology and biological efficacy of a recombinant, humanized, single-chain antibody C5 complement inhibitor in patients undergoing coronary artery bypass graft surgery with cardiopulmonary bypass. Circulation. 1999;100:2499–2506. doi: 10.1161/01.cir.100.25.2499. [DOI] [PubMed] [Google Scholar]
- 20.Keshavjee S, Davis RD, Zamora MR, et al. A randomized, placebo-controlled trial of complement inhibition in ischemia-reperfusion injury after lung transplantation in human beings. J Thorac Cardiovasc Surg. 2005;129:423–428. doi: 10.1016/j.jtcvs.2004.06.048. [DOI] [PubMed] [Google Scholar]
- 21.Hillmen P, Hall C, Marsh JCW, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350:552–559. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 22.Nunn MA, Sharma A, Paesen GC, et al. Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J Immunol. 2005;174:2084–2091. doi: 10.4049/jimmunol.174.4.2084. [DOI] [PubMed] [Google Scholar]
- 23.Paesen GC, Adams PL, Harlos K, et al. Tick histamine-binding proteins: isolation, cloning, and three-dimensional structure. Mol Cell. 1999;3:661–671. doi: 10.1016/s1097-2765(00)80359-7. [DOI] [PubMed] [Google Scholar]
- 24.Hepburn NJ, Williams AS, Nunn MA, et al. In vivo characterization and therapeutic efficacy of a C5-specific inhibitor from the soft tick Ornithodoros moubata. J Biol Chem. 2007;282:8292–8299. doi: 10.1074/jbc.M609858200. [DOI] [PubMed] [Google Scholar]
- 25.Fredslund F, Laursen NS, Roversi P, et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat Immunol. 2008;9:753–760. doi: 10.1038/ni.1625. [DOI] [PubMed] [Google Scholar]
- 26.Roversi P, Lissina O, Johnson S, et al. The structure of OMCI, a novel lipocalin inhibitor of the complement system. J Mol Biol. 2007;369:784–793. doi: 10.1016/j.jmb.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. 1980;285:238–240. doi: 10.1038/285238a0. [DOI] [PubMed] [Google Scholar]
- 28.Coligan JE. Current protocols in immunology. New York: John Wiley & Sons; 1996. p. 3. [Google Scholar]
- 29.Kabet EA, Mayer MM. Experimental Immunochemistry. In: Thomas Charles C., editor. Science. 2. Vol. 134. Springfield; IL: 1961. pp. 149–153. [Google Scholar]
- 30.Wu JT, Astill M, Lloyd C, Salmon VC. Rhabdomyosarcoma cell line can be used for the isolation of soluble acetylcholine receptor and for assaying blocking and modulating autoantibodies. J Clin Lab Anal. 1993;7:11–18. doi: 10.1002/jcla.1860070104. [DOI] [PubMed] [Google Scholar]
- 31.Childs LA, Harrison R, Lunt GG. Complement-mediated muscle damage produced by myasthenic sera. Ann N Y Acad Sci. 1987;505:180–193. doi: 10.1111/j.1749-6632.1987.tb51291.x. [DOI] [PubMed] [Google Scholar]
- 32.Lyons BW, Wu LL, Astill ME, Wu JT. Development of an assay for modulating anti-acetylcholine receptor autoantibodies using human rhabdomyosarcoma cell line. J Clin Lab Anal. 1998;12:315–319. doi: 10.1002/(SICI)1098-2825(1998)12:5<315::AID-JCLA12>3.0.CO;2-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Baets M, Stassen M, Losen M, et al. Immunoregulation in experimental autoimmune myasthenia gravis—about T cells, antibodies, and endplates. Ann NY Acad Sci. 2003;998:308–317. doi: 10.1196/annals.1254.033. [DOI] [PubMed] [Google Scholar]
- 34.Christadoss P, Tuzun E, Li J, et al. Classical complement pathway in experimental autoimmune myasthenia gravis pathogenesis. Ann N Y Acad Sci. 2008;1132:210–219. doi: 10.1196/annals.1405.009. [DOI] [PubMed] [Google Scholar]
- 35.Wikel SK, Bergman D. Tick-host immunology: significant advances and challenging opportunities. Parasitol Today. 1997;13:383–389. doi: 10.1016/s0169-4758(97)01126-5. [DOI] [PubMed] [Google Scholar]
- 36.Kao I, Drachman DB. Myasthenic immunoglobulin accelerates acetylcholine receptor degradation. Science. 1977;196:527–529. doi: 10.1126/science.850793. [DOI] [PubMed] [Google Scholar]
- 37.Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci. 1987;505:326–332. doi: 10.1111/j.1749-6632.1987.tb51301.x. [DOI] [PubMed] [Google Scholar]