

Abstract
Mental retardation—known more commonly nowadays as intellectual disability—is a severe neurological condition affecting up to 3% of the general population. As a result of the analysis of familial cases and recent advances in clinical genetic testing, great strides have been made in our understanding of the genetic etiologies of mental retardation. Nonetheless, no treatment is currently clinically available to patients suffering from intellectual disability. Several animal models have been used in the study of memory and cognition. Established paradigms in Drosophila have recently captured cognitive defects in fly mutants for orthologs of genes involved in human intellectual disability. We review here three protocols designed to understand the molecular genetic basis of learning and memory in Drosophila and the genes identified so far with relation to mental retardation. In addition, we explore the mental retardation genes for which evidence of neuronal dysfunction other than memory has been established in Drosophila. Finally, we summarize the findings in Drosophila for mental retardation genes for which no neuronal information is yet available. All in all, this review illustrates the impressive overlap between genes identified in human mental retardation and genes involved in physiological learning and memory.
Keywords: Drosophila, mental retardation, neurological disorders, genetics, development, treatment
Introduction
Mental retardation: A clinical description
Attempts at studying the genetic basis of mental retardation date back from the beginning of the 19th century,1 but it is only recently that a massive amount of clinical data has informed us about the molecular basis of mental retardation (MR also referred to as intellectual disabilities in the clinical literature).2 This enormous amount of information was compiled by Dr. Victor A. McKusick and is now available freely online in the Online Mendelian Inheritance of Man (OMIM). We have indicated the OMIM reference when available. The World Wide Web address at the moment of this review is: http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim. Mental retardation affects 1–3% of the population3–7 and consists of significantly sub-average general intellectual functioning accompanied by limitations in adaptive functioning in at least two of the following areas: communication, self care, home living, social/interpersonal skills, use of community resources, self-direction, functional academic skills, work, leisure, health and safety. The onset must be before the age of 18.8,9 Mental retardation is described as an intellectual quotient (IQ) of less than 70. The IQ represents the relative performance of an individual (composite verbal, non-verbal) compared with the age equivalent general population performance (deviation IQ). Based on the IQ score Mental retardation can be classified as mild (50–70), moderate (35–50) or severe (less than 35). Mild mental retardation is the most common form. Males are overrepresented with a ratio of 1:1.3 to 1:1.9 due to many X-linked cases.10 Most cases are diagnosed between the ages of 3 and 9. Various classifications have been used in mental retardation.11 In general mental retardation is divided between syndromic and non-syndromic. Another classification relates to the inheritance pattern in multiple multiplex families: autosomal versus X-linked. X-linked mental retardation, also known as MRX, is further divided into syndromic, neuromuscular and non-specific.10,12
Syndromic mental retardation is diagnosed in a patient when mental retardation is associated with dysmorphic or other neurological features.13 On the other hand, non-syndromic mental retardation is diagnosed when no hallmark features are observed in association with mental retardation.
Mental retardation’s associated diseases (co-morbidity) include: cerebral palsy, epilepsy, severe hearing impairment or deafness, severe vision impairment or blindness, hydrocephalus, autism and psychiatric disorders.5
For many years, research on mental retardation consisted of clinical descriptions of the recurrent association of clinical signs and symptoms lumped into syndromes. Development of karyotyping through microscopic visualization of the chromosomes- allowed the linkage of mental retardation and abnormal facial or body appearance (dysmorphic features) in patients to chromosomal defects. For example, the trisomy of chromosome 21 in Down syndrome patients14,15 represents one of many chromosomal abnormalities that can be identified on a routine karyotype analysis. In addition to variation in chromosome number, deletions, duplications, translocations and ring chromosomes can be identified by karyotyping. Increasing G-Banding resolution allowed identification of smaller deletions or duplications (the resolution being approximately 5 to 10 Mb). Interestingly, karyotype analysis of cells grown in growth media deficient in folate revealed the chromosomal “fragility” of the X chromosome in patients with Martin-Bell syndrome.16 This is why Martin Bell syndrome is now known as Fragile X syndrome.
The next advance was made using fluorescent in situ hybridization (FISH). FISH surpassed the resolution of light microscopy and led to the identification of microdeletions in several syndromes. This was followed by the identification of additional microdeletions in subtelomeric regions in several patients with mental retardation.17,18
More recently, array-based comparative genomic hybridization (aCGH) has created a boom in the number of chromosomal aberrations discovered.19 Multiple probes consisting of bacterial artificial chromosomes (BAC) clones of the human genome spaced by 1–1.4 megabase (Mb) are used in arrays to compare the relative number of copies for each probe in a patient compared to control genomic DNA. Targeted aCGH using more probes in regions known for human diseases have replaced the randomly distributed probes for clinical testing.20,21 Although similar to FISH in resolution, aCGH allows for screening of more regions at once by having probes covering the entire genome. Variations in gene copy number (loss or gain) have been identified in complex cognitive disorders such as autism,22–24 schizophrenia25,26 and cases of mental retardation27–32 where sporadic cases did not allow for traditional linkage-analysis based genetics.33,34 In addition to leading to diagnosis in cases in which karyotype analysis did not pick up defects, aCGH, with its higher resolution, allows better genotype-phenotype correlation than the karyotype. Nevertheless, in clinical practice, aCGH will frequently identify deletions or duplications that involve more that one gene, making causality judgment difficult at first.
Thus far, mutations identified at the molecular level in human patients can be grouped by function: cytoskeleton modification (RhoGTPases such as GDI, PAK3, ARHGEF6, OPHN1), protein synthesis modulation (TSC1, TSC2, FMR1), chromatin remodeling (RPS6KA3, ATRX, CBP), synaptic vesicle formation and dynamics (SYN, SLC6A8, NLGN4) and transcription factors (ZNF41, ZNF81, ZNF674).2 Importantly, these molecular functions are also key pathways in memory formation.
Several etiologies have been identified in mental retardation.5,35,36 Despite variability in the causative lesion, however, mental retardation is associated with deficits in (i) learning new information, (ii) understanding complex information, (iii) memorizing information, (iv) transferring information from one context to another, and (v) elaborate thinking based on multiple pieces of information. This probably explains why several mutations in Drosophila genes related to human MR have been reported to cause memory defects37–39 (Fig. 1).
Figure 1.

Memory genes identified in model systems overlap with genes identified in human mental retardation. Protein synthesis dysregulation, abnormal gene regulation and spine structure anomalies are commonly found in mental retardation. Interestingly, several of the genes involved in the signaling pathway affecting these phenotypes are responsible for learning or memory defects.
Drosophila as an animal model of cognitive disorders
Despite the neuro-anatomical divergence between flies and humans, the molecular mechanisms underlying learning and memory seem to be conserved.40,41 An emerging concept is that memory can be used as an endophenotype of cognition and its pathological counterpart, MR. More to the point, about 87% of the genes known to be involved in human mental retardation have orthologs in Drosophila.62 As noted by Restifo,41 this correspondence is higher than for other classes of genes.42,43
Drosophila is an important tool in understanding the molecular-genetic basis of mental retardation. First, Drosophila offers the possibility of studying the genetic network of a given gene. Second, it is well suited for dissection of the differential function of a gene during development and adulthood. Third, given the economy of scale, Drosophila is the perfect in vivo system in which to study pharmacological rescue. Finally, flies can be submitted to various learning paradigms (classical, operant conditioning) that can reveal task specific requirements for the gene of interest. We will now review three paradigms developed in Drosophila to study associative learning and memory: olfactory classical conditioning, operant visual conditioning and courtship memory.
Pavlovian olfactory conditioning
Initial observations and conceptualizations by psychologists such as Ribot44 and Ebbinghaus45 were formalized in the early 20th century with the systematic study of animal learning and memory. Learning has traditionally been identified as the early phase of new information storage. In addition, learning will sometimes be used to refer to a very early post acquisition time point for testing the new acquisition. Information stored for longer time or a later time point for testing performance related to the acquisition will be referred to as memory.46 Memory can be then subdivided between short-term, medium term and long-term memory depending on the timing relative to the acquisition. Ivan Pavlov,47 in particular, established experimental paradigms to study “associative” learning, a change in a behavioral response caused by the temporal association of two stimuli. Pavlov’s associative learning can be considered an elemental building block of more complex forms of “contingency” learning.48,49 In the canonical form of associative learning, an animal is presented with a neutral “conditioned stimulus” (CS), which itself does not normally elicit a behavioral response. When presentation of the CS is paired for several trials with an “unconditioned stimulus” (US), which has an inherent reward or punishment value and accordingly elicits an “unconditioned response” (UR), the CS comes to elicit a response similar to the UR. From this basic observation, Pavlov derived the general principle of stimulus substitution, which we can now think of as a basic component of more complex forms of learning. Another major behavioral psychology model for learning was defined as operant conditioning. In the operant conditioning model described by Skinner,50 the stimulus (S) is followed by a response that is then rewarded or not (R), depending on its correspondence to the response desired by the observer. This experimental paradigm derives from the Thorndike law of effect,51 which attributes the maintenance of a response to the effect of that response.
In Drosophila, Seymour Benzer pioneered the behavioral genetics of cognition in Drosophila.52–54 In Benzer’s laboratory at CalTech, Quinn et al.55 were the first to show data illustrating that the Dipteran, Drosophila melanogaster, was capable of associating odors with footshock punishment. They used a pair of odors and subsequent reinforcement following one of the odors selectively. Using a set of tubes to which the flies had first to walk [they were attracted by a light (S)], flies were successively exposed to two odors in two different tubes (2 and 3) with footshock associated with one of the odors (R) (tube 2). Odor avoidance was then quantified by exposing the trained flies to each odor successively in the absence of footshock (tubes 4 and 5). Later, this operant odor-shock avoidance task was modified into a classical conditioning task by (i) trapping flies into the training chamber and (ii) presenting both odors simultaneously to trained flies in a T-maze.56 Learning retention measured immediately after one of these Pavlovian training sessions is robust. With repetitive training, flies show memory retention lasting more than a week (Fig. 2).
Figure 2.
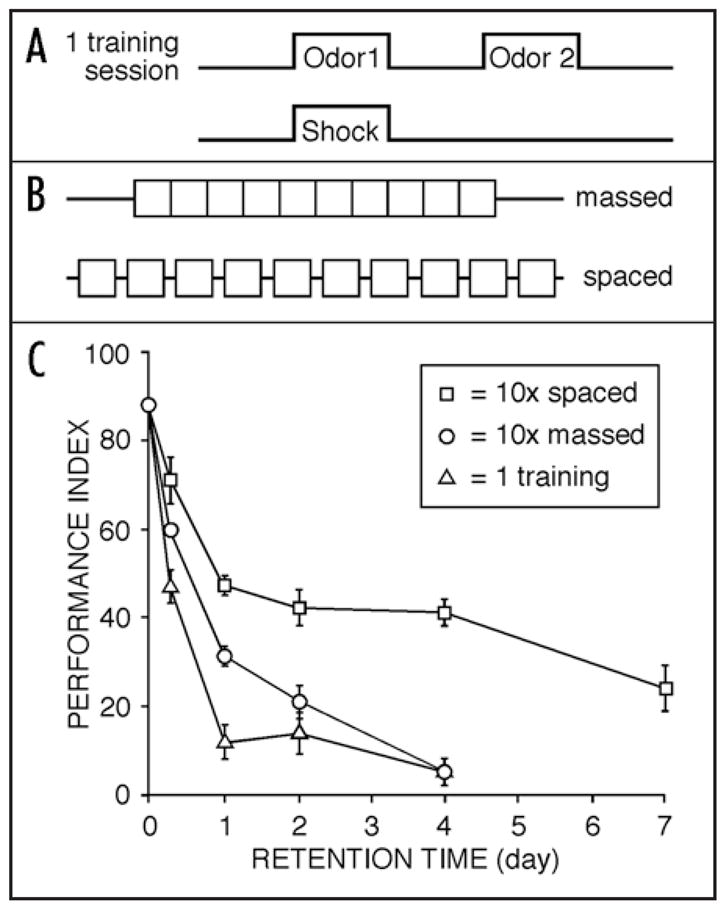
Classical olfactory conditioning leads to long-term memory in Drosophila. (A) Drosophila can be trained to remember the association between an odor (CS) and a footshock (US). (B) Repeated training (10 training sessions) can be performed without rest intervals (massed training) or with 15-minute rest intervals between training sessions (spaced training). (C) Performance index curve after a single training session (△) spaced training (□) or massed training (○). Single training leads to a memory that decays rapidly but persists at low levels for up to four days. Repeated training without rest intervals leads to a higher level of performance, but also lasts approximately four days. In contrast, the same amount of repeated training separated by rest intervals leads to a memory lasting for at least one week at a higher performance level (Modified after Tully et al.75).
The development of a learning/memory task in Drosophila brought to bear the power of fly genetics to the discovery of genes involved in cognitive plasticity. It also showed that as psychologists could divide new acquisition storage into successive learning and memory phases, geneticists of memory could identify genes required for each of these phases from learning to long-term memory. The first two genes discovered as single-gene mutants, dunce57 and rutabaga, both participate in the cyclic adenosine monophosphate (cAMP) intracellular signaling pathway (Fig. 3) and were required for learning. dunce (dnc) encodes cAMP-specific phosphodiesterase (PDE), an enzyme that is responsible for the degradation of cAMP,58 while rutabaga (rut) encodes Ca2+-Calmodulin sensitive adenylyl cyclase (AC), an enzyme that is responsible for the synthesis of cAMP.59,60 Because these single-gene mutants were randomly generated and independently screened for defects in the same behavioral task (olfactory learning), these two “hits” in the same biochemical pathway most certainly underlined the importance of the cAMP pathway. Thus, cAMP signaling clearly appeared to underlie associative60 learning in Drosophila.
Figure 3.

Learning and short-term memory depend on the cAMP pathway. After the activation of a G protein coupled receptor by a ligand (L), adenylyl cyclase (AC) synthesizes cAMP, which in turn is hydrolyzed by phosphodiesterase (PDE). The Drosophila mutants rutabaga (AC) and dunce (PDE) were among the first mutants isolated with heritable defects in learning/memory. Increased levels of cAMP lead to the activation of Protein Kinase A (PKA). After dissociation from its regulatory subunit, the catalytic subunit phosphorylates many targets: (1) potassium channels, which regulate neural activity during short-term memory phases; and (2) the cyclic AMP responsive element binding protein (CREB) transcription factor, which regulates the expression of other genes during long-term memory formation.
In a subsequent study of these cAMP mutants, Tully and Quinn76 obtained data that would raise an important therapeutic question: is the absolute level of a factor such as cAMP important for learning or is the critical factor its variation in relation to activity? They observed that despite near normal basal levels of cAMP in dnc, rut double-mutants, the learning deficit of these flies was in fact worse than that of single-gene mutants for either dnc or rut.56 This observation suggested that learning depends on more than the restoration of normal levels of cAMP. This finding raises challenging therapeutic concerns, as drugs that restore normal levels of a given factor may not rescue the activity-dependent variation.
Production of cAMP depends on G protein signaling.61,62 In Drosophila, recent studies have shown learning in response to footshock (punishment) and sucrose (reward) stimuli during classical conditioning to be mediated by dopamine and octopamine, respectively.63 Both neurotransmitters bind to G-protein coupled receptors. When ligand binds the receptor, GTP-bound Gα protein is free to interact with AC. This interaction is then terminated when the intrinsic GTPase activity of the Gα protein hydrolyzes GTP to GDP. G proteins are made of three subunits (α, β, γ), and the α subunit can either be stimulatory (Gs) or inhibitory (Gi). Connolly et al.64 showed that constitutively active Gs disrupted Drosophila olfactory learning.
The importance of the cAMP pathway in learning was reaffirmed by the conservation of its key molecules across different species. Indeed, using a sensitization procedure in Aplysia for the gill withdrawal reflex in which an electric shock to the tail provokes a withdrawal of the gill, Kandel and coworkers65,66 had uncovered the role of the cAMP pathway in learning and memory. Castellucci et al.67,68 and Kupfermann et al.85 established a minimal monosynaptic neural circuitry involved in sensitization of the gill withdrawal reflex. This allowed Brunelli et al.69 to show that this simple form of learning produced an increase of the second messenger cAMP, which then led to a cAMP-dependent protein kinase A (PKA)-dependent presynaptic enhancement of neurotransmitter release at this synapse. The mechanism of increased neurotransmitter release was later shown to depend on the serotonin sensitive (S-type) K+ channel.70 After changes in such potassium channel currents, a given stimulus produced a stronger calcium influx in the presynaptic (sensory) neuron, thereby increasing the release probabilities of the neurotransmitter.71
Further dissection of the genetics of learning and memory in Drosophila uncovered even more conservation with Kandel’s work in Aplysia (Fig. 3). In addition, behavioral genetics in Drosophila illustrated that moving downstream in the cAMP pathway correlated with performance defects downstream of learning: memory. The cAMP-dependent protein kinase (PKA) was shown to be involved in Drosophila olfactory medium-term memory. PKA is a tetramer, composed of two regulatory subunits and two catalytic subunits. In the absence of cAMP, the regulatory dimer binds the catalytic dimer and inhibits its kinase activity. In the presence of cAMP, the regulatory subunits fail to bind the catalytic dimer and thus, activate the kinase. Drain et al.72 overexpressed an inducible transgenic construct encoding a mutant form of the regulatory subunit, which no longer was able to bind cAMP. Consequently, this transgene had a dominant-negative effect on PKA activity. When expression was induced in adult flies, a defect in memory resulted.
An important question remained, is there a link between the cAMP pathway and the gene transcription known to be required for the formation of long-term memory? Transcription inhibitors had been shown to disrupt memory in Aplysia73 and mammals.74 Since transcription factors (TFs) are key regulators of this process, Yin et al.90 embarked on identifying TFs required for long-term memory. Drosophila could be trained in a single training session or could receive multiple training sessions. Tully et al.75 showed that ten training sessions separated by 15 minutes rest intervals (spaced training) led to a non-decaying long-term memory. In the other hand, the same amount of training but without rest intervals (massed training) would lead to a memory that decayed completely after four days. Guided by early studies in Drosophila, which used mutant screens to identify the learning mutants dunce (PDE) and rutabaga (AC), by subsequent reverse-genetics methods used to dissect other components of the cAMP signaling pathway, and by the behavior paradigm for spaced and massed training, Yin et al.76 disrupted the cAMP response element binding gene (creb) and confirmed the hypothesis that long-term memory formation would specifically be blocked. CREB is a transcription factor that binds as a dimer to a specific DNA sequence (TGACGTCA) known as a cAMP response element (CRE) that is present in the promoter or enhancer regions of CREB-regulated genes. Several genes with CRE sites are involved in neuronal plasticity. CREB has also been shown to be required in mammals. Indeed, CREB inhibition impairs memory in mice,77–79 and injection of antisense oligonucleotides to CREB into the hippocampus or amygdala of normal rats also produces memory deficits.78 More recently, Wagatsuma et al.80 showed that CREB was required presynaptically for synaptic enhancement.
Impey et al.81 showed that CREB-dependent gene transcription (as reported by a CRE-β-galactosidase transgene) was induced after protocols that yielded late long term potentiation (LTP) but not early LTP. LTP (Fig. 4) is a cellular model of learning and memory. Bliss and Lomo82 initially described facilitation of synaptic transmission in the perforant pathway of the dendate gyrus (a region of the hippocampus) after a high frequency tetanic stimulation. LTP protocols have now been established in neurons of multiple regions of the brain. LTP is divided by N-methyl-D-aspartic acid (NMDA) receptor dependence. In the CA1 region of the hippocampus, long-term potentiation was discovered to require NMDA receptors.83 Recently Xia et al.84 demonstrated a role for NDMA receptors in Drosophila memory, again emphasizing the evolutionarily conservation in molecular mechanisms of associative learning. NMDA receptors are composed of an obligatory NR1 subunit and a variable NR2 subunit (NR2A-D).85 Variation in NR2 subunits and subsequently the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor is associated with potentiation.86,87 An NMDA receptor-independent form of LTP can be induced in the mossy fiber-CA3 region by glutamatergic activation of kainate receptors. This form of LTP is cAMP-dependent and appears to be presynaptic.
Figure 4.

Long-term potentiation and depression are caused by differential signaling in response to different levels of elevated calcium. (A) Long-term depression is induced after a mild intracellular rise in Calcium in response to NMDA receptor activation. This minimal entry of calcium usually results from low-frequency stimulation of the synapse. A reduction in the number of AMPA receptors containing the subunits 1 and 3 is then observed, causing a decrease in synaptic strength. (B) In contrast, a high-frequency stimulation will lead to massive calcium entry through NMDA receptors leading to de novo AMPA receptor insertion at the synapse. The addition of these new receptors will cause a net increase in synaptic strength.
The converse phenomenon is known as long-term depression (LTD) (Fig. 4). LTD can be triggered by low frequency stimulation or chemically by metabotropic glutamate receptor agonists.103 All forms of LTD seem to involve decreased postsynaptic AMPA signaling.88,89 Long-term synaptic depression (LTD) has been associated with loss of AMPA receptors via endocytosis. The process is associated with clathrin-coated vesicles and is dependent on dynamin.
The relationship between LTP, LTD and the changes in the brains of animals involved in behavior paradigms such as conditioning in Drosophila remains incomplete. Several molecules, as well as transcription and protein synthesis, seem to be required in both models. Moreover, LTP-like changes in CA1 neurons of the hippocampus were observed following one-trial avoidance learning in rats.90 Further studies will be required to localize LTP or LTD like process to learning and memory in Drosophila.
Operant visual conditioning
Operant conditioning is another prominent model for human learning and intelligence. Skinner50 conceptualized this learning paradigm where an action (CS) is followed by a consequence (US), which itself serves as a positive or negative reinforcement. In flies, visual operant learning was established by Gotz91 and then optimized by Heisenberg.92,93 Tethered flies are placed in the center of a cylindrical arena where visual information is displayed.94 Flies are trained to respond to the visual cues that are displayed. The fly wing movement reflects the response (CS) and is transmitted via the motor control unit that, in turn, leads to the rotation of the arena. Reinforcement (US) is produced by exposing the fly to the noxious heat generated by a light beam directed to them.
Development of multiple behavior paradigms revealed that, as in humans, not all learning/memory genes are required for all forms of leaning/memory. First, Gong et al.95 showed that the olfactory learning mutant dnc has normal operant visual learning. Second, task specialization is also reflected at the neurotransmitter level using another operant conditioning paradigm. Operant conditioning has also been developing with reference to spatial localization utilizing a hot chamber and is known as operant place learning. Sitaraman et al.96 showed that serotonin is required for operant place learning whereas dopamine is not. On the other hand Schwaerzel et al.63 documented that aversive olfactory conditioning depends on dopamine. Third, this task specificity extends beyond molecules to anatomical circuits. Indeed, it was shown early on that the Drosophila central complex, a circular structure located in the middle of the fly brain, is required for visual learning.97,98 In olfactory classical conditioning, most reports have identified another structure called the mushroom body as the site for memory.99–101 An exception is the recent report by Wu et al,99 that showed that NMDA signaling in the central complex is involved in olfactory learning. Overall, these data illustrate that in the fly as well as in humans, different forms of memory may require different genes and neuroanatomical structures.
Courtship memory
Finally, some researchers have aimed to train flies to modify an innate behavior: for example, courtship. Drosophila displays a stereotyped behavior during courtship.102–104 Siegel and Hall105 took advantage of this tractable behavior to study courtship conditioning. They showed that a male exposed to a female will pursue her to engage in a mating song behavior. Once mated, a female will reject the male. Typically, the male will be exposed to a mated female for 1 hour (training). During this time, he will progressively decrease the amount of time spent in courtship, resulting in suppression of courtship behavior in the last 10 minutes of the training compared to the first 10 minutes (identified as learning). Retention of this suppression can be tested when, after a given rest interval, the same male is exposed to another female (memory). The degree of courtship suppression is then measured.105–107 There are multiple mechanisms involved in courtship conditioning.106,108–110
As for operant conditioning, courtship conditioning widens our understanding of the neuronal basis of learning and memory. Kane et al.111 showed that males mutant for pkc did not decrease their courtship during the training period with a mated female, but did perform similarly to controls 10 minutes and 2 hours after training. In other words, they presented evidence of a courtship-learning deficit, but appeared to have normal memory, a phenomenon rarely seen in olfactory conditioning. Also, important plasticity molecules involved in LTP such as CAMKII and ORB, have been identified thus far as involved in courtship memory only. The inhibition of calcium/calmodulin-dependent protein kinase II (CamKII) using heat-shock induction of a peptide inhibitor leads to the disruption of courtship conditioning.112 More recently Keleman et al.113 showed a defect in courtship memory in orb2 mutants, the Drosophila cytoplasmic polyadenylation element binding protein (CPEB) homolog. In summary, even in a single animal, utilization of multiple behavior paradigms is likely to help form a more comprehensive view of the ensemble of genes responsible for learning and memory.
Genes Identified in Human Mental Retardation and Shown to have a Drosophila Learning or Memory Phenotype
Albright hereditary osteodystrophy (Gs protein)
Connolly et al.64 used Drosophila genetics to test whether G protein-mediated signaling is involved in Pavlovian olfactory learning. They generated transgenic flies carrying a mutant Gs subunit that could no longer hydrolyze GTP to GDP. Thus, this mutant G protein would irreversibly bind to adenylate cyclase (AC), rendering it constitutively active. Associative learning was completely abolished in these transgenic flies. In humans, interestingly, an imprinting defect for a gene encoding Gs is known to cause Albright hereditary osteodystrophy (OMIM #103580).114 This syndrome is characterized by short stature, obesity, round faces, subcutaneous ossifications, brachydactyly, other skeletal anomalies and mental retardation.115
Mental retardation autosomal recessive-MRT1 (Neurotrypsin)
Mutations in the human PRSS12 gene, which encodes the protéase Neurotrypsin, lead to non-syndromic autosomal recessive mental retardation (OMIM #606709).116 Didelot et al.37 identified long-term memory defects in flies with reduced expression of tequila, the Drosophila ortholog of Neurotrypsin. Importantly, this was the first demonstration that a gene involved in human mental retardation has an acute role in memory formation. Furthermore, Didelot et al. were able to localize the requirement for tequila expression to the mushroom bodies. The role of proteases in neuronal migration, axon outgrowth117,118 and synapse elimination has been reviewed by Molinari et al.119
Fragile X mental retardation syndrome (Fmr1)
Several mental retardation conditions due to single gene defects have been identified to date. Fragile X mental retardation syndrome (OMIM # 309550) is the most common cause of single gene mental retardation in males (1/4000) and is a leading cause in females (1/8000). Mental retardation (MR) is moderate to severe.120,121 Fragile X syndrome is associated with autism and epilepsy. Initially localized to the X chromosome based on karyotype defects,122 the precise molecular lesion was later linked to a trinucleotide (CGG) repeat expansion above 200.123,124 Expansion in the 5′ untranslated region (UTR) of Fmr1 leads to DNA methylation and lack of FMR protein (FMRP) expression.125 In some cases, however, deletions126,127 and even point mutation128 are responsible for Fragile X syndrome. Age-dependent changes in the FMR phenotype and developmental effects have also been observed.129,130 Although caused by a single-gene mutation, understanding the cognitive phenotype in Fragile X syndrome is complicated because of the multiple protein-protein and protein-mRNA interactions of FMRP.131–133 Biochemically, FMRP has been shown to be involved in translational control.134,135
Drosophila has a homolog of Fmr1 known as dFmr1.136 At the protein level, conservation of important domains varies between 25 and 75%.137 Initial studies of Fragile X mutant flies identified a defect in circadian rhythm, a debilitating symptom commonly found in patients with Fragile X.138,139 The overlap in neuronal dendritic spine abnormalities is yet another example of similarities between flies132,140 and humans in the Fragile X mutant phenotype.141–143 Fragile X patients144,145 and FMR1 knock-out (KO) mice display spatial learning deficits.146–148 Also, enhanced hippocampal long-term depression (LTD)149 and defective cortical long-term potentiation (LTP)150,151 are found in FMR KO mice.
In Drosophila McBride et al.152 have shown that the Drosophila 3-hour courtship conditioning memory is impaired in dFmr1 mutants. Moreover, they demonstrated a pharmacological rescue of the phenotype using the metabotropic glutamate receptor 5 (mGluR5)-specific antagonist 2-methyl-6-phenylethynyl-pyridine (MPEP) and Lithium. We have shown that the defect in long-term memory (assessed at one day) is specific to memory formed after spaced training.38 Indeed, 1-day memory performance after massed training was normal in dFmr1 mutants. Spaced training induces protein synthesis-dependent memory in addition to a protein synthesis-independent memory, whereas massed training involves only protein synthesis-independent memory.75 We therefore reasoned that a disruption in the control of protein synthesis in dFmr1 mutants was causing a negative impact on memory and rescued it with use of protein synthesis inhibitors such as cycloheximide and puromycin.38 We also demonstrated an interaction between FMRP and staufen in long-term memory measured at 1 day. Staufen was initially identified in translational control in the fly oocyte. As staufen was previously identified in our laboratory in a screen for genes required for long-term memory formation,153 the genetic interaction between dFmr1 and staufen in LTM behavior reinforces the link between physiological memory genes and mental retardation genes. It also established for the first time a role at the cognitive level for the interaction between staufen and dFmr1. This observation was also supported by the previous co-localization of Staufen and dFmr1 observed in subcellular structures involved in mRNA modulation known as processing bodies (p-bodies).154
Consistent with the idea that RNA processing, and RNA interference in particular, is involved in memory, FMRP was shown biochemically to be part of the RNA-induced silencing complex (RISC)155 and we found that dFMRP was acutely required in long-term memory.38 In addition, Drosophila armitage mutants were shown to be defective in one-day memory after olfactory spaced training.156 Armitage codes for a silencing defective 3 (SDE-3) class RNA helicase responsible for the unwinding of double stranded RNA (dsRNA) and its subsequent loading onto RISC.157
RNA interference can be further divided in several pathways. We sought to identify modifiers of dFmr1 among known RNA interference molecules. Jin et al.158 had demonstrated a genetic interaction between dFmr1 and argonaute 1 (AGO1), an important molecule for RNA interference. Indeed, flies heterozygous mutant for dFmr1 and AGO1 displayed abnormal morphology of neuromuscular junctions. We identified an interaction between ago1 and dFmr1 in long-term memory specifically.38 In addition, we rescued the memory deficit observed in ago1/+, dFmr1/+ double heterozygous mutants using protein synthesis inhibitors such as cycloheximide and puromycin.38 Taken together, these results suggest that dFmr1 mutants may be defective in ago1-dependent translational control. These mutants would have excessive baseline general protein synthesis that could be occluding the normal activity-dependent protein synthesis required for memory. A similar rescue of memory using another protein synthesis inhibitor, rapamycin, was obtained in a mouse model of tuberous sclerosis of Bourneville, in which there is also an excess of protein synthesis.159 Since both Fragile X and Tuberous sclerosis syndrome also cause autism, these findings suggests that social cognition and autism may be related to protein synthesis control.160
Kelley et al.161 expanded on previous reports of altered cAMP levels in Fragile X patient cells162,163 by showing that the induction of cAMP is reduced in humans, mice and flies defective in Fragile X. Another connection with early research on learning and memory was the observation that the circadian rhythm disruption may be linked to abnormal CREB level fluctuations in Fragile X Drosophila mutants.138
Neurofibromatosis type 1 (NF1)
Neurofibromatosis type 1 (OMIM #162200) is clinically characterized by multiple tumors (optic neuroma, schanomas, plexiform neurofibroma), skin anomalies (café au lait spots, axilary freckling), increased head size (macrocephaly) and learning disability.164 At the molecular level, the Neurofibromin protein contains a Ras-specific guanosine triphosphatase-activating (Ras-GAP) domain. GAP proteins enhance the intrinsic hydrolytic activity of Ras proteins, enhancing the GTP (active) to GDP (inactive) transition. Persistent activation of Ras has been suggested as an explanation for multiple tumors observed in NF1 patients.
nf1 mutant flies have decreased body size and deficits in Pavlovian olfactory learning.165 Interaction between NF1 and the cAMP pathway165 is suggested by rescue of nf1 mutant body size and learning defects with PKA overexpression. In addition, mutation of nf1 blocks the mammalian pituitary adenylate cyclase activating peptide (PACAP)-induced enhancement of potassium current at the larval NMJ.166 PACAP stimulates cAMP synthesis via two different pathways: the rutabaga AC and the Ras-Raf pathway. Moreover, Quinn et al.167 identified a PACAP homolog mutant, amnesiac, with normal learning but defective medium-term memory.168,169 Further biochemical dissection of the NF1 protein showed that a region outside of the Ras-GAP domain is responsible for the growth defect present in NF1 mutants.170 Recently, Ho showed that the one-day memory defect is associated with the altered NF1-regulated Ras activity and not cAMP levels.171 In summary, NF1 defects in learning and memory seem to be linked to dysfunction in different signaling pathway by different domains within the NF1 protein.
Angelman syndrome (UBE3A)
Angelman syndrome (OMIM #105830) was described in 1965 in children with severe mental retardation, fits of laughter, epilepsy, ataxia and specific dysmorphic features. A special behavioral characteristic was the appearance of a happy demeanor, which led to them being described as “happy puppet”. The patient also presents microcephaly, especially when the disease is related to a deletion in the 15q11.2-q13 region. Patients lack the maternal active copy of the ubiquitin ligase E3A. UBE3A is a type of E3 ubiquitin-protein ligase characterized by a HECT (homologous E6-AP carboxyl terminus) domain. The HECT domain is required to transfer ubiquitin to proteins targeted for degradation via the ubiquitin-proteosome pathway.172 Mice with maternal inactivation of the Ube3a genes present impaired long-term potentiation and a deficit in context-dependent fear conditioning one day after training.173 In addition to UBE3A, the ubiquitin proteosome pathway has been linked to memory in the case of proteosome-dependent degradation of Armitage (a protein required for olfactory memory).156
We observed that the Drosophila dube3a mutants showed a significant defect in one-day performance after spaced training when compared with appropriate genetic controls.39 One-day memory after massed training was similar to controls. In addition to supporting a role in long-term memory as shown previously in mice,173 the specific impairment of memory after spaced training (which is transcription and translation-dependent75) in dube3a mutant flies suggests that the role of UbeE3A in cognition could be related to transcriptional regulation174 and/or protein degradation.175
Periventricular nodular heterotopia (Filamin A)
Filamin A mutations have been found in patients with periventricular nodular heterotopia (OMIM #300017), and these patients suffer from cognitive dysfunction and epilepsy.176,177 The Filamin A gene is involved in actin cytoskeleton remodeling.178,179 Filamin A is expressed in neurites of embryonic rat hippocampal neurons.176 Disruption of Filamin A impairs neuronal migration, probably because ligand binding no longer induces actin reorganization.176,180 Drosophila’s ortholog of Filamin A is cheerio. A behavioral screen in our laboratory identified joy, a mutant which carries a P-element insertion within cheerio.153 Joy has normal immediate learning but is defective in one-day memory after spaced training. It remains unclear to which extent this phenotype is developmentally regulated.
Coffin-Lowry syndrome (RSKII)
CREB is thought to be constitutively bound to DNA but inactive until phosphorylated. Phosphorylation at serine 133 promotes transcription.181 Several kinases are known to target serine 133 of CREB for phosphorylation, among these are PKA, CaMKIV and MAPK-activated ribosomal S6 kinase (Rsk). In humans, the mutation of Rsk 2 is known to cause Coffin-Lowry syndrome (OMIM#303600). This syndrome is characterized by mental retardation, a peculiar pugilistic nose, large ears, tapered fingers, drumstick terminal phalanges on x-ray and pectus carinatum. Four RSK isoforms are present in humans, whereas Drosophila has only one. Putz et al.182 used visual operant and classical olfactory conditioning to identify the requirement for Rsk in each paradigm. They showed that Drosophila null for rsk were deficient in olfactory learning, whereas rsk P-element derived hypomorphic mutants were defective in visual learning. The N-terminal region of the protein was required in both paradigms.
C.A.D.A.S.I.L. (Notch)
Mutations in Notch 3 have been identified in cerebral arteriopathy, autosomal dominant with subcortical infarct and leukodystrophy (C.A.D.A.S.I.L.) (OMIM #125310). CADASIL is clinically characterized by recurrent migraine and strokes, progressive focal neurological signs (pseudobulbar signs), seizures and dementia. Notch is another “developmental gene” previously known to be involved in cellular differentiation during neuronal development that serves both as a transmembrane receptor and a transcription factor. It has been documented that Notch in Drosophila has a role in long-term memory formation.183 Presente et al.183 used a temperature-sensitive allele to limit the possible contribution of developmental defects to the phenotype observed. Similarly, Costa et al.184 showed that Notch mutant mice display memory defects in a water maze task. Mice expressing a Notch anti-sense transgene exhibit normal development but have a decreased level of LTP.185
Notch is also connected with dementia by its interaction with its ligands Delta, Serrate or Lag-2 (DSL). Notch is known to be cleaved by a furin-like convertase and to insert in the cellular membrane. Binding with DSL then triggers a gamma secretase-dependent cleavage, thereby releasing an intracellular fragment that then translocates to the nucleus. Presenilin, for which mutations have been identified in some patients with Alzheimer’s disease, is a component of gamma-secretase.186 Furthermore, mutations in presenilin were shown to abolish Notch signaling by blocking its access to the nucleus.187
MRX58 (Integrin)
Mutations in transmembrane 4 superfamily 2 (TM4SF2) have been linked to X-linked mental retardation 58 (MRX58) (OMIM # 300096), which was initially reported in an Austrian family.188 A behavioral screen for long-term memory mutants in our laboratory identified an allele of a tetraspanin gene (TSp42Ef).153 TM4SF2 is a member of the Tetraspanin family of genes, which is involved in the β-1-integrin pathway. β-1-integrins are usually localized presynatically and join α-integrin that are localized post-synaptically. Alpha-integrin mutants have impaired short-term memory in Drosophila.189
CRASH syndrome (L1-CAM)
L1 cell adhesion molecule (L1-CAM) is involved in mammalian memory formation.190,191 L1 was originally identified in patients with X-linked hydrocephalus. Further investigation of these patients has revealed a more complex affliction known as CRASH syndrome (OMIM #308840). CRASH disease stands for corpus callosum hypoplasia, mental retardation, adducted thumbs, spastic paraplegia, and hydrocephalus.164
Neuroglian is the Drosophila homolog of L1-CAM. Mutations in neuroglian (nrg) were identified in a screen for learning and locomotor activity.192,193 De Belle and Heisenberg showed that nrg mutants, referred to as central brain deranged, had neuroanatomical defects in the central complex. Liu et al. showed that short-term visual memory was defective in nrg mutants.194 Mutants for nrg849 have decreased microtubules at the active zone of giant neurons, resulting in an abnormal synaptic terminal morphology. In addition to these structural defects, these mutants also display functional defects in synaptic responses.
Down syndrome (DYRK1A)
Trisomy of chromosome 21 leads to Down syndrome (OMIM #190685), which is characterized by mental retardation and dysmorphic features. Down syndrome is the most common cause of mental retardation.195 Most patients (90–95%) become fully trisomic because of a non-disjunction in meiosis of chromosome 21. Some patients (2–4%) will have mosaicism for trisomy 21. Since chromosome 21 contains an estimated 225 genes, it is complicated to draw genotype-phenotype correlations.196 Nonetheless, some patients presenting with Down syndrome have partial chromosome 21 trisomy by inheriting translocations (2–4%).197 This partial trisomy has allowed the definition of a “critical region”. The dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A) is encoded by a gene located in the critical region of chromosome 21 for Down syndrome (OMIM #190685).198 DYRK1A phosphorylates multiple targets: signal transducer and activator of transcription 3 (STAT3), the ε subunit of eukaryotic initiation factor 2B (eIF2Bε), Tau, forkhead family transcription factor (FKHR), dynamin, glycogen synthase and cyclin L2.199
The Drosophila ortholog of DYRK1A is known as minibrain gene (mnb).200 Heisenberg has shown that defects in mnb lead to olfactory learning defects but preservation of learning of color discrimination or visual based operant learning.201 mnb is involved in postembryonic neurogenesis and mutations are associated with an abnormal spacing of neuroblasts.202 This results in reduced optic lobe and central brain hemisphere size. These results suggest a role for minibrain in cognition but more work is required to link an increase in DYRK1A copy number to the cognitive symptoms in Down syndrome.
Epilepsy, X-linked, with variable learning disabilities and behavior disorders (Synapsin)
A mutation in Synapsin was identified in a family with epilepsy and learning disabilities (OMIM* 313440).203 Synapsin enhances the GTPase activity of Rab3a and Rab3a recruitment to the synaptic vesicle membrane.204,205 In Drosophila synapsin mutants, Godenschwege et al.206 showed defects in learning and memory and faster habituation in the olfactory jump response.
Mice mutant for Synapsin 1 have delayed synapse formation.207 Chin et al.208 also demonstrated abnormal synaptic vesicle clustering in these mice. In addition to these developmental abnormalities, they identified abnormal recovery after high frequency stimulation in synapsin adult mice.
Genes Identified in Human Mental Retardation and Shown to have Neuronal Phenotype Other than Memory in Drosophila
In this section, we review genes involved in human mental retardation but for which we could not identify in the literature a cognitive phenotype.
Walker-Warburg syndrome (POMT1)
Protein-O-mannosyl transferase 1 (POMT1) is involved in Walker-Warburg syndrome (OMIM #236670) and limb girdle muscular dystrophy type 2K (OMIM #609308).209 Walker-Warburg is characterized by mental retardation in addition to cobblestone lissencephaly, congenital muscular dystrophy, cataracts and other anomalies.210 Limb-girdle muscular dystrophy 2K is associated with mild proximal weakness starting at an early age, mild calves or thigh pseudohypertrophy, elevated CK, microcephaly, and mental retardation.211 The location of the mutation within the gene influences the phenotype.212
The Drosophila ortholog of POMT1 is encoded by rotated abdomen (rt). Drosophila mutants are viable but present a clockwise rotation of the abdomen, which causes mating defects.213 Recently, the Drosophila rt mutation was identified in a screen for synaptic mutants in Drosophila.224 The mutants displayed decreased neuromuscular synaptic release probability and changes in glutamate receptor subunit composition. Indeed, DGluRIIA is absent and the levels of DGluRIIB are decreased in flies containing a P-element insertion within rt. The decreased probability of release seems to be due to a defect in the glycosylation of Dystroglycan.
Down syndrome (SIM2)
As noted above, trisomy of chromosome 21 leads to Down syndrome (OMIM #190685), which is characterized by mental retardation and dysmorphic features. SIM2 is one of the genes in a critical region of chromosome 21 for Down syndrome.210 The ortholog of SIM2 is named Single-minded (Sim) in Drosophila. Sim is involved in neurogenesis and midline cell fate determination.215 Sim is a transcription factor that is part of a family that contains a domain similar to the basic helix-loop-helix (bHLH) motif. Another member of this family is the circadian rhythm protein Period.216 Sim activates midline gene transcription 217 while repressing the lateral gene expression.218,219 After heterodimerization with Tango, Sim migrates to the nucleus where it binds the CNS-Midline Element sequence, which leads to hedgehog transcription.220 The role of Sim is not limited to the brain, it is also required in eye development.221 Locomotor deficits have been observed in sim mutants. Interestingly, the mutants can only walk in circles.222 This behavioral shortcoming is attributed to a defect in the central complex.223,224
Down syndrome (Dscam)
Another gene previously identified as present in three copies in Down syndrome patients with congenital heart disease is Dscam.225 Dscam is a member of the immunoglobulin superfamily and constitutes a new class of neural cell adhesion molecules.226 The fly ortholog was identified227 and shown to be able to generate multiple isoforms by alternative splicing. Hattori et al.228 engineered a Dscam gene that could not be alternatively spliced and therefore produced a single isoform. They observed that the embryonic ventral cord wiring was severely disrupted. Also, they showed abnormal crossing of the B-lobe of the mushroom body across the brain midline.
α-thalassemia/mental retardation syndrome (XNP/ATRX)
A mutation in the XNP/ATRX gene causes mental retardation syndromes associated with facial dysmophic features, urogenital defects and α-thalassemia (OMIM #301040).239 XNP/ATRX encodes an SNF2 family zinc-finger ATPase/helicase protein.240 It has been shown to be involved in DNA methylation,240 chromatin remodeling,241 transcription,242 the cell cycle and apoptosis.
dATR-X is the Drosophila homolog of the disease gene causing α-thalassemia/mental retardation X-linked.232–238 Sun et al.249 identified dATRX as an enhancer of jing in axon scaffolding formation by glia cells. Lee et al.240 isolated another Drosophila homolog of XNP/ATRX that they named dXNP, and they showed that ectopic overexpression of dXNP in the Drosophila eye led to apoptosis. The fly and human proteins are most conserved in the helicase and SNF2 domains.239 Sun et al.239 showed that in flies, dATRX is expressed both in neurons and glial cells. In addition, overexpression of dATRX led to thinner longitudinal connectives and reduced spacing between the anterior and posterior commissures. dATRX RNAi expression in neurons results in neuronal crossing and disruption of the longitudinal fibers.
Mental retardation, hypopigmentation (WOC)
Van der Maarel et al.241 reported the case of a female patient with mental retardation, hypopigmentation over the abdomen and scoliosis that presented with a mutation in the 5′ UTR region of the zinc finger gene ZNF261. ZNF261 is part of a polypeptide complex also containing the histone deactylase 1 and 2 (HDAC1/HDAC2).242 without children (woc) encodes the Drosophila homolog of ZNF261. Mutant males and females are sterile.243 Raffa et al.244 showed that Woc protein is localized to telomeres and serves as a transcription factor with a telomere-capping function. Raffa et al. also showed that woc mutants have frequent telomeric fusions in Drosophila brain cells.
Genes Identified in Human Mental Retardation and Shown to have Extra-Neuronal Phenotype in Drosophila
In this section, we review genes involved in human mental retardation but for which we could not identify in the literature a cognitive or neuronal phenotype.
Cornelia de Lange (NIPPEDB)
A mutation in Nipped-B-Like (NIPBL) (OMIM #122470) has been identified in approximately 50% of Cornelia de Lange syndrome patients.245 More rarely, mutations in SMC1L1 are identified (OMIM #300590).246 Cornelia de Lange syndrome (CdLS) is characterized by synophrys, variable mental retardation, growth retardation, hirsutism, and upper limb reduction defects that range from subtle phalangeal abnormalities to oligodactyly.247 NIPBL is involved in sister chromatid cohesion via the cohesin complex, and is also involved in long range enhancer-promoter interaction.248 Interestingly, a mutation in the cohesin complex (OMIM #610759) has been identified in a third Cornelia de Lange group. Recently Gause et al.249 have shown in Drosophila that NIPBL is co-localized with cohesin in somatic and meiotic cells. Furthermore, in meiotic cells, it is co-localized with the synaptonemal complex.
Rubinstein-Taybe syndrome (CBP)
Finally, we return to the cAMP pathway and CREB. Once phosphorylated at serine 133, CREB recruits CREB binding protein (CBP). CBP is a histone acetyltransferase (HAT) that leads to chromatin opening, exposure of DNA to polymerase II and transcription. Deletions within CBP are responsible for Rubinstein-Taybi syndrome (RTS) (OMIM #180849). This syndrome is characterized by mental retardation (IQ average 51), seizures (23% of patients), microcephaly and various dysmorphic features, such as downward slanted palpebral fissures, hypoplastic maxilla with narrow palate, small mouth opening, broad thumb with radial angulations and broad great toes. Oike et al.250,251 first established a mouse model of RTS and showed defective long-term memory in passive avoidance and fear conditioning tasks. Bourtchouladze et al.252 went on to show that mutant CBP mice have a selective deficit in long-term memory but normal short-term memory. In accordance with the previously established cAMP pathway, phosphodiesterase inhibitors ameliorated this long-term memory defect, presumably by increasing cAMP levels and CREB phosphorylation after training. Importantly, this outcome in the mouse model of RTS suggests that (i) some forms of mental retardation result from functional (biochemical) rather than structural (developmental) deficits in brain function, and (ii) the former will respond to traditional drug therapy.253 The role of CBP has not been studied for memory in Drosophila, but Jung et al.254 recently showed that CBP mutant flies experience an increase in CAG repeat instability. In addition, CBP has been shown to regulate the hedgehog pathway.255 Mutations in Sonic hedgehog have been identified in patients with holoprosencephaly,256,257 a cerebral malformation causing severe mental retardation. It remains unclear if the cognitive defect is solely due to the developmental cerebral malformation.
Conclusion
We have reviewed several single gene human mental retardation disorders for which mutations in the Drosophila ortholog result in deficient learning and long-term memory. We have also shown that synaptic dysfunction is present in Drosophila mutants for genes involved in human mental retardation. This reinforces the notion that, although divergent from a neuroanatomic perspective, flies and humans have conserved molecular pathways for plasticity, memory and cognition. It also supports the idea of using Drosophila as a screening tool for studying genetic interactions, determining spatiotemporal requirements for a given gene and identifying pharmacological agents to treat intellectual disabilities. The next challenge will be to understand the genetic network in which a given gene is operating in vivo for cognitive processes and to treat intellectual disabilities using a multi-target approach that minimizes the risk of side effects or paradoxical responses.
Acknowledgments
We would like to dedicate this review to Dr. Seymour Benzer (1921–2007) for his pioneering work in the field. We would like to thank Mr. Michel Bolduc, Mme. Susan Van Nispen, Mr. Cory Rosenfelt and Mr. James Duffy for their help with manuscript preparation.
References
- 1.Penrose L. A Clinical and Genetic Study of 1280 Cases of Mental Defect. London: 1938. [Google Scholar]
- 2.Raymond FL, Tarpey P. The genetics of mental retardation. Hum Mol Genet. 2006;15(Spec No 2):R110–6. doi: 10.1093/hmg/ddl189. [DOI] [PubMed] [Google Scholar]
- 3.Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 4.Hagberg B, Hagberg G, Lewerth A, Lindberg U. Mild mental retardation in Swedish school children. I. Prevalence. Acta Paediatr Scand. 1981;70:441–4. doi: 10.1111/j.1651-2227.1981.tb05720.x. [DOI] [PubMed] [Google Scholar]
- 5.McLaren J, Bryson SE. Review of recent epidemiological studies of mental retardation: prevalence, associated disorders, and etiology. Am J Ment Retard. 1987;92:243–54. [PubMed] [Google Scholar]
- 6.Yeargin-Allsopp M, Boyle C. Overview: the epidemiology of neurodevelopmental disorders. Ment Retard Dev Disabil Res Rev. 2002;8:113–6. doi: 10.1002/mrdd.10030. [DOI] [PubMed] [Google Scholar]
- 7.Roeleveld N, Zielhuis GA, Gabreels F. The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol. 1997;39:125–32. doi: 10.1111/j.1469-8749.1997.tb07395.x. [DOI] [PubMed] [Google Scholar]
- 8.Raymond FL. X linked mental retardation: a clinical guide. J Med Genet. 2006;43:193–200. doi: 10.1136/jmg.2005.033043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Association AP. Diagnostic and statistical manual of mental disorders DSM IV. 1994. [Google Scholar]
- 10.Kleefstra T, Hamel BC. X-linked mental retardation: further lumping, splitting and emerging phenotypes. Clin Genet. 2005;67:451–67. doi: 10.1111/j.1399-0004.2005.00434.x. [DOI] [PubMed] [Google Scholar]
- 11.Moog U. The outcome of diagnostic studies on the etiology of mental retardation: considerations on the classification of the causes. Am J Med Genet A. 2005;137:228–31. doi: 10.1002/ajmg.a.30841. [DOI] [PubMed] [Google Scholar]
- 12.Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. Eur J Hum Genet. 2008;16:422–34. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- 13.Frints SG, Froyen G, Marynen P, Fryns JP. X-linked mental retardation: vanishing boundaries between non-specific (MRX) and syndromic (MRXS) forms. Clin Genet. 2002;62:423–32. doi: 10.1034/j.1399-0004.2002.620601.x. [DOI] [PubMed] [Google Scholar]
- 14.Lejeune J, Turpin R, Gautier M. Chromosomic diagnosis of mongolism. Arch Fr Pediatr. 1959;16:962–3. [PubMed] [Google Scholar]
- 15.Lejeune J, Turpin R, Gautier M. Mongolism; a chromosomal disease (trisomy) Bull Acad Natl Med. 1959;143:256–65. [PubMed] [Google Scholar]
- 16.Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science. 1977;197:265–6. doi: 10.1126/science.877551. [DOI] [PubMed] [Google Scholar]
- 17.Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE. The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet. 1995;9:132–40. doi: 10.1038/ng0295-132. [DOI] [PubMed] [Google Scholar]
- 18.Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, et al. Subtelomere FISH analysis of 11,688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43:478–89. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–23. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 20.Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–67. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- 21.Bejjani BA, Theisen AP, Ballif BC, Shaffer LG. Array-based comparative genomic hybridization in clinical diagnosis. Expert Rev Mol Diagn. 2005;5:421–9. doi: 10.1586/14737159.5.3.421. [DOI] [PubMed] [Google Scholar]
- 22.Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, et al. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci USA. 2007;104:12831–6. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebat J. Major changes in our DNA lead to major changes in our thinking. Nat Genet. 2007;39:S3–5. doi: 10.1038/ng2095. [DOI] [PubMed] [Google Scholar]
- 24.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizuguchi T, Hashimoto R, Itokawa M, Sano A, Shimokawa O, Yoshimura Y, et al. Microarray comparative genomic hybridization analysis of 59 patients with schizophrenia. J Hum Genet. 2008;53:914–9. doi: 10.1007/s10038-008-0327-6. [DOI] [PubMed] [Google Scholar]
- 26.Moon HJ, Yim SV, Lee WK, Jeon YW, Kim YH, Ko YJ, et al. Identification of DNA copy-number aberrations by array-comparative genomic hybridization in patients with schizophrenia. Biochem Biophys Res Commun. 2006;344:531–9. doi: 10.1016/j.bbrc.2006.03.156. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg C, Knijnenburg J, Bakker E, Vianna-Morgante AM, Sloos W, Otto PA, et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet. 2006;43:180–6. doi: 10.1136/jmg.2005.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vissers LE, de Vries BB, Osoegawa K, Janssen IM, Feuth T, Choy CO, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–70. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41:241–8. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid BM, Nordenskjold M. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J Med Genet. 2005;42:699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tyson C, Harvard C, Locker R, Friedman JM, Langlois S, Lewis ME, et al. Submicroscopic deletions and duplications in individuals with intellectual disability detected by array-CGH. Am J Med Genet A. 2005;139:173–85. doi: 10.1002/ajmg.a.31015. [DOI] [PubMed] [Google Scholar]
- 32.Miyake N, Shimokawa O, Harada N, Sosonkina N, Okubo A, Kawara H, et al. BAC array CGH reveals genomic aberrations in idiopathic mental retardation. Am J Med Genet A. 2006;140:205–11. doi: 10.1002/ajmg.a.31098. [DOI] [PubMed] [Google Scholar]
- 33.Shevell MI, Bejjani BA, Srour M, Rorem EA, Hall N, Shaffer LG. Array comparative genomic hybridization in global developmental delay. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1101–8. doi: 10.1002/ajmg.b.30730. [DOI] [PubMed] [Google Scholar]
- 34.Baris HN, Tan WH, Kimonis VE, Irons MB. Diagnostic utility of array-based comparative genomic hybridization in a clinical setting. Am J Med Genet A. 2007;143A:2523–33. doi: 10.1002/ajmg.a.31988. [DOI] [PubMed] [Google Scholar]
- 35.Hagberg B, Hagberg G, Lewerth A, Lindberg U. Mild mental retardation in Swedish school children. II. Etiologic and pathogenetic aspects. Acta Paediatr Scand. 1981;70:445–52. doi: 10.1111/j.1651-2227.1981.tb05721.x. [DOI] [PubMed] [Google Scholar]
- 36.Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–80. doi: 10.1212/01.wnl.0000031431.81555.16. [DOI] [PubMed] [Google Scholar]
- 37.Didelot G, Molinari F, Tchenio P, Comas D, Milhiet E, Munnich A, et al. Tequila, a neurotrypsin ortholog, regulates long-term memory formation in Drosophila. Science. 2006;313:851–3. doi: 10.1126/science.1127215. [DOI] [PubMed] [Google Scholar]
- 38.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–5. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Bolduc FV, Bell K, Tully T, Fang Y, Sehgal A, et al. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci USA. 2008;105:12399–404. doi: 10.1073/pnas.0805291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayford M, Kandel ER. Genetic approaches to memory storage. Trends Genet. 1999;15:463–70. doi: 10.1016/s0168-9525(99)01846-6. [DOI] [PubMed] [Google Scholar]
- 41.Restifo LL. Mental retardation genes in Drosophila: New approaches to understanding and treating developmental brain disorders. Ment Retard Dev Disabil Res Rev. 2005;11:286–94. doi: 10.1002/mrdd.20083. [DOI] [PubMed] [Google Scholar]
- 42.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–25. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fortini ME, Bonini NM. Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer. Trends Genet. 2000;16:161–7. doi: 10.1016/s0168-9525(99)01939-3. [DOI] [PubMed] [Google Scholar]
- 44.Ribot T. L’Heredite Psychologique. Paris: 1882. [Google Scholar]
- 45.Ebbinghaus H. Memory: A Contribution to Experimental Psychology. New Yrok: Teachers College, Columbia University; 1885. [Google Scholar]
- 46.Dubnau J, Chiang AS, Tully T. Neural substrates of memory: from synapse to system. J Neurobiol. 2003;54:238–53. doi: 10.1002/neu.10170. [DOI] [PubMed] [Google Scholar]
- 47.Pavlov I. Conditioned Reflexes. New York: 1927. [Google Scholar]
- 48.Rescorla RA. Effect of a stimulus intervening between CS and US in autoshaping. J Exp Psychol Anim Behav Process. 1982;8:131–41. [PubMed] [Google Scholar]
- 49.Rescorla RA. Simultaneous second-order conditioning produces S-S learning in conditioned suppression. J Exp Psychol Anim Behav Process. 1982;8:23–32. [PubMed] [Google Scholar]
- 50.Skinner B. The Behavior of Organisms. New York: Appleton-Century Company; 1938. [Google Scholar]
- 51.Thorndike E. The influence of improvement in one mental function upon the efficiency of other functions. Psychological Review. 1901;8:247–61. [Google Scholar]
- 52.Benzer S. Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc Natl Acad Sci USA. 1967;58:1112–9. doi: 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benzer S. Genetic dissection of behavior. Sci Am. 1973;229:24–37. doi: 10.1038/scientificamerican1273-24. [DOI] [PubMed] [Google Scholar]
- 54.Hotta Y, Benzer S. Mapping of behavior in Drosophila mosaics. Symp Soc Dev Biol. 1973;31:129–67. doi: 10.1016/b978-0-12-612975-5.50010-x. [DOI] [PubMed] [Google Scholar]
- 55.Quinn WG, Harris WA, Benzer S. Conditioned behavior in Drosophila melanogaster. Proc Natl Acad Sci USA. 1974;71:708–12. doi: 10.1073/pnas.71.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol [A] 1985;157:263–77. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- 57.Dudai Y, Jan YN, Byers D, Quinn WG, Benzer S. dunce, a mutant of Drosophila deficient in learning. Proc Natl Acad Sci USA. 1976;73:1684–8. doi: 10.1073/pnas.73.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis RL, Davidson N. Isolation of the Drosophila melanogaster dunce chromosomal region and recombinational mapping of dunce sequences with restriction site polymorphisms as genetic markers. Mol Cell Biol. 1984;4:358–67. doi: 10.1128/mcb.4.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Livingstone MS, Sziber PP, Quinn WG. Loss of calcium/calmodulin responsiveness in adenylate cyclase of rutabaga, a Drosophila learning mutant. Cell. 1984;37:205–15. doi: 10.1016/0092-8674(84)90316-7. [DOI] [PubMed] [Google Scholar]
- 60.Levin LR, Han PL, Hwang PM, Feinstein PG, Davis RL, Reed RR. The Drosophila learning and memory gene rutabaga encodes a Ca2+/Calmodulin-responsive adenylyl cyclase. Cell. 1992;68:479–89. doi: 10.1016/0092-8674(92)90185-f. [DOI] [PubMed] [Google Scholar]
- 61.Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–8. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 62.Spiegel A. G Proteins. Austin, Tx: R.G. Landes Company; 1994. [Google Scholar]
- 63.Schwaerzel M, Monastirioti M, Scholz H, Friggi-Grelin F, Birman S, Heisenberg M. Dopamine and octopamine differentiate between aversive and appetitive olfactory memories in Drosophila. J Neurosci. 2003;23:10495–502. doi: 10.1523/JNEUROSCI.23-33-10495.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Connolly JB, Roberts IJ, Armstrong JD, Kaiser K, Forte M, Tully T, O’Kane CJ. Associative learning disrupted by impaired Gs signaling in Drosophila mushroom bodies. Science. 1996;274:2104–7. doi: 10.1126/science.274.5295.2104. [DOI] [PubMed] [Google Scholar]
- 65.Kandel ER. Search of Memory: The Emergence of a New Science of Mind. New York: W.W. Norton; 2006. [Google Scholar]
- 66.Kandel ER. The molecular biology of memory storage: A dialogue between genes and synapses. Science. 2001;294:1030–8. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 67.Castellucci V, Pinsker H, Kupfermann I, Kandel ER. Neuronal mechanisms of habituation and dishabituation of the gill-withdrawal reflex in Aplysia. Science. 1970;167:1745–8. doi: 10.1126/science.167.3926.1745. [DOI] [PubMed] [Google Scholar]
- 68.Kupfermann I, Castellucci V, Pinsker H, Kandel E. Neuronal correlates of habituation and dishabituation of the gill-withdrawal reflex in Aplysia. Science. 1970;167:1743–5. doi: 10.1126/science.167.3926.1743. [DOI] [PubMed] [Google Scholar]
- 69.Brunelli M, Castellucci V, Kandel ER. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science. 1976;194:1178–81. doi: 10.1126/science.186870. [DOI] [PubMed] [Google Scholar]
- 70.Klein M, Camardo J, Kandel ER. Serotonin modulates a specific potassium current in the sensory neurons that show presynaptic facilitation in Aplysia. Proc Natl Acad Sci USA. 1982;79:5713–7. doi: 10.1073/pnas.79.18.5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kandel ER, Schwartz JH. Molecular biology of learning: modulation of transmitter release. Science. 1982;218:433–43. doi: 10.1126/science.6289442. [DOI] [PubMed] [Google Scholar]
- 72.Drain P, Folkers E, Quinn WG. cAMP-dependent protein kinase and the disruption of learning in transgenic flies. Neuron. 1991;6:71–82. doi: 10.1016/0896-6273(91)90123-h. [DOI] [PubMed] [Google Scholar]
- 73.Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–54. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- 74.Barondes SH, Jarvik ME. The Influence of Actinomycin-D on Brain Rna Synthesis and on Memory. J Neurochem. 1964;11:187–95. doi: 10.1111/j.1471-4159.1964.tb06128.x. [DOI] [PubMed] [Google Scholar]
- 75.Tully T, Preat T, Boynton SC, Del Vecchio M. Genetic dissection of consolidated memory in Drosophila. Cell. 1994;79:35–47. doi: 10.1016/0092-8674(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 76.Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, et al. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 77.Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, et al. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–62. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- 78.Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci USA. 1997;94:2693–8. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 80.Wagatsuma A, Azami S, Sakura M, Hatakeyama D, Aonuma H, Ito E. De Novo synthesis of CREB in a presynaptic neuron is required for synaptic enhancement involved in memory consolidation. J Neurosci Res. 2006;84:954–60. doi: 10.1002/jnr.21012. [DOI] [PubMed] [Google Scholar]
- 81.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–82. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 82.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xia S, Miyashita T, Fu TF, Lin WY, Wu CL, Pyzocha L, et al. NMDA receptors mediate olfactory learning and memory in Drosophila. Curr Biol. 2005;15:603–15. doi: 10.1016/j.cub.2005.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 86.Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–53. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- 87.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–79. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 88.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 89.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 90.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–7. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 91.Gotz KG. Optomoter studies of the visual system of several eye mutants of the fruit fly Drosophila. Kybernetik. 1964;2:77–92. doi: 10.1007/BF00288561. [DOI] [PubMed] [Google Scholar]
- 92.Heisenberg M. Initial activity and voluntary behavior in animals. Naturwissenschaften. 1983;70:70–8. doi: 10.1007/BF00365500. [DOI] [PubMed] [Google Scholar]
- 93.Wolf R, Heisenberg M. Basic organization of operant behavior as revealed in Drosophila flight orientation. J Comp Physiol [A] 1991;169:699–705. doi: 10.1007/BF00194898. [DOI] [PubMed] [Google Scholar]
- 94.Brembs B, Heisenberg M. Conditioning with compound stimuli in Drosophila melanogaster in the flight simulator. J Exp Biol. 2001;204:2849–59. doi: 10.1242/jeb.204.16.2849. [DOI] [PubMed] [Google Scholar]
- 95.Gong Z, Xia S, Liu L, Feng C, Guo A. Operant visual learning and memory in Drosophila mutants dunce, amnesiac and radish. J Insect Physiol. 1998;44:1149–58. doi: 10.1016/s0022-1910(98)00076-6. [DOI] [PubMed] [Google Scholar]
- 96.Sitaraman D, Zars M, Laferriere H, Chen YC, Sable-Smith A, Kitamoto T, et al. Serotonin is necessary for place memory in Drosophila. Proc Natl Acad Sci USA. 2008;105:5579–84. doi: 10.1073/pnas.0710168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bausenwein B, Muller NR, Heisenberg M. Behavior-dependent activity labeling in the central complex of Drosophila during controlled visual stimulation. J Comp Neurol. 1994;340:255–68. doi: 10.1002/cne.903400210. [DOI] [PubMed] [Google Scholar]
- 98.Ilius M, Wolf R, Heisenberg M. The central complex of Drosophila melanogaster is involved in flight control: studies on mutants and mosaics of the gene ellipsoid body open. J Neurogenet. 1994;9:189–206. doi: 10.3109/01677069409167279. [DOI] [PubMed] [Google Scholar]
- 99.Wu CL, Xia S, Fu TF, Wang H, Chen YH, Leong D, et al. Specific requirement of NMDA receptors for long-term memory consolidation in Drosophila ellipsoid body. Nat Neurosci. 2007;10:1578–86. doi: 10.1038/nn2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pascual A, Preat T. Localization of long-term memory within the Drosophila mushroom body. Science. 2001;294:1115–7. doi: 10.1126/science.1064200. [DOI] [PubMed] [Google Scholar]
- 101.Krashes MJ, Keene AC, Leung B, Armstrong JD, Waddell S. Sequential use of mushroom body neuron subsets during Drosophila odor memory processing. Neuron. 2007;53:103–15. doi: 10.1016/j.neuron.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Greenspan RJ, Ferveur JF. Courtship in Drosophila. Annu Rev Genet. 2000;34:205–32. doi: 10.1146/annurev.genet.34.1.205. [DOI] [PubMed] [Google Scholar]
- 103.Hall JC. The mating of a fly. Science. 1994;264:1702–14. doi: 10.1126/science.8209251. [DOI] [PubMed] [Google Scholar]
- 104.Dickson BJ. Wired for sex: the neurobiology of Drosophila mating decisions. Science. 2008;322:904–9. doi: 10.1126/science.1159276. [DOI] [PubMed] [Google Scholar]
- 105.Siegel RW, Hall JC. Conditioned responses in courtship behavior of normal and mutant Drosophila. Proc Natl Acad Sci USA. 1979;76:3430–4. doi: 10.1073/pnas.76.7.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ejima A, Smith BP, Lucas C, Levine JD, Griffith LC. Sequential learning of pheromonal cues modulates memory consolidation in trainer-specific associative courtship conditioning. Curr Biol. 2005;15:194–206. doi: 10.1016/j.cub.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gailey DA, Jackson FR, Siegel RW. Conditioning mutations in Drosophila melanogaster affect an experience-dependent behavioral modification in courting males. Genetics. 1984;106:613–23. doi: 10.1093/genetics/106.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cook RM. Courtship processing in Drosophila melanogaster. II. An adaptation to selection for receptivity to wingless males. Anim Behav. 1973;21:349–58. doi: 10.1016/s0003-3472(73)80077-6. [DOI] [PubMed] [Google Scholar]
- 109.Cook RM. Courtship processing in Drosophila melanogaster. I. Selection for receptivity to wingless males. Anim Behav. 1973;21:338–48. doi: 10.1016/s0003-3472(73)80076-4. [DOI] [PubMed] [Google Scholar]
- 110.Wicker-Thomas C, Jallon JM. Control of female pheromones in Drosophila melanogaster by homeotic genes. Genet Res. 2001;78:235–42. doi: 10.1017/s0016672301005377. [DOI] [PubMed] [Google Scholar]
- 111.Kane NS, Robichon A, Dickinson JA, Greenspan RJ. Learning without performance in PKC-deficient Drosophila. Neuron. 1997;18:307–14. doi: 10.1016/s0896-6273(00)80270-6. [DOI] [PubMed] [Google Scholar]
- 112.Griffith LC, Verselis LM, Aitken KM, Kyriacou CP, Danho W, Greenspan RJ. Inhibition of calcium/calmodulin-dependent protein kinase in Drosophila disrupts behavioral plasticity. Neuron. 1993;10:501–9. doi: 10.1016/0896-6273(93)90337-q. [DOI] [PubMed] [Google Scholar]
- 113.Keleman K, Kruttner S, Alenius M, Dickson BJ. Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat Neurosci. 2007;10:1587–93. doi: 10.1038/nn1996. [DOI] [PubMed] [Google Scholar]
- 114.Farfel Z, Bourne HR, Iiri T. The expanding spectrum of G protein diseases. N Engl J Med. 1999;340:1012–20. doi: 10.1056/NEJM199904013401306. [DOI] [PubMed] [Google Scholar]
- 115.Fitch N. Albright’s hereditary osteodystrophy: a review. Am J Med Genet. 1982;11:11–29. doi: 10.1002/ajmg.1320110104. [DOI] [PubMed] [Google Scholar]
- 116.Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J, Bacq D, Briault S, Vekemans M, Munnich A, Attie-Bitach T, Sonderegger P, Colleaux L. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–81. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 117.Shiosaka S. Serine proteases regulating synaptic plasticity. Anat Sci Int. 2004;79:137–44. doi: 10.1111/j.1447-073x.2004.00080.x. [DOI] [PubMed] [Google Scholar]
- 118.Hirata A, Yoshida S, Inoue N, Matsumoto-Miyai K, Ninomiya A, Taniguchi M, et al. Abnormalities of synapses and neurons in the hippocampus of neuropsin-deficient mice. Mol Cell Neurosci. 2001;17:600–10. doi: 10.1006/mcne.2000.0945. [DOI] [PubMed] [Google Scholar]
- 119.Molinari F, Meskanaite V, Munnich A, Sonderegger P, Colleaux L. Extracellular proteases and their inhibitors in genetic diseases of the central nervous system. Hum Mol Genet. 2003;12(Spec No 2):R195–200. doi: 10.1093/hmg/ddg276. [DOI] [PubMed] [Google Scholar]
- 120.Hinton VJ, Halperin JM, Dobkin CS, Ding XH, Brown WT, Miezejeski CM. Cognitive and molecular aspects of fragile X. J Clin Exp Neuropsychol. 1995;17:518–28. doi: 10.1080/01688639508405142. [DOI] [PubMed] [Google Scholar]
- 121.Skinner M, Hooper S, Hatton DD, Roberts J, Mirrett P, Schaaf J, et al. Mapping nonverbal IQ in young boys with fragile X syndrome. Am J Med Genet A. 2005;132:25–32. doi: 10.1002/ajmg.a.30353. [DOI] [PubMed] [Google Scholar]
- 122.Lubs HA. A marker X chromosome. Am J Hum Genet. 1969;21:231–44. [PMC free article] [PubMed] [Google Scholar]
- 123.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 124.Oostra BA, Jacky PB, Brown WT, Rousseau F. Guidelines for the diagnosis of fragile X syndrome. National Fragile X Foundation. J Med Genet. 1993;30:410–3. doi: 10.1136/jmg.30.5.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hornstra IK, Nelson DL, Warren ST, Yang TP. High resolution methylation analysis of the FMR1 gene trinucleotide repeat region in fragile X syndrome. Hum Mol Genet. 1993;2:1659–65. doi: 10.1093/hmg/2.10.1659. [DOI] [PubMed] [Google Scholar]
- 126.Trottier Y, Imbert G, Poustka A, Fryns JP, Mandel JL. Male with typical fragile X phenotype is deleted for part of the FMR1 gene and for about 100 kb of upstream region. Am J Med Genet. 1994;51:454–7. doi: 10.1002/ajmg.1320510431. [DOI] [PubMed] [Google Scholar]
- 127.Wohrle D, Kotzot D, Hirst MC, Manca A, Korn B, Schmidt A, et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet. 1992;51:299–306. [PMC free article] [PubMed] [Google Scholar]
- 128.De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3:31–5. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 129.Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–46. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–64. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 131.Zalfa F, Bagni C. Molecular insights into mental retardation: multiple functions for the Fragile X mental retardation protein? Curr Issues Mol Biol. 2004;6:73–88. [PubMed] [Google Scholar]
- 132.Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 133.Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24:5798–809. doi: 10.1523/JNEUROSCI.1102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–83. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–38. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 136.Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the Fragile X mental retardation protein. Mol Cell Biol. 2000;20:8536–47. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gao FB. Understanding Fragile X syndrome: insights from retarded flies. Neuron. 2002;34:859–62. doi: 10.1016/s0896-6273(02)00740-7. [DOI] [PubMed] [Google Scholar]
- 138.Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–84. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 139.Inoue S, Shimoda M, Nishinokubi I, Siomi MC, Okamura M, Nakamura A, et al. A role for the Drosophila Fragile X-related gene in circadian output. Curr Biol. 2002;12:1331–5. doi: 10.1016/s0960-9822(02)01036-9. [DOI] [PubMed] [Google Scholar]
- 140.Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–70. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- 141.Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol (Berl) 1985;67:289–95. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- 142.Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289–94. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- 143.Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–7. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 144.Cornish KM, Munir F, Cross G. Spatial cognition in males with Fragile-X syndrome: evidence for a neuropsychological phenotype. Cortex. 1999;35:263–71. doi: 10.1016/s0010-9452(08)70799-8. [DOI] [PubMed] [Google Scholar]
- 145.Cianchetti C, Sannio-Fancello G, Fratta AL, Manconi F, Orano A, Pischedda MP, et al. Neuropsychological, psychiatric, and physical manifestations in 149 members from 18 fragile X families. Am J Med Genet. 1991;40:234–43. doi: 10.1002/ajmg.1320400222. [DOI] [PubMed] [Google Scholar]
- 146.Fmr1 knockout mice: a model to study Fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- 147.Maes B, Fryns JP, Van Walleghem M, Van den Berghe H. Cognitive functioning and information processing of adult mentally retarded men with fragile-X syndrome. Am J Med Genet. 1994;50:190–200. doi: 10.1002/ajmg.1320500211. [DOI] [PubMed] [Google Scholar]
- 148.Kooy RF, D’Hooge R, Reyniers E, Bakker CE, Nagels G, De Boulle K, et al. Transgenic mouse model for the fragile X syndrome. Am J Med Genet. 1996;64:241–5. doi: 10.1002/(SICI)1096-8628(19960809)64:2<241::AID-AJMG1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 149.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–50. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–51. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- 151.Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25:7385–92. doi: 10.1523/JNEUROSCI.1520-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–64. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 153.Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, et al. The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol. 2003;13:286–96. doi: 10.1016/s0960-9822(03)00064-2. [DOI] [PubMed] [Google Scholar]
- 154.Barbee SA, Estes PS, Cziko AM, Hillebrand J, Luedeman RA, Coller JM, et al. Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron. 2006;52:997–1009. doi: 10.1016/j.neuron.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002;16:2491–6. doi: 10.1101/gad.1025202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ashraf SI, McLoon AL, Sclarsic SM, Kunes S. Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell. 2006;124:191–205. doi: 10.1016/j.cell.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 157.Cook HA, Koppetsch BS, Wu J, Theurkauf WE. The Drosophila SDE3 homolog armit-age is required for oskar mRNA silencing and embryonic axis specification. Cell. 2004;116:817–29. doi: 10.1016/s0092-8674(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 158.Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, et al. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7:113–7. doi: 10.1038/nn1174. [DOI] [PubMed] [Google Scholar]
- 159.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–6. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 161.Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS ONE. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Berry-Kravis E, Hicar M, Ciurlionis R. Reduced cyclic AMP production in fragile X syndrome: cytogenetic and molecular correlations. Pediatr Res. 1995;38:638–43. doi: 10.1203/00006450-199511000-00002. [DOI] [PubMed] [Google Scholar]
- 163.Berry-Kravis E, Sklena P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am J Med Genet. 1993;45:81–7. doi: 10.1002/ajmg.1320450120. [DOI] [PubMed] [Google Scholar]
- 164.Swaiman . Pediatric Neurology: Principles and Practice. Philadelphia: 2006. [Google Scholar]
- 165.Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. 2000;403:895–8. doi: 10.1038/35002593. [DOI] [PubMed] [Google Scholar]
- 166.Zhong Y. Mediation of PACAP-like neuropeptide transmission by coactivation of Ras/Raf and cAMP signal transduction pathways in Drosophila. Nature. 1995;375:588–92. doi: 10.1038/375588a0. [DOI] [PubMed] [Google Scholar]
- 167.Quinn WG, Sziber PP, Booker R. The Drosophila memory mutant amnesiac. Nature. 1979;277:212–4. doi: 10.1038/277212a0. [DOI] [PubMed] [Google Scholar]
- 168.Feany MB, Quinn WG. A neuropeptide gene defined by the Drosophila memory mutant amnesiac. Science. 1995;268:869–73. doi: 10.1126/science.7754370. [DOI] [PubMed] [Google Scholar]
- 169.Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U. Ethanol intoxication in Drosophila: Genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell. 1998;93:997–1007. doi: 10.1016/s0092-8674(00)81205-2. [DOI] [PubMed] [Google Scholar]
- 170.Hannan F, Ho I, Tong JJ, Zhu Y, Nurnberg P, Zhong Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum Mol Genet. 2006;15:1087–98. doi: 10.1093/hmg/ddl023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ho IS, Hannan F, Guo HF, Hakker I, Zhong Y. Distinct functional domains of neurofibromatosis type 1 regulate immediate versus long-term memory formation. J Neurosci. 2007;27:6852–7. doi: 10.1523/JNEUROSCI.0933-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci USA. 1995;92:5249. doi: 10.1073/pnas.92.11.5249-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 174.Lonard DM, O’Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125:411–4. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 175.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 176.Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–25. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 177.Battaglia G, Granata T, Farina L, D’Incerti L, Franceschetti S, Avanzini G. Periventricular nodular heterotopia: epileptogenic findings. Epilepsia. 1997;38:1173–82. doi: 10.1111/j.1528-1157.1997.tb01213.x. [DOI] [PubMed] [Google Scholar]
- 178.Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–45. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 179.Flanagan LA, Chou J, Falet H, Neujahr R, Hartwig JH, Stossel TP. Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J Cell Biol. 2001;155:511–7. doi: 10.1083/jcb.200105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bellenchi GC, Gurniak CB, Perlas E, Middei S, Ammassari-Teule M, Witke W. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007;21:2347–57. doi: 10.1101/gad.434307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70:375–87. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 182.Putz G, Bertolucci F, Raabe T, Zars T, Heisenberg M. The S6KII (rsk) gene of Drosophila melanogaster differentially affects an operant and a classical learning task. J Neurosci. 2004;24:9745–51. doi: 10.1523/JNEUROSCI.3211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Presente A, Boyles RS, Serway CN, de Belle JS, Andres AJ. Notch is required for long-term memory in Drosophila. Proc Natl Acad Sci USA. 2004;101:1764–8. doi: 10.1073/pnas.0308259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Costa RM, Honjo T, Silva AJ. Learning and memory deficits in Notch mutant mice. Curr Biol. 2003;13:1348–54. doi: 10.1016/s0960-9822(03)00492-5. [DOI] [PubMed] [Google Scholar]
- 185.Wang Y, Chan SL, Miele L, Yao PJ, Mackes J, Ingram DK, et al. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci USA. 2004;101:9458–62. doi: 10.1073/pnas.0308126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Verdile G, Gandy SE, Martins RN. The role of presenilin and its interacting proteins in the biogenesis of Alzheimer’s beta amyloid. Neurochem Res. 2007;32:609–23. doi: 10.1007/s11064-006-9131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–5. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 188.Holinski-Feder E, Chahrockh-Zadeh S, Rittinger O, Jedele KB, Gasteiger M, Lenski C, et al. Nonsyndromic X-linked mental retardation: mapping of MRX58 to the pericentromeric region. Am J Med Genet. 1999;86:102–6. doi: 10.1002/(sici)1096-8628(19990910)86:2<102::aid-ajmg2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 189.Grotewiel MS, Beck CD, Wu KH, Zhu XR, Davis RL. Integrin-mediated short-term memory in Drosophila. Nature. 1998;391:455–60. doi: 10.1038/35079. [DOI] [PubMed] [Google Scholar]
- 190.Luthl A, Laurent JP, Figurov A, Muller D, Schachner M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature. 1994;372:777–9. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- 191.Bliss T, Errington M, Fransen E, Godfraind JM, Kauer JA, Kooy RF, et al. Long-term potentiation in mice lacking the neural cell adhesion molecule L1. Curr Biol. 2000;10:1607–10. doi: 10.1016/s0960-9822(00)00865-4. [DOI] [PubMed] [Google Scholar]
- 192.Strauss R, Heisenberg M. A higher control center of locomotor behavior in the Drosophila brain. J Neurosci. 1993;13:1852–61. doi: 10.1523/JNEUROSCI.13-05-01852.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.de Belle JS, Heisenberg M. Expression of Drosophila mushroom body mutations in alternative genetic backgrounds: a case study of the mushroom body miniature gene (mbm) Proc Natl Acad Sci USA. 1996;93:9875–80. doi: 10.1073/pnas.93.18.9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liu G, Seiler H, Wen A, Zars T, Ito K, Wolf R, Heisenberg M, Liu L. Distinct memory traces for two visual features in the Drosophila brain. Nature. 2006;439:551–6. doi: 10.1038/nature04381. [DOI] [PubMed] [Google Scholar]
- 195.Report. MaMW. The brain in Down syndrome (Trisomy 21) Morb Mortal Wkly Rep. 1994:617–22. [Google Scholar]
- 196.Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–9. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 197.Mikkelsen M. Down syndrome: cytogenetical epidemiology. Hereditas. 1977;86:45–50. doi: 10.1111/j.1601-5223.1977.tb01211.x. [DOI] [PubMed] [Google Scholar]
- 198.Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, et al. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet. 1993;1:114–24. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- 199.Galceran J, de Graaf K, Tejedor FJ, Becker W. The MNB/DYRK1A protein kinase: genetic and biochemical properties. J Neural Transm Suppl. 2003:139–48. doi: 10.1007/978-3-7091-6721-2_12. [DOI] [PubMed] [Google Scholar]
- 200.Guimera J, Casas C, Pucharcos C, Solans A, Domenech A, Planas AM, et al. A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum Mol Genet. 1996;5:1305–10. doi: 10.1093/hmg/5.9.1305. [DOI] [PubMed] [Google Scholar]
- 201.Heisenberg M, Borst A, Wagner S, Byers D. Drosophila mushroom body mutants are deficient in olfactory learning. J Neurogenet. 1985;2:1–30. doi: 10.3109/01677068509100140. [DOI] [PubMed] [Google Scholar]
- 202.Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, et al. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 203.Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, et al. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet. 2004;41:183–6. doi: 10.1136/jmg.2003.013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Giovedi S, Darchen F, Valtorta F, Greengard P, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles. II. Functional effects of the Rab3A-synapsin I interaction. J Biol Chem. 2004;279:43769–79. doi: 10.1074/jbc.M404168200. [DOI] [PubMed] [Google Scholar]
- 205.Giovedi S, Vaccaro P, Valtorta F, Darchen F, Greengard P, Cesareni G, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles. I. Identification and characterization of the synapsin I-Rab3 interactions in vitro and in intact nerve terminals. J Biol Chem. 2004;279:43760–8. doi: 10.1074/jbc.M403293200. [DOI] [PubMed] [Google Scholar]
- 206.Godenschwege TA, Reisch D, Diegelmann S, Eberle K, Funk N, Heisenberg M, et al. Flies lacking all synapsins are unexpectedly healthy but are impaired in complex behavior. Eur J Neurosci. 2004;20:611–22. doi: 10.1111/j.1460-9568.2004.03527.x. [DOI] [PubMed] [Google Scholar]
- 207.Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9235–9. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9230–4. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–24. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 210.Warburg M. Ocular malformations and lissencephaly. Eur J Pediatr. 1987;146:450–2. doi: 10.1007/BF00441592. [DOI] [PubMed] [Google Scholar]
- 211.Dincer P, Balci B, Yuva Y, Talim B, Brockington M, Dincel D, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycan. Neuromuscul Disord. 2003;13:771–8. doi: 10.1016/s0960-8966(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 212.Taniguchi K, Kobayashi K, Saito K, Yamanouchi H, Ohnuma A, Hayashi YK, et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet. 2003;12:527–34. doi: 10.1093/hmg/ddg043. [DOI] [PubMed] [Google Scholar]
- 213.Martin-Blanco E, Garcia-Bellido A. Mutations in the rotated abdomen locus affect muscle development and reveal an intrinsic asymmetry in Drosophila. Proc Natl Acad Sci USA. 1996;93:6048–52. doi: 10.1073/pnas.93.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wairkar YP, Fradkin LG, Noordermeer JN, DiAntonio A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci. 2008;28:3781–9. doi: 10.1523/JNEUROSCI.0478-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Crews ST, Thomas JB, Goodman CS. The Drosophila single-minded gene encodes a nuclear protein with sequence similarity to the per gene product. Cell. 1988;52:143–51. doi: 10.1016/0092-8674(88)90538-7. [DOI] [PubMed] [Google Scholar]
- 216.Crews ST. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev. 1998;12:607–20. doi: 10.1101/gad.12.5.607. [DOI] [PubMed] [Google Scholar]
- 217.Nambu JR, Lewis JO, Wharton KA, Jr, Crews ST. The Drosophila single-minded gene encodes a helix-loop-helix protein that acts as a master regulator of CNS midline development. Cell. 1991;67:1157–67. doi: 10.1016/0092-8674(91)90292-7. [DOI] [PubMed] [Google Scholar]
- 218.Xiao H, Hrdlicka LA, Nambu JR. Alternate functions of the single-minded and rhomboid genes in development of the Drosophila ventral neuroectoderm. Mech Dev. 1996;58:65–74. doi: 10.1016/s0925-4773(96)00559-x. [DOI] [PubMed] [Google Scholar]
- 219.Estes P, Mosher J, Crews ST. Drosophila single-minded represses gene transcription by activating the expression of repressive factors. Dev Biol. 2001;232:157–75. doi: 10.1006/dbio.2001.0174. [DOI] [PubMed] [Google Scholar]
- 220.Liu C, Goshu E, Wells A, Fan CM. Identification of the downstream targets of SIM1 and ARNT2, a pair of transcription factors essential for neuroendocrine cell differentiation. J Biol Chem. 2003;278:44857–67. doi: 10.1074/jbc.M304489200. [DOI] [PubMed] [Google Scholar]
- 221.Umetsu D, Murakami S, Sato M, Tabata T. The highly ordered assembly of retinal axons and their synaptic partners is regulated by Hedgehog/Single-minded in the Drosophila visual system. Development. 2006;133:791–800. doi: 10.1242/dev.02253. [DOI] [PubMed] [Google Scholar]
- 222.Pielage J, Steffes G, Lau DC, Parente BA, Crews ST, Strauss R, et al. Novel behavioral and developmental defects associated with Drosophila single-minded. Dev Biol. 2002;249:283–99. doi: 10.1006/dbio.2002.0770. [DOI] [PubMed] [Google Scholar]
- 223.Nambu JR, Franks RG, Hu S, Crews ST. The single-minded gene of Drosophila is required for the expression of genes important for the development of CNS midline cells. Cell. 1990;63:63–75. doi: 10.1016/0092-8674(90)90288-p. [DOI] [PubMed] [Google Scholar]
- 224.Thomas JB, Crews ST, Goodman CS. Molecular genetics of the single-minded locus: a gene involved in the development of the Drosophila nervous system. Cell. 1988;52:133–41. doi: 10.1016/0092-8674(88)90537-5. [DOI] [PubMed] [Google Scholar]
- 225.Barlow GM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, Celle L, et al. Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genet Med. 2001;3:91–101. doi: 10.1097/00125817-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 226.Yamakawa K, Huot YK, Haendelt MA, Hubert R, Chen XN, Lyons GE, et al. DSCAM: a novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum Mol Genet. 1998;7:227–37. doi: 10.1093/hmg/7.2.227. [DOI] [PubMed] [Google Scholar]
- 227.Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, et al. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101:671–84. doi: 10.1016/s0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- 228.Hattori D, Demir E, Kim HW, Viragh E, Zipursky SL, Dickson BJ. Dscam diversity is essential for neuronal wiring and self-recognition. Nature. 2007;449:223–7. doi: 10.1038/nature06099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Villard L, Fontes M. Alpha-thalassemia/mental retardation syndrome, X-Linked (ATR-X, MIM #301040, ATR-X/XNP/XH2 gene MIM #300032) Eur J Hum Genet. 2002;10:223–5. doi: 10.1038/sj.ejhg.5200800. [DOI] [PubMed] [Google Scholar]
- 230.Gibbons RJ, McDowell TL, Raman S, O’Rourke DM, Garrick D, Ayyub H, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet. 2000;24:368–71. doi: 10.1038/74191. [DOI] [PubMed] [Google Scholar]
- 231.Berube NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–67. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tang J, Wu S, Liu H, Stratt R, Barak OG, Shiekhattar R, et al. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J Biol Chem. 2004;279:20369–77. doi: 10.1074/jbc.M401321200. [DOI] [PubMed] [Google Scholar]
- 233.Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome) Cell. 1995;80:837–45. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 234.Villard L, Gecz J, Mattei JF, Fontes M, Saugier-Veber P, Munnich A, Lyonnet S. XNP mutation in a large family with Juberg-Marsidi syndrome. Nat Genet. 1996;12:359–60. doi: 10.1038/ng0496-359. [DOI] [PubMed] [Google Scholar]
- 235.Villard L, Lacombe D, Fontes M. A point mutation in the XNP gene, associated with an ATR-X phenotype without alpha-thalassemia. Eur J Hum Genet. 1996;4:316–20. doi: 10.1159/000472225. [DOI] [PubMed] [Google Scholar]
- 236.Villard L, Toutain A, Lossi AM, Gecz J, Houdayer C, Moraine C, Fontes M. Splicing mutation in the ATR-X gene can lead to a dysmorphic mental retardation phenotype without alpha-thalassemia. Am J Hum Genet. 1996;58:499–505. [PMC free article] [PubMed] [Google Scholar]
- 237.Abidi F, Schwartz CE, Carpenter NJ, Villard L, Fontes M, Curtis M. Carpenter-Waziri syndrome results from a mutation in XNP. Am J Med Genet. 1999;85:249–51. doi: 10.1002/(sici)1096-8628(19990730)85:3<249::aid-ajmg12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 238.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA. 2003;100:10635–40. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sun X, Morozova T, Sonnenfeld M. Glial and neuronal functions of the Drosophila homolog of the human SWI/SNF gene ATR-X (DATR-X) and the jing zinc-finger gene specify the lateral positioning of longitudinal glia and axons. Genetics. 2006;173:1397–415. doi: 10.1534/genetics.106.057893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lee NG, Hong YK, Yu SY, Han SY, Geum D, Cho KS. dXNP, a Drosophila homolog of XNP/ATRX, induces apoptosis via Jun-N-terminal kinase activation. FEBS Lett. 2007;581:2625–32. doi: 10.1016/j.febslet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 241.van der Maarel SM, Scholten IH, Huber I, Philippe C, Suijkerbuijk RF, Gilgenkrantz S, et al. Cloning and characterization of DXS6673E, a candidate gene for X-linked mental retardation in Xq13.1. Hum Mol Genet. 1996;5:887–97. doi: 10.1093/hmg/5.7.887. [DOI] [PubMed] [Google Scholar]
- 242.Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. 2003;278:7234–9. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 243.Wismar J, Habtemichael N, Warren JT, Dai JD, Gilbert LI, Gateff E. The mutation without children(rgl) causes ecdysteroid deficiency in third-instar larvae of Drosophila melanogaster. Dev Biol. 2000;226:1–17. doi: 10.1006/dbio.2000.9811. [DOI] [PubMed] [Google Scholar]
- 244.Raffa GD, Cenci G, Siriaco G, Goldberg ML, Gatti M. The putative Drosophila transcription factor woc is required to prevent telomeric fusions. Mol Cell. 2005;20:821–31. doi: 10.1016/j.molcel.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 245.Borck G, Redon R, Sanlaville D, Rio M, Prieur M, Lyonnet S, et al. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet. 2004;41:e128. doi: 10.1136/jmg.2004.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006;38:528–30. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 247.Ptacek LJ, Opitz JM, Smith DW, Gerritsen T, Waisman HA. The Cornelia De Lange Syndrome. J Pediatr. 1963;63:1000–20. doi: 10.1016/s0022-3476(63)80234-6. [DOI] [PubMed] [Google Scholar]
- 248.Dorsett D. Adherin: key to the cohesin ring and cornelia de Lange syndrome. Curr Biol. 2004;14:R834–6. doi: 10.1016/j.cub.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 249.Gause M, Webber HA, Misulovin Z, Haller G, Rollins RA, Eissenberg JC, et al. Functional links between Drosophila Nipped-B and cohesin in somatic and meiotic cells. Chromosoma. 2008;117:51–66. doi: 10.1007/s00412-007-0125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–96. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 251.Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, et al. Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood. 1999;93:2771–9. [PubMed] [Google Scholar]
- 252.Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA. 2003;100:10518–22. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–77. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- 254.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 255.Akimaru H, Chen Y, Dai P, Hou DX, Nonaka M, Smolik SM, et al. Drosophila CBP is a co-activator of cubitus interruptus in hedgehog signalling. Nature. 1997;386:735–8. doi: 10.1038/386735a0. [DOI] [PubMed] [Google Scholar]
- 256.Hatziioannou AG, Krauss CM, Lewis MB, Halazonetis TD. Familial holoprosencephaly associated with a translocation breakpoint at chromosomal position 7q36. Am J Med Genet. 1991;40:201–5. doi: 10.1002/ajmg.1320400216. [DOI] [PubMed] [Google Scholar]
- 257.Cordero D, Marcucio R, Hu D, Gaffield W, Tapadia M, Helms JA. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114:485–94. doi: 10.1172/JCI19596. [DOI] [PMC free article] [PubMed] [Google Scholar]