

Abstract
Aggregate culture provides a three-dimensional (3-D) environment for differentiating or differentiated cells; it is particularly useful to study in vitro chondrogenesis and cartilage biology. We have recently ported this method from a conical tube-based format to a 96-well plate format for the study of mesenchymal stem cell (MSC) chondrogenesis. The microplate format has greatly reduced the workload and materials cost, while maintaining reproducible chondrogenic differentiation. A long-term goal is to fully automate aggregate culture—this requires critically identifying all the indispensable steps of the protocol. Robotic laboratory equipment for manipulating microplate assays are commercially available; however, centrifugation steps are difficult to implement automatically. We, therefore, tested whether the centrifugation step can be eliminated, thus significantly streamlining the assay work-flow. By comparing aggregates prepared from human bone marrow-derived MSCs (hMSCs) that were formed either through centrifugation or through free sedimentation, we found that both methods produce aggregates with similar formation kinetics, and that there was no perceptible difference in the timing of the appearance of markers of chondrogenesis. Thus, it appears safe to eliminate the centrifugation step from the aggregate culture protocol. This results in significant time and effort savings and paves the way for future full automation of the aggregate assay.
INTRODUCTION
Aggregate culture is a cell culture technique that has been used widely to provide a three-dimensional (3-D) environment for differentiating cells, particularly to study in vitro chondrogenesis. This system was used in the 1960s to study organ-cultured chondrocyte aggregates on nutrient agar (1). A version of this culture system, in which the aggregates are established in 15-mL polypropylene centrifuge tubes, has been used to study terminal differentiation of rat (2,3) and rabbit chondrocytes (4). More recently, the chondrogenic potential of rabbit (5) and human bone marrow-derived mesenchymal progenitor cells (6) and rat periosteum-derived progenitor cells (7) have also been studied using this culture technique. In this approach, an aliquot of cell suspension is centrifuged in a 15-mL tube, and the resulting cell pellet is left undisturbed at the bottom of the tube. The cells form free-floating cell aggregates in which cell-cell, rather than cell-substrate, interactions take place. Polypropylene tubes are a critical component of this assay, because the cells do not adhere to the tube walls, but rather to one another. Our group is investigating factors that contribute to the chondrogenic differentiation of human mesenchymal stem cells (hMSCs) in vitro (8). These experiments require us to prepare aggregates in large numbers. Generating large numbers of aggregates using 15-mL conical tubes is both time-consuming and expensive. We have recently simplified the aggregate culture method by substituting polypropylene 96-well plates for the conical tubes (9). The microplate-based methodology has enabled us to greatly reduce work load and materials costs, while maintaining reproducible chondrogenic differentiation. It can now be used to efficiently study combinatorial effects of growth factors and cytokines on the chondrogenic potential of hMSCs (10) using large sample sizes.
Fully automating this culture system seems a logical next step. Robotic laboratory equipment for handling, stacking, shaking, sampling, and washing wells in the microplate format are commercially available. Although centrifuges as part of a robotic workstation do exist, they are complex and expensive. In this study, we tested whether the centrifugation step could be eliminated outright, thus significantly streamlining the workflow in this assay, saving time and effort, and making automation simpler.
MATERIALS AND METHODS
Materials
Cell culture medium [Dulbecco’s modified Eagle’s medium (DMEM) with either 4.5 g/L DMEM-HG or 1.5 g/L glucose DMEM-LG], trypsin, L-glutamine, antibiotic/antimycotic (105 U/mL penicillin G sodium, 10 mg/mL streptomycin sulfate, and 25 μg/mL amphotericin B in 0.85% saline), nonessential amino acids, and sodium pyruvate were obtained from Invitrogen (Carlsbad, CA, USA) or Mediatech (Herndon, VA, USA). Transforming growth factor β-1 (TFG-β1) was obtained from PeproTech (Rocky Hill, NJ, USA), and recombinant human fibroblast growth factor-2 (FGF-2) was generously provided by the Biological Resources Branch of the National Cancer Institute (www.ncifcrf.gov/research/brb/site/home.asp). ITS+ Premix was obtained from Collaborative Biomedical Products (Bedford, MA, USA), and ascorbate-2-phosphate from Wako Chemicals (Richmond, VA, USA). Fetal bovine serum (FBS) was obtained from Invitrogen, after screening as described by Lennon et al. (11). Bovine calf serum (BCS) was from Hyclone (Logan, UT, USA). Collagenase was from Worthington Chemical Corporation (Lakewood, NJ, USA). Percoll®, Hoechst 33258 dye, dexamethasone, papain, and Tyrode’s salt solution were obtained from Sigma-Aldrich (St. Louis, MO, USA). DNA standards were from Amersham Biosciences (Piscataway, NJ, USA), while chondroitin sulfate C standards were from Seikagaku America (East Falmouth, MA, USA). All cell culture plasticware was from BD Biosciences (San Jose, CA, USA), except for the polypropylene 96-well plates, which were sourced from Phenix Research Products (Hayward, CA, USA). Antibodies to collagen type I were from Sigma-Aldrich, anti-collagen type II from the Developmental Studies Hybridoma Bank at the University of Iowa (dshb.biology.uiowa.edu), and anti-collagen type × was a gift from Dr. Gary J. Gibson. Mouse pre-immune immunoglobulin G (IgG) was purchased from Vector Laboratories (Burlingame, CA, USA). Software packages used for data acquisition and analysis include ImageJ (rsb.info. nih.gov/ij), Microsoft® Excel®, and Sigmaplot/Sigmastat (Systat Software, San Jose, CA, USA).
Cells
hMSCs were obtained from four healthy adult volunteer donors. The marrow was collected using a procedure reviewed and approved by the University Hospitals of Cleveland Institutional Review Board; informed consent was obtained from all donors. The cells were prepared as described by Haynesworth et al. (12). Briefly, 10–15 mL bone marrow aspirate were combined with DMEM-LG supplemented with 10% FBS and centrifuged at 500× g for 5 min. The cell pellet was resuspended in 5 mL DMEM-LG supplemented with 10% FBS from a selected lot (11) and layered over 35 mL 63% (v/v) Percoll in Tyrode’s salt solution, adjusted to 100 mM final NaCl concentration. After centrifugation at 460× g, the top 25% of the gradient was transferred to a 50-mL tube. The volume was increased to 50 mL with serum-containing medium and recentrifuged at 500× g. The resulting pellet was resuspended in serum-supplemented medium; the cells were seeded onto 10-cm diameter plates at 1.8 × 105 nucleated cells/cm2 and incubated for 4 days. At this point, nonadherent cells were removed by replacing the medium, which was now supplemented with 10 ng/mL FGF-2, since it had been previously shown that FGF-2 improves chondrogenesis (13). Subsequently, the medium was replaced every 3 days for approximately 2 weeks, after which they were subcultured using trypsin (14) and replated at 5 × 103 cells/cm2.
Chondrogenic Medium
We used a defined chondrogenic medium composed of DMEM-HG supplemented with 1% ITS+ Premix, 100 μM ascorbate-2-phosphate, 10−7 M dexamethasone, and 10 ng/mL TGF-β1 (5). Additional supplements, such as L-glutamine, antibiotic/antimycotic, nonessential amino acids, and sodium pyruvate were also added at 1%.
Aggregate Culture
Aggregates were prepared as described by Ballock et al. (3), modified as described previously (5,9). At the end of first passage, culture-expanded hMSCs were released using 0.05% trypsin EDTA; the trypsin was then inhibited using calf serum. The cell suspension was mixed by gentle pipeting to ensure homogeneity, and the cells were then suspended in chondrogenic medium at a final concentration of 1.25 × 106 cells/mL. Two hundred microliters of cell suspension were then pipeted into each well of an autoclaved 96-well, V-bottom, 300-μL polypropylene microplate. The plates were capped with polypropylene lids. Half of the plates were centrifuged for 5 min at 500 × g and then placed in a cell culture incubator (37°C; humidified atmosphere of 95% air and 5% CO2). The other half of the plates was placed in the incubator immediately. Twenty-four hours later, the newly formed aggregates were released from the bottom of the wells by gently squirting a small volume of the existing medium with an 8-channel pipet to ensure that they float freely (9). Although in most cases this release step was not necessary for the freely sedimented cells, some few aggregates remained partially attached so it should be retained. The chondrogenic medium was replaced every 3 days.
Harvest and Analysis
Every 2–3 days, nine aggregates from each group were harvested. Six aggregates were used to measure DNA and glycosaminoglycan (GAG) content, and three were used for histology.
Histology samples were fixed with 10% neutral-buffered formalin, paraffin-embedded, and sectioned. Adjacent sections were either stained with toluidine blue O to evaluate proteoglycan content or by an indirect immunofluorescence assay using a panel of antibodies against various collagen isoforms. In the latter case, antigen unmasking was performed with a solution of 1 mg/mL pronase in phosphate-buffered saline (PBS) for 15 min. Sections were then blocked with 5% bovine serum albumin (BSA) in PBS for 30 min. The primary antibody against collagen type I, type II, or type X, diluted in 1% BSA in PBS, was applied to the sections for 1 h. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG, diluted in 1% BSA in PBS, was applied for 45 min. Between steps in the protocol, the sections were washed with PBS for 5 min. Mouse pre-immune IgG and no primary antibody were used as negative controls. The slides were then wet mounted using 5% N-propyl gallate in glycerol, and photographed with a SPOT RT digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) with a 10× objective, using a Leica fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany). A montage of the entire construct was made from individual images (Figure 1) (15). The montage was analyzed using Image J image analysis software. Biochemical analyses included determining the cellularity of six replicate aggregates by measuring the DNA content. Aggregates were digested with 200 μL papain solution (25 μg/mL papain, 2 mM cysteine, 50 mM NaH2PO4, and 2 mM EDTA, pH 6.5) (16) at 65°C. The papain-digested extracts were then combined with 400 μL 0.1 N NaOH and incubated at room temperature for 30 min. The extracts were neutralized with 400 μL 0.1 N HCl in 5 M NaCl and 100 mM NaH2PO4. In a 96-well microplate, 100 μL extracts were combined with 100 μL 0.7 μg/mL Hoechst 33258 dye in water. Fluorescence was read using a GENios Pro™ micro-plate reader (Tecan, Durham, NC, USA; λEx = 340 nm, λEm = 465 nm) and compared to that of a certified calf thymus DNA standard.
Figure 1. Toluidine blue O stained histological sections of centrifuged (indicated as C) and uncentrifuged (indicated as U) aggregates harvested at various time points.
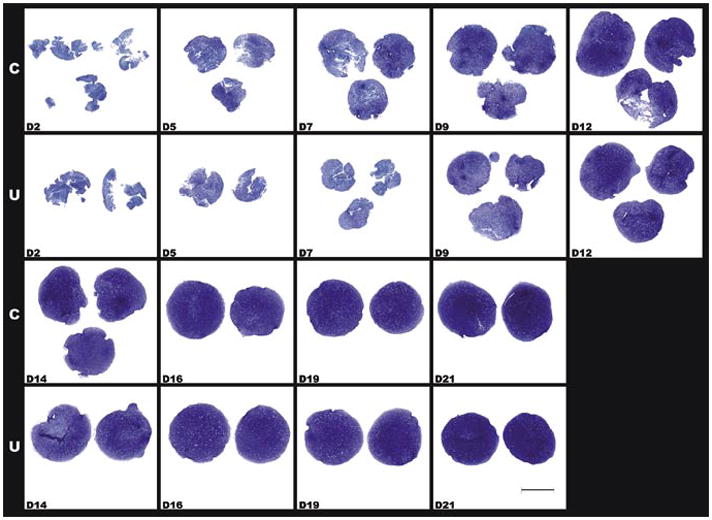
D2 indicates day 2 and so on through day 21. Scale bar, 1 mm.
For glycosaminoglycan quantification, 0.45 μm pore-size nitrocellulose membranes were prewetted with distilled water and placed into a dot-blot apparatus. A 250-μL aliquot of 0.02% Safranin O in 50 mM sodium acetate, pH 4.8, was pipeted into each well, and 25-μL aliquots of the papain-digested extracts or standards (shark cartilage chondroitin sulfate C) (17) were then pipeted into the Safranin O. Vacuum was applied until the samples moved through the filter. The wells were rinsed with distilled water, and the filter removed from the apparatus. The individual dots were cut out, transferred to microcentrifuge tubes, and eluted for 20 min in 10% cetylpyridinium chloride at 37°C. The absorbance of the elutates was read at 530 nm in the microplate reader.
RESULTS AND DISCUSSION
Compared to the practice of using conical tubes, the microplate aggregate culture technique described in our previous report eliminates many manual steps; however, it is still relatively labor-intensive (9). Aggregate culture is initiated by sedimenting a cell suspension in a polypropylene conical-bottomed vessel, and as a matter of routine, this sedimentation has been accelerated by centrifuging the tubes or plates containing the cell suspension. In an effort to further simplify this culture technique, we investigated whether the centrifugation step was, in fact, necessary.
In this side-by-side comparison of centrifuged and freely sedimented hMSCs in the 96-well plate format, aggregates formed within 24 h in both the centrifuged and uncentrifuged plates. Overall, there was a trend for centrifuged aggregates to be slightly (approximately 5%) larger in diameter than donor- and time point-matched uncentrifuged aggregates, but this trend failed to reach statistical significance (P = 0.107, N = 186 sections). We did not observe a difference in the histological aspect or timing of the appearance of toluidine blue metachromatic staining at around day 5 (Figure 1), or in the expression and distribution patterns of the collagen types (Figure 2). In both centrifuged and uncentrifuged aggregates, type II collagen expression was undetectable by immunohistochemistry in day 5 samples, but was ubiquitous by day 7 (Figure 2). Similarly, type × collagen was undetectable at day 6, but simultaneously appeared by day 8, and was widespread by day 10 in both types of aggregates (Figure 2). Aggregates made with FGF-2-treated cells, such as these, express barely detectable type I collagen in the first few days, but it is undetectable after a week (Figure 2). Also, in donor- and time point-matched comparisons, DNA content was identical (1.008:1, P = 0.29, N = 27), suggesting that the aggregate formation itself occurred with equivalent efficiency. While there was a trend toward a slight (4%–10% depending on the cell preparation) difference in GAG content, this was mostly due to a lag in GAG production in the first 3–5 days in the uncentrifuged samples, and it failed to reach statistical significance (P = 0.06, N = 27). GAG normalized to DNA content was slightly but statistically significantly (P = 0.035) higher in centrifuged aggregates, again due to the slight lag in GAG production in the first week (Figure 3). We speculate that this lag is due to the additional time required for the aggregates to form in the free-sedimenting format. Cell sedimentation and initial contraction to an aggregate takes 8–24 h after the cell suspension is placed into the multi-well plate, whereas the centrifuged cells are forced into the aggregate configuration immediately. This offset likely accounts for much of the early lag in GAG production. Overall, our results show that the aggregates produced by both methods undergo chondrogenic differentiation in the same temporal sequence, yielding substantially equivalent results under otherwise identical culture conditions. Ten to fifteen minutes of preparation time were saved per batch of four plates. Eliminating this step has several advantages: first, it reduces human intervention, resulting in cost, labor, and time savings, and also reducing or eliminating the chance for operator error associated with these interventions (e.g., contamination or mislabeling). Depending on the size of the experiment and the capacity of the centrifuge, the time savings can add up to an hour or more with the time involved in loading the centrifuge, the actual centrifugation time, and waiting for the rotor to stop and unloading the centrifuge.
Figure 2. Immunohistochemically stained sections of centrifuged (indicated as C) and uncentrifuged (indicated as U) aggregates.
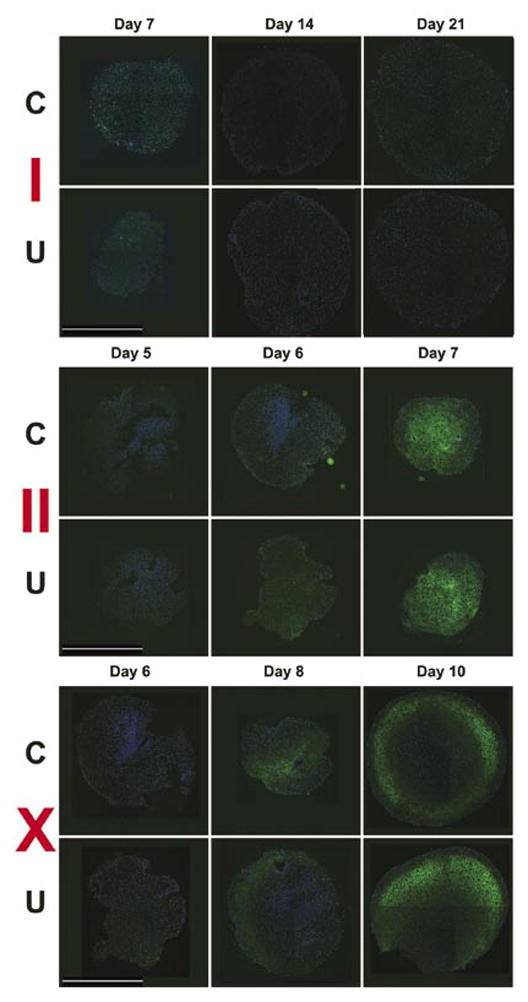
Immunoreactivity for collagen types I, II, and X was detected using a fluorescein isothiocyanate (FITC)-labeled secondary (green) antibody; 4′,6-diamidino-2-phenyl-indole (DAPI) counter-stain of the nuclei (blue). Scale bar, 1 mm.
Figure 3. Microgram glycosaminoglycan (GAG) per microgram of DNA measured in centrifuged (black bars) and uncentrifuged (gray bars) aggregates harvested at the indicated days.
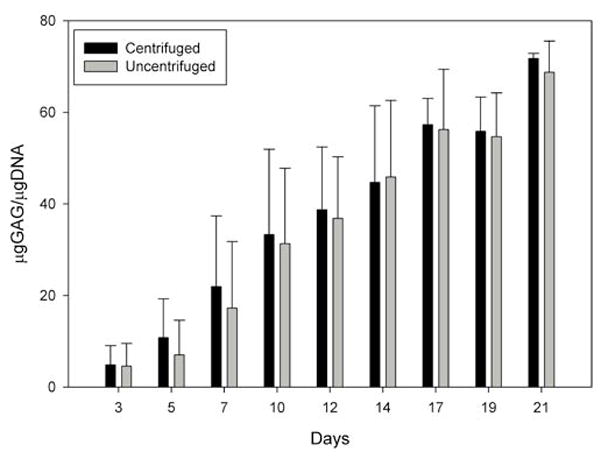
Mean ± sd, average from three preps with six internal replicates each.
Second, additional equipment may be required if using the microplate technique, as not all centrifuges are equipped with microplate-capable rotors and adapters.
Third, exposure to elevated g forces is likely not an inert process; it has the potential to stimulate or inhibit cellular events in ways which may interfere with the processes being studied.
Lastly, eliminating the centrifugation step would greatly facilitate full automation of the aggregate-culture system for a true robotic high-throughput method. Such a fully automated system would be useful in drug screening and testing for joint disease therapy, development of culture media, or toxicology tests.
The bulk of the equipment and methods for such automation in the 96-well microplate format already exist with different applications [e.g., in automated enzyme-linked immunosorbent assay (ELISA) systems or hybridoma screening systems]. The components of such a system would include robotic handling of micro-well plate stacks, automatic fluid handling, a sterile and controlled environment (temperature, gas, etc.), and a provision for online quality control at critical points. As a research tool, full automation would further improve upon the versatility and throughput capability of the aggregate culture technique, allowing the screening of extensive grids of experimental variables. In a clinical setting for providers of cell-based or tissue-engineered products and services, end-to-end automation of the process would partly fulfill the goal of GLP/GMP-in-a-box, where all the manipulations and documentation thereof are done in a controlled environment, within a closed system, requiring minimal or no human intervention, without the possibility of cross-contamination between specimens from different patients.
Acknowledgments
This work was funded in part by grants from the Arthritis Foundation (to J.F.W. and L.A.S.) and the National Institutes of Health (5R01-AR050208 to J.F.W.). The authors wish to thank Dr. Arnold Caplan and the Skeletal Research Center Mesenchymal Stem Cell Facility (supported in part by the Center for Stem Cell and Regenerative Medicine with funding from the Ohio Third Frontier Program) for supplying the hMSCs used in this study, and Ms. Margie Harris for her dedication in the preparation of the primary cell cultures. We also acknowledge Ms. Lisa Walsh for preparing the histology sections. Anti-collagen type X antibody was a gift from Dr. Gary J. Gibson, Breech Research Laboratory, Bone and Joint Center, Henry Ford Hospital and Medical Centers, and the human FGF-2 was provided by the Biological Resources Branch of the National Cancer Institute.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
To purchase reprints of this article, contact: Reprints@BioTechniques.com
References
- 1.Holtzer H, Abbott J, Lash J, Holtzer S. The loss of phenotypic traits by differentiated cells in vitro, I. dedifferentiation of cartilage cells. Proc Natl Acad Sci USA. 1960;46:1533–1542. doi: 10.1073/pnas.46.12.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballock RT, Heydemann A, Wakefield LM, Flanders KC, Roberts AB, Sporn MB. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytes: regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- 3.Ballock RT, Reddi AH. Thyroxine is the serum factor that regulates morphogenesis of columnar cartilage from isolated chondrocytes in chemically defined medium. J Cell Biol. 1994;126:1311–1318. doi: 10.1083/jcb.126.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato Y, Iwamoto M, Koike T, Suzuki F, Takano Y. Terminal differentiation and calcification in rabbit chondrocyte cultures grown in centrifuge tubes: regulation by transforming growth factor beta and serum factors. Proc Natl Acad Sci USA. 1988;85:9552–9556. doi: 10.1073/pnas.85.24.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp Cell Res. 1998;238:265–272. doi: 10.1006/excr.1997.3858. [DOI] [PubMed] [Google Scholar]
- 6.Yoo JU, Barthel TS, Nishimura K, Solchaga L, Caplan AI, Goldberg VM, Johnstone B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J Bone Joint Surg Am. 1998;80:1745–1757. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Hanada K, Solchaga LA, Caplan AI, Hering TM, Goldberg VM, Yoo JU, Johnstone B. BMP-2 induction and TGF-beta 1 modulation of rat periosteal cell chondrogenesis. J Cell Biochem. 2001;81:284–294. doi: 10.1002/1097-4644(20010501)81:2<284::aid-jcb1043>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 8.Solchaga LA, V, Goldberg M, Mansour JM, Caplan AI, Welter JF. Cartilage tissue engineering: in vitro generation of large cartilaginous constructs. Trans Orthop Res Soc. 2003;28:944. [Google Scholar]
- 9.Penick KJ, Solchaga LA, Welter JF. High-throughput aggregate culture system to assess the chondrogenic potential of mesenchymal stem cells. Biotechniques. 2005;39:687–691. doi: 10.2144/000112009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solchaga L, Penick K, Goldberg V, Caplan A, Welter J. Regulation of lineage progression of human mesenchymal stem cell-derived chondrocytes. Proceedings of the 6th Symposium of the International Cartilage Repair Society; San Diego, CA. 2006. pp. 4–51. [Google Scholar]
- 11.Lennon DP, Haynesworth SE, Bruder SP, Jaiswal N, Caplan AI. Human and animal mesenchymal progenitor cells from bone marrow: Identification of serum for optimal selection and proliferation. In Vitro Cell Dev Biol. 1996;32:602–611. [Google Scholar]
- 12.Haynesworth SE, Goshima J, Goldberg VM, Caplan AI. Characterization of cells with osteogenic potential from human marrow. Bone. 1992;13:81–88. doi: 10.1016/8756-3282(92)90364-3. [DOI] [PubMed] [Google Scholar]
- 13.Solchaga L, V, Goldberg M, Mishra R, Caplan A, Welter J. FGF-2 modifies the gene expression profile of bone marrow-derived human mesenchymal stem cells. Trans Orthop Res Soc. 2004;29:777. [Google Scholar]
- 14.Solchaga LA, Welter JF, Lennon DP, Caplan AI. Generation of pluripotent stem cells and their differentiation to the chondrocytic phenotype. Methods Mol Med. 2004;100:53–68. doi: 10.1385/1-59259-810-2:053. [DOI] [PubMed] [Google Scholar]
- 15.Solchaga L, Penick K, Welter J. A “manual” mosaicking approach to generating large, high-resolution digital images of histological sections. Proc Roy Microsc Soc. 2004;39:313–320. [Google Scholar]
- 16.Ponticiello MS, Schinagl RM, Kadiyala S, Barry FP. Gelatin-based resorbable sponge as a carrier matrix for human mesenchymal stem cells in cartilage regeneration therapy. J Biomed Mater Res. 2000;52:246–255. doi: 10.1002/1097-4636(200011)52:2<246::aid-jbm2>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 17.Carrino DA, Arias JL, Caplan AI. A spectrophotometric modification of a sensitive densitometric Safranin O assay for glycosaminoglycans. Biochem Int. 1991;24:485–495. [PubMed] [Google Scholar]