

Abstract
Resistance (R) protein recognizes molecular signature of pathogen infection and activates downstream hypersensitive response signalling in plants. R protein works as a molecular switch for pathogen defence signalling and represent one of the largest plant gene family. Hence, understanding molecular structure and function of R proteins has been of paramount importance for plant biologists. The present study is aimed at predicting structure of R proteins signalling domains (CC-NBS) by creating a homology model, refining and optimising the model by molecular dynamics simulation and comparing ADP and ATP binding. Based on sequence similarity with proteins of known structures, CC-NBS domains were initially modelled using CED- 4 (cell death abnormality protein) and APAF-1 (apoptotic protease activating factor) as multiple templates. The final CC-NBS structural model was built and optimized by molecular dynamic simulation for 5 nanoseconds (ns). Docking of ADP and ATP at active site shows that both ligand bind specifically with same residues and with minor difference (1 Kcal/mol) in binding energy. Sharing of binding site by ADP and ATP and low difference in their binding site makes CC-NBS suitable for working as molecular switch. Furthermore, structural superimposition elucidate that CC-NBS and CARD (caspase recruitment domains) domain of CED-4 have low RMSD value of 0.9 A° Availability of 3D structural model for both CC and NBS domains will . help in getting deeper insight in these pathogen defence genes.
Background
Plants are constantly attacked throughout their life cycle by a range of phytopathogens that includes viruses, mycoplasma, bacteria, fungi, nematodes, protozoa and other parasites. In the first line of defence, presence of an invading pathogen is detected by host immune system through pathogen–associated molecular patterns (PAMPs), such as bacterial flagellin, lipopolysaccharides and fungal-oomycete cellulosebinding elicitor proteins [1]. However, some pathogens have developed a way to evade PAMPs mediated pathogen defence mechanisms. For instance, Agrobaterium tumefaciens circumvents recognition by Arabidopsis flagellin receptor by domain modification and Pseudomonas syringae effectors proteins by suppression of basal immune system in Arabidopsis [2, 3]. In the second line of defence ‘gene for gene resistance’ gets activated which is governed by resistance (R) genes that recognize pathogen race specific effecter (Avr) proteins [4]. The R gene family is one of the largest known plants gene families that mediate elicitor recognition and activate downstream signalling response leading to disease resistance by localized cell death (hypersensitive response) [5]. R proteins monitor the integrity of PAMPs mediated basal immune system and any perturbation of these components by effector proteins could trigger activation of R proteins. Pathogen recognition elicits nucleotide-dependent conformational changes that might induce oligomerisation, thereby providing a scaffold for activation of downstream signalling components [6].
Most of the R proteins contain C-terminal leucine-rich repeats (LRRs), a central nucleotide-binding site (NBS) and a variable amino-terminal domain. The LRRs are mainly involved in recognition, where as the amino-terminal domain determines signalling specificity. The NBS domain that presumably functions as a molecular switch consists of two subdomains: nucleotide binding (NB) and ARC (common in APAF-1 in human, R protein in plants and CED-4 in Caenorhabditis elegans). NBARC is a member of the P-loop NTPase domain superfamily that is characterized by conserved nucleotide phosphate-binding motif, also referred to as the Walker A motif, and the Walker B motif. The Walker A and B motifs bind the beta-gamma phosphate moiety of the bound nucleotide (typically ATP or GTP) and the Mg2+ cation, respectively
Molecular structure of plant R protein homologoues in human APAF-1 and C. elegans CED-4 have been resolved by X-ray crystallography [7, 8]. In plants 3D structure of NBS domain with conserved P loop NTPase motif has been modelled for tomato, Arabidopsis and flex [9], however such information for coil-coil (CC) domain is lacking. Moreover the previously predicted 3D models are static in nature that do not reflect true and most stable confirmation as observed in dynamic conditions. In the present work, sequence for CC-NBS domain of R protein was cloned from brinjal (Solanum melongena) and a structural model was developed by homology modeling that was further optimized and refined by molecular dynamic simulations for 5 ns. The refined structural model was used for docking with ADP and ATP. Docking studies confirmed specific but differential affinity of ADP and ATP binding at active site.
Methodology
Target protein
The brinjal (Solanum melongena L.) R protein sequence, for which only one sequence coding for CC-NBS domain is available in database was retrieved from NCBI GenBank (accession number EU573702) and used for modeling.
Template identification
For identification of a suitable template and homology modeling different web servers, 3D-Jury [10], ESyPred3D [11], I-TASSER [12], MODEB (http://salilab.org/modweb), PHYRE [13], (PS)2 [14] and SWISS-MODEL [15].
Homology modelling
Sequence alignment provided by I-TASSER was manually improved and used as an input for homology modeling by HOMMER (http://protein.cribi.unipd.it/homer/) and Swiss-Model [15]. The 3-D model was energy minimized by SPDBV [15] and quality evaluation was done on the basis of Ramachandran’s plot and energetics using VERIFY- 3D by PROCHECK [16]. Protein 3D structure were aligned and RMSD calculated by TM-Align [17].
Molecular Dynamics Simulations
The model was further refined and optimized by 5 ns molecular dynamics (MD) simulation. MD simulations were done using Amber 10 with the AMBERff99 forcefield and the SPC/E water model [18]. The predicted model was solvated in a truncated octahedron periodic boxThe system was then energy minimised using a steepest gradient method for 300 steps followed by conjugate gradient method. Initial velocities for each atom were assigned from a Boltzmann distribution at 298K followed by a constant volume MD simulation. A 5 ns MD simulation was run in periodic water box to establish the equilibrium behaviour of protein. All covalent bonds containing hydrogen were fixed at equilibrium lengths using the SHAKE algorithm. A 1 femotoseconds integration time step was used and configurations were collected every 1 picoseconds for subsequent analysis. A real space non-bonded cut off and particle-mesh Ewald summation method was used to compute long range electrostatic energy and force corrections. MD trajectories were analysed using ptraj module of AMBER visualized using VMD [19].
Prediction of protein function
Protein cavities were identified by SiteHound programme [20]. The residues involved in ligand interaction were identified by PDBsum [21]. Docking of ligand to the protein structure was done using GLIDE [22].
Docking of ADP and ATP
The geometry of ADP and ATP was optimized by molecular mechanics using IMPACT in a dynamic environment using standard TIP4P water model. The energy minimization was done using Optimized Potentials for Liquid Simulations 2005 force field; using Polak-Ribier conjugate gradient and Truncated Newton conjugate gradient algorithms. The convergence threshold used was rms gradient of 0.01.
Docking of the ATP and ADP ligand with CC-NBS was carried out using extra precision (XP) method called GLIDE (Grid-based Ligand Docking with Energetics). The ligands were prepared for docking using LIGPREP. The receptor grid generation for docking was done using the centroid of selected active site residues as well as blind docking. The different conformations of the compounds were docked flexibly and 1000 poses per compound were obtained. The analysis of the poses, complexes and the binding affinities between the receptor and ligands were analyzed using Schrodinger's suite.
Discussion
Template identification and sequence alignments
PDB-BLAST using found human APAF-1 as the closest match and aligned a segment of 112 amino acids (187-299) that share a sequence identity of 32%. Similarly all five homology modeling programs selected either chain A of human apoptotic protease-activating factor 1 (APAF-1) (PDB ID 1Z6T) or C. elegans CED-4 chain B (PDB ID 2A5Y, involved in programmed cell death) as a template [12, 13] (Table 1 see Table 1). However, none of these programmes were able to align S. melongena CC-NBS sequence with either APAF-1 or CED-4 templates completely. Most of these programs except I-TASSER and PHYRE could match only the conserved P-loop containing ‘nucleoside triphosphate hydrolase’ region, but not the entire CC-NBS sequence. This was quite possible because of very low sequence similarity between the two sequences.
Structural modelling
Initial CC-NBS structure was modelled with I-TASSER using APAF-1 and CED-4 as multiple templates. Ramachandran plot analysis of I-TASSSER model showed that only 80 percent residues were present in most favoured regions and 5 percent residues in disallowed regions (Table 2 see Table 2). Although I-TASSER failed to develop a satisfactory model but the predicted model guided sequence alignment of S. melongena CC-NBS with CED-4 chain B became an valuable input for further 3D structure modeling with HOMMER and SwissModel (Figure 1).
Figure 1.
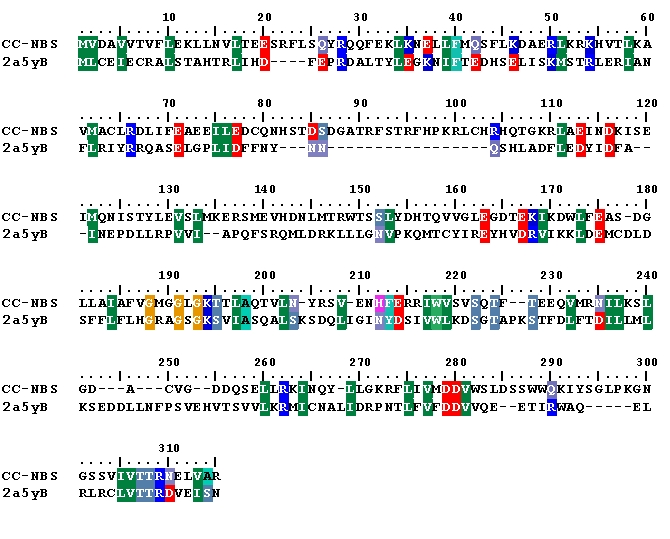
Alignment of S. melongena CC-NBS (query) with CED-4 (template), for homology modeling.
Quality of homology model
The final structure was subsequently checked by VERIFY-3D graph. The compatibility score above zero in the VERIFY-3D graph corresponded to acceptable side chain environments. All residues appeared to be reasonable and therefore the structure of S. melongena CC-NBS can be considered relibale. Validation of the model was further carried out using Ramachandran's plot calculations computed with PROCHECK program. The phi and psi distributions of the Ramachandran's plot of non-glycine, non-proline residues are summarized in Table 2 (see Table 2). Ramachandran's plot analysis further shows that 99.9% main chain bond length and 95.6 bond angles are within limit with overall PROCHECK G factor of -0.01.
Optimisation of the model by MD simulation
The MD trajectories obtained were analysed to study the behaviour of the structure. Figure 2A shows energy plot with respect to time for the complete 5 ns simulation. Comparison of the initial model obtained from homology modeling with the frames from MD trajectories shows that there are pronounced structural fluctuations with root mean square deviations of 2.5-3.0Å (Figure 2B). The frames were then taken from the production phase and analysed further. RMSD for backbone atoms was >1.2 Å revealing that structural fluctuations are not highly pronounced and consistent over frames obtained from production phase (Figure 2C). Overlapping the different conformations from the latter half of the trajectories shows that the molecule is relatively rigid, an indicative of structure stability. The average structure from the production phase frames was taken as the final model. The structural superimposition of Ca trace of CED-4 template and S. melongena CC-NBS shows RMSD of 0.9 A° with an identity score of 0.14 and TM score of 0.52 (Figure 3A).
Figure 2.
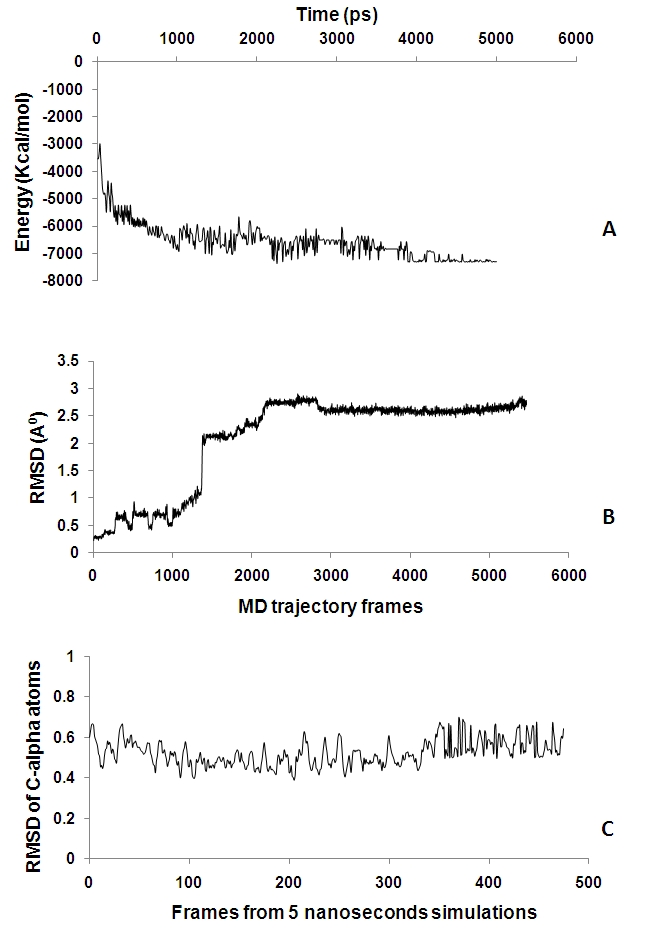
A. Plot of potential energy vs. time in ps for 5 ns simulation. B. Plot of root mean square deviation (RMSD) of Cα vs. frames from MD trajectories. The graph shows the RMSD of backbone atoms (Cα, C, N) during 5ns simulation. C. Plot of RMSD of Cα atoms vs. frames from MD trajectories. The graph represents the variations in Cα atoms for 5ns simulation for frames chosen from production phase.
Figure 3.
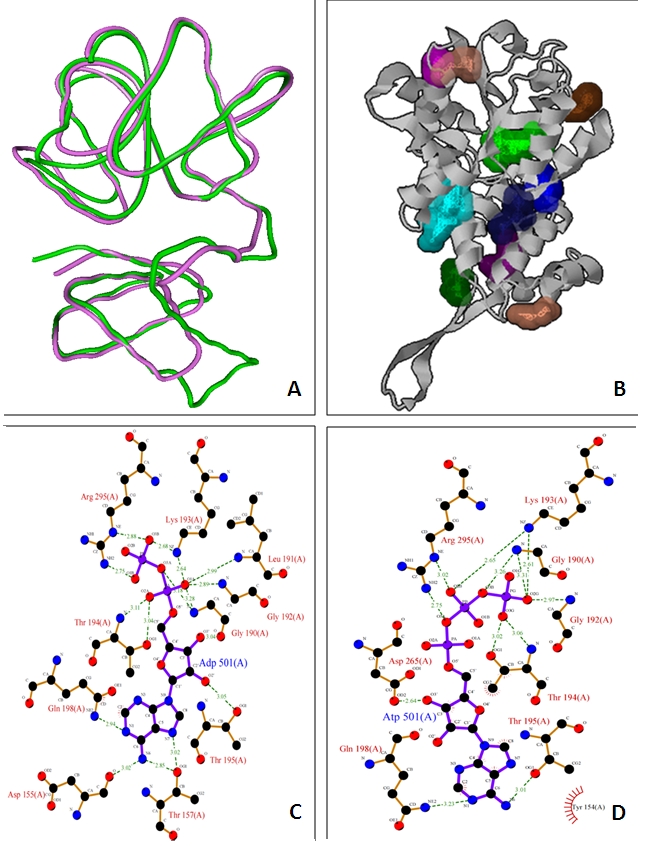
A. Superimposition of main chain alpha trace atoms of S. melongena CC-NBS (green) and C. elegans apoptosis regulator (magenta); B. Identification of cavities on CC-NBS surface; C-D. Interactions of conserved residues present in active site of S. melongena CC-NBS with ADP (C) and ATP (D) using Ligplot.
Active site prediction
The SiteHound program revealed 10 possible ligand binding sites (Figure 3B) and residues in the site1, Val160, Gly 190, Gly192, Lys193, Thr194, Thr195, Arg295 were found conserved with the active site of CED-4 . Thus, site1 was chosen as the most favourable binding site to dock ADP and ATP and the remaining 9 sites were excluded from the study.
Predicted model functionality
In the docked CC-NBS ligand ADP and ATP are located in the centre of the active site and stabilized by hydrogen bond interactions Figure 3 C-D. In the docked complex H-bond/electrostatic interaction was observed between ligand and residues Asp155, Gly190, Lys193, Thr194 and Arg295. Interestingly, these residues are in the active site of the protein as predicted by the active site detection program. In the docked complex ADP has slightly lower (-83 kcal/mol) energy than ATP (-82 kcal/mol). It indicates that ADP has slightly competitive edge over ATP in binding to the active site.
Conclusion
The CC-NBS domains have specific binding site for ADP and ATP, with minor difference in their binding affinity that makes it a suitable molecular switch. Presence of slightly lower binding energy of ADP (-84 kcal/mol) in comparison to ATP (-83 kcal/mol) may have a special significance in the context of their binding functioning as a molecular switch. In the ground state there will be more ADP binding because of its lower binding energy. However, in the activated state ADP has to be swapped with ATP, for downstream signalling [5, 6]. Had there been large difference between ADP and ATP binding energies it would have become hard to exchange the two molecules. These studies confirm that CC-NBS domains are perfectly suited for working as molecular switch. Availability of 3D structure of CC-NBS domains will further help us in understanding evolution, structure and function of these proteins and help in designing novel proteins for pathogen resistance management.
Supplementary material
Acknowledgments
SS wishes to acknowledge Supercomputing Facility for Bioinformatics and Computational Biology (SCFBio), IIT Delhi for providing access to their facility to carry out molecular dynamics simulations.
Footnotes
Citation:Shrivastava et al, Bioinformation 5(8): 326-330 (2011
References
- 1.Zipfel C, et al. Curr Opin Plant Biol. 2009;14:14. doi: 10.1016/j.pbi.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Gomez L, et al. Plant J. 1999;18:277. doi: 10.1046/j.1365-313x.1999.00451.x. [DOI] [PubMed] [Google Scholar]
- 3.Nurnberger T, et al. Immunol Rev. 2004;198:249. doi: 10.1111/j.0105-2896.2004.0119.x. [DOI] [PubMed] [Google Scholar]
- 4.Van der Biezen EA, Jones JD. Trends Biochem Sci. 1998;23:454. doi: 10.1016/s0968-0004(98)01311-5. [DOI] [PubMed] [Google Scholar]
- 5.Moffett P, et al. Embo J. 2002;21:4511. doi: 10.1093/emboj/cdf453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takken FL, et al. Curr Opin Plant Biol. 2006;9:383. doi: 10.1016/j.pbi.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Yan N, et al. Nature. 2005;437:831. doi: 10.1038/nature04002. [DOI] [PubMed] [Google Scholar]
- 8.Riedl SJ, et al. Nature. 2005;434:926. doi: 10.1038/nature03465. [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyaya R, Pal A. J Biomol Struct Dyn. 2008;25:357. doi: 10.1080/07391102.2008.10507184. [DOI] [PubMed] [Google Scholar]
- 10.Ginalski K, et al. Bioinformatics. 2003;19:1015. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 11.Lambert C, et al. Bioinformatics. 2002;18:1250. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, et al. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley LA, Sternberg MJ. Nat Protoc. 2009;4:363. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 14.Chen CC, et al. Nucleic Acids Res. 2006;34:W152. doi: 10.1093/nar/gkl187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski RA, et al. Journal of Applied Crystallography. 1993;26:283. [Google Scholar]
- 17.Zhang Y, Skolnick J. Nucleic Acids Res. 2005;33:2302. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Case DA, et al. J Comput Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphrey W, et al. J Mol Graph. 1994;14:33-8, 27. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez M, et al. Nucleic Acids Res. 2009;37:W413. doi: 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laskowski RA, et al. Nucleic Acids Res. 2009;37:D355. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friesner RA, et al. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.