

Abstract
Nasopharyngeal carcinoma (NPC) is a head and neck cancer rare throughout most of the world but common in certain geographic areas, such as southern Asia. While environmental factors and genetic susceptibility play important roles in NPC pathogenesis, the Epstein–Barr virus in particular has been implicated in the molecular abnormalities leading to NPC. There is upregulation of cellular proliferation pathways such as the Akt pathway, mitogen-activated protein kinases, and the Wnt pathway. Cell adhesion is compromised due to abnormal E-cadherin and β-catenin function. Aberrations in cell cycle are due to dysregulation of factors such as p16, cyclin D1, and cyclin E. Anti-apoptotic mechanisms are also upregulated. There are multiple abnormalities unique to NPC that are potential targets for novel treatments.
Keywords: nasopharyngeal carcinoma (NPC), Epstein, Barr virus (EBV), LMP1, tumorigenesis, review
Nasopharyngeal carcinoma (NPC) is a squamous cell carcinoma that usually develops around the ostium of the Eustachian tube in the lateral wall of the nasopharynx.1 Though rare among whites, NPC is particularly common in the southern Chinese population of Guangdong, Inuits of Alaska, and native Greenlanders.2,3 Chinese emigrants continue to have a high incidence of the disease, but the rate of NPC among ethnic Chinese born in North America is considerably lower than those born in China.4 This epidemiologic evidence implies that both environmental factors and genetic susceptibility play roles in the development of NPC. The environmental factors may include exposure to nitrosamines in salted and pickled foods.5 Certain human leukocyte antigen subtypes have been associated with NPC, as they have various genetic polymorphisms.6
The World Health Organization classifies NPC based on histology.7 Type 1, keratinizing squamous carcinoma, is characterized by well-differentiated cells that produce keratin. Type 2, nonkeratinizing squamous carcinoma, varies in cell differentiation but does not produce keratin. Type 3 is also nonkeratinizing, but is less differentiated, with highly variable cell types (clear cell, spindle cell, anaplastic). Types 2 and 3 NPC are Epstein–Barr virus (EBV) associated and have better prognoses than type 1; EBV infection is generally absent in type 1, especially in nonendemic areas.8 However, more recent data state that almost all NPC tumors, regardless of histologic subtype, have comorbid EBV infections, which is strong evidence for EBV as the etiology of NPC.9 This close association with EBV is what makes NPC unique from other head and neck cancers.
Standard treatment for NPC is radiotherapy, but concurrent adjuvant chemotherapy improves survival rates.10 As with other cancers, the prognosis of NPC depends upon tumor size, lymph node involvement, and distant metastasis (TMN staging).11 But NPC, in contrast to other head and neck malignancies, is highly sensitive to radiation and chemotherapy.12,13 High survival rates are reported for stage 1 and 2 diseases, but the prognosis for metastatic disease remains poor even with combined radiation and chemotherapy treatment, with disease relapse rates as high as 82%.14,15 Unfortunately, the majority of NPC is diagnosed at an advanced stage because of non-specific presenting symptoms (cervical nodal enlargement, headache, nasal and aural dysfunction), delay in seeking treatment after the onset of symptoms, and the difficulty of a thorough nasopharyngeal exam.16 In light of this, more targeted treatments of NPC need to be developed. To do so, the molecular changes that lead to NPC tumorigenesis must first be clarified. This review provides an overview of the major molecular mechanisms underlying the development of NPC. Figure 1 gives a summary of the pathways and their downstream effects on the tumorigenesis of NPC. Tables 1 and 2 provide a list of proteins aberrantly expressed and the percentage of NPC tumors that have these abnormalities.
FIGURE 1.
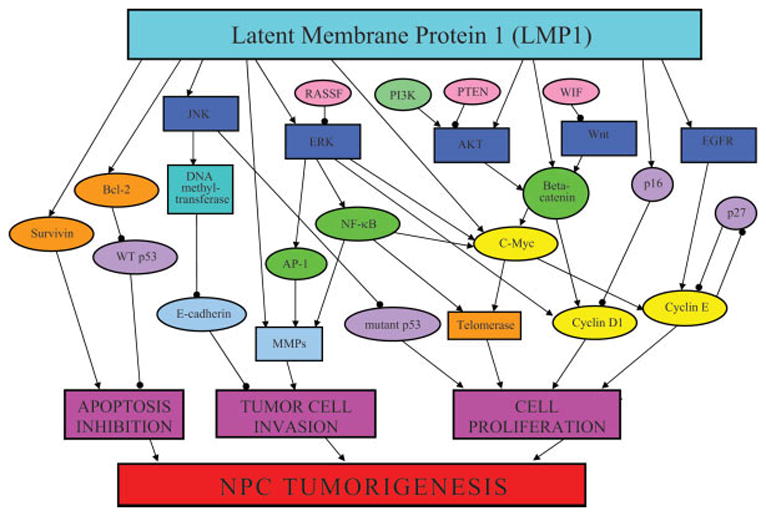
Overview of the molecular mechanisms involved in nasopharyngeal carcinoma (NPC) development. NPC development begins with the upregulation of pathways that promote cellular proliferation: Akt pathway, mitogen-activated protein kinases (JNK, ERK), Wnt pathway, and EGFR signaling. Subsequent increases of transcription factors such as NF-κB and β-catenin lead to cellular proliferation via cell cycle dysregulation (high c-myc, cyclin D1, and cyclin E expression) and inhibition of tumor suppressors (p16, p27, and wild-type p53). In addition, cell adhesion abilities are compromised because of low E-cadherin levels and high expression of MMPs. Also, antiapoptotic mechanisms such as bcl-2, survivin, and telomerase are upregulated. , Stimulatory effect;
, Stimulatory effect;  , inhibitory effect; orange color indicates apoptosis regulators; light blue color indicates cell adhesion proteins; yellow color indicates cell cycle regulators; dark blue color indicates proliferative pathways; green color indicates transcription factors; purple color indicates tumor suppressors; EGFR, epidermal growth factor receptor; ERK, extracellular signal related kinase; JNK, c-Jun N-terminal kinase; LMP1, latent membrane protein 1; MMP, matrix metalloproteinase; NPC, nasopharyngeal carcinoma; PTEN, phosphatase and tensin homolog; PI3K, phosphoinositol-3-kinase; RASSF, Ras association domain family; WIF, Wnt inhibitory factor; WT p53, wild-type p53. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
, inhibitory effect; orange color indicates apoptosis regulators; light blue color indicates cell adhesion proteins; yellow color indicates cell cycle regulators; dark blue color indicates proliferative pathways; green color indicates transcription factors; purple color indicates tumor suppressors; EGFR, epidermal growth factor receptor; ERK, extracellular signal related kinase; JNK, c-Jun N-terminal kinase; LMP1, latent membrane protein 1; MMP, matrix metalloproteinase; NPC, nasopharyngeal carcinoma; PTEN, phosphatase and tensin homolog; PI3K, phosphoinositol-3-kinase; RASSF, Ras association domain family; WIF, Wnt inhibitory factor; WT p53, wild-type p53. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Table 1.
Proteins overexpressed in NPC.
| Abnormal protein | % NPC with overexpression | Reference no. |
|---|---|---|
| NF-κB | 100 | 17,18 |
| Survivin | 100 | 18 |
| Intranuclear β-catenin | 92 | 19 |
| Wnt protein | 93 | 20 |
| hTERT | 91 | 21 |
| Telomerase | 85 | 21,22 |
| Bcl-2 | 75–86 | 18,23,24 |
| C-myc | 90 | 25 |
| Cyclin D1 | 66 | 26 |
| ERK | 53 | 27 |
| EGFR | 49 | 23 |
| Wild-type p53 | 95 | 24 |
Abbreviations: NF-κB, nuclear factor kappa B; hTERT, human telomerase reverse transcriptase; ERK, extracellular signal-regulated kinase; EGFR, epidermal growth factor receptor.
Table 2.
Proteins underexpressed in NPC.
| Abnormal protein | % NPC with underexpression | Reference no. |
|---|---|---|
| WIF | 75 | 18 |
| RASSF2A | 80 | 28 |
| p27 | 68 | 29 |
| p16 | 65–70 | 26,30 |
| CHFR | 61 | 31 |
| C-myc | 60 | 29 |
| PTEN | 48 | 32 |
| E-cadherin | 42 | 33 |
Abbreviations: WIF, Wnt inhibitory factor; RASSF2A, Ras association domain family 2A; CHFR, checkpoint with forkhead-associated and RING finger domains; PTEN, phosphatase and tensin homolog.
EPSTEIN–BARR VIRUS
EBV is a γ-herpes virus34 present in over 90% of adults worldwide.35 Though the infection is lifelong, it usually remains harmless unless the balance between host and virus is altered. Diseases associated with EBV include those of lymphocytic origin (infectious mononucleosis, Hodgkin’s disease, and Burkitt’s lymphoma) and epithelial origin (NPC, oral hairy leukoplakia, and undifferentiated gastric carcinoma).36 At least 95% of NPC tumors are EBV associated. Additionally, the severity of EBV infection varies with carcinoma type, with undifferentiated carcinomas having the highest EBV titers.37
EBV has tumorigenic potential due to a unique set of latent genes: latent membrane proteins (LMP1, LMP2A, and LMP2B) and EBV-determined nuclear antigens (EBNA1 and EBNA2) are the proteins predominantly expressed in NPC.38 LMP1 is the principal oncogene of NPC—it is required for cell immortalization and is present in 80%to 90%of NPC tumors.39 The LMP1 molecule includes 6 transmembrane domains and a car-boxy-terminus containing 2 signaling domains called C-terminal activating regions 1 and 2 (CTAR 1 and 2). The transmembrane domains allow LMP1 to associate with the host membrane, whereas the CTAR regions directly activate a number of signaling pathways including nuclear factor κ-B (NF-κB), mitogen-activated protein (MAP) kinases, and phosphoinositol-3-kinase (PI3K).40 Although the basic role of LMP1 is to prevent apoptosis, it has other important functions in cancer development. LMP1-positive cells have greater mobility, leading to higher metastatic potential41 and faster disease progression.42 LMP1 is also involved in suppressing immunogenic responses against NPC; for example, LMP1 has intrinsic T-cell inhibitory properties43 and mediates downregulation of CD99,44 an important component of the anti-NPC immune response. The importance of LMP1 in tumorigenesis is illustrated by numerous studies that show the inhibition of LMP1 results in increased tumor cell sensitivity to chemotherapy.45
Less is known about LMP2 and EBNA than LMP1, but with recent research their role in EBV-induced tumorigenesis is becoming more understood. Previously it was thought that LMP2 probably only mediated tumor cell survival46; however, newer data show that LMP2, particularly LMP2A, has more diverse functions. LMP2A downregulates the NF-κB transcription factor and can decrease LMP1 expression.47 Additionally, LMP2A expression causes NPC cells to become migratory and invasive.48 EBNA1 is an unusual protein that binds the EBV genome to host chromosomes, and thus mediates equal partitioning of viral DNA into daughter cells during cell division49 and may play a role in immune evasion.17 EBNA2 may be involved in the transactivation of LMP1.50 The roles of these EBV latent genes are summarized in Figure 2.
FIGURE 2.
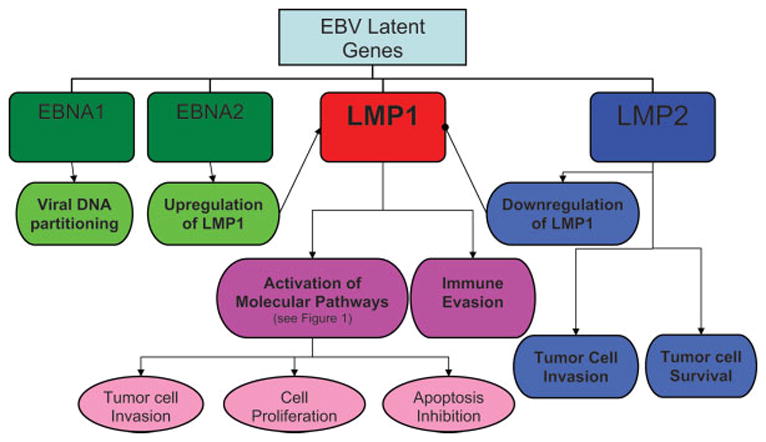
Mechanisms of Epstein-Barr virus (EBV) latent proteins in nasopharyngeal carcinoma (NPC) development. NPC tumorigenesis depends on the activity of latent proteins LMP1 and 2 and EBNA 1 and 2. The majority of cancer development is propagated by LMP1, which is responsible for the activation of various molecular pathways (see Figure 1) and immune evasion. LMP1 is regulated positively by LMP2 and negatively by EBNA 2. The other functions of LMP2 include mediation of tumor cell survival and invasion. Finally, EBNA 1 is critical for viral DNA partitioning during replication. , Stimulatory effect; , inhibitory effect; LMP, latent membrane protein; EBNA, EBV-determined nuclear antigen. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
CRITICAL PROLIFERATIVE SIGNALS
EBV infection appears to be necessary but not sufficient for tumorigenesis in NPC. Clearly, other mechanisms are required. Although there are many aberrations that contribute to tumorigenesis, the critical signals in NPC development are the Wnt pathway and transcription factors NF-κB and β-catenin.
Wnt Pathway Is Upregulated and Critical to the Intranuclear Accumulation of β-Catenin
The Wnt signaling pathway is important for normal development but is aberrantly activated in cancer. Wnt proteins bind to receptors belonging to the frizzled (Fz) family. In the canonical pathway, this activates an intracellular cascade that involves the inhibition of glycogen synthase kinase 3β (GSK-3β) and the adenomatous polyposis coli (APC) protein, ultimately resulting in the stabilization and nuclear translocation of cytoplasmic β-catenin. Nuclear β-catenin then interacts with various transcription factors to cause cellular proliferation and differentiation. Cytoplasmic β-catenin also has a role in the normal cell by binding to the intracellular domain of E-cadherin to maintain cellular adhesion.51
Abnormal Wnt signaling has been implicated in a number of cancers including head and neck carcinoma, lung cancer, colorectal cancer, melanoma, and leukemia.52 Prolonged Wnt signaling activates Akt, which together with disheveled phosphorylates GSK-3β.53 Phosphorylated GSK-3β is inactive, thus allowing β-catenin accumulation. Although the role of the Wnt pathway in NPC has not been fully explored, there is abundant evidence that aberrant Wnt signaling plays a role in NPC development. A gene expression study of NPC found the upregulation of the Fz 7 receptor and claudin 1 (a gene positively regulated by β-catenin) and the underexpression of axin 2, an inhibitory protein of the Wnt pathway.54 Most NPC tumors exhibit Wnt pathway protein dysregulation—93%have increased Wnt protein expression and 75% of tumors have decreased expression Wnt inhibitory factor (WIF), an endogenous Wnt antagonist.18,20 The expression of WIF has been shown to be silenced via promoter hypermethylation in NPC cell lines.55 These results indicate that aberrant Wnt signaling is a critical component of NPC.
Increased β-Catenin Is Critical to NPC Proliferation
β-Catenin is the key mediator of canonical signaling in the Wnt pathway. In normal cells, a protein complex of axin, APC, and GSK-3β phosphorylates β-catenin, marking it for subsequent ubiquitin-mediated degradation. Wnt pathway activation leads to deactivation of this protein complex and thus increased levels of β-catenin. Nuclear β-catenin levels are increased in 92% of NPC tumors, making nuclear β-catenin 1 of the more important components of NPC development.19,20 EBV infection increases the level of cytoplasmic β-catenin and causes its nuclear localization via increased GSK-3β.56 Mutations of the β-catenin gene are rare in NPC cells, indicating that GSK-3β is underactive because of down-regulation by Wnt or Akt.52,57,58 Interestingly, while EBV infection upregulates Akt in both Hodgkin’s lymphoma and NPC, only NPC cells showed increased levels of phosphorylated GSK-3β and intranuclear β-catenin.19 This suggests that the Wnt pathway has a more critical role in the dysregulation of β-catenin in NPC than in other EBV-associated diseases.
Intranuclear β-catenin contributes to NPC development in several ways. It activates numerous downstream proliferation signals, including c-myc and cyclin D1.52 Beta-catenin binds to the interleukin (IL)-8 promoter site and increasing IL-8 levels in NPC; IL-8 is an important angiogenic factor in NPC.59 Intranuclear β-catenin also downregulates RASSF1A expression.60 Normal RASSF1A is critical for microtubule stabilization and regulation of mitotic events; its downregulation leads to abnormal mitotic spindles and micro-tubule organization, ultimately resulting in aneuploidy and facilitation of transformation of cancerous NPC cells.60 Despite these developments, the full impact of aberrant β-catenin in the development of NPC remains to be completely understood. Given the importance of the Wnt pathway and β-catenin to NPC tumorigenesis, a greater understanding of these pathways is likely to contribute to the development of novel, targeted treatments for NPC.
High Levels of NF-κB Mediate Cell Immortalization
NF-κB normally plays 2 main roles in the cell: regulation of cell growth and modulation of inflammation. Constitutive NF-κB signaling is required for cell growth and proliferation in carcinogenesis of numerous neoplasms.61–63 Mechanisms responsible for NF-κB activation are diverse—for example, in head and neck squamous cell carcinoma (HNSCC), interleukin 1 directly induces NF-κB binding to DNA, whereas oncoproteins activate NF-κB in breast carcinoma.64,65 In NPC, LMP1 activates NF-κB through the binding of tumor necrosis factor receptor-associated factors (TRAFs).66 NF-κB dysregulation is 1 of the most important components of NPC tumorigenesis, illustrated by the fact that almost all NPC tumors exhibit NF-κB overexpression.17,18 NF-κB activation in turn leads to a myriad of different and sometimes conflicting cellular pathways. Upregulation of NF-κB results in the activation of a number of proliferative signals, including Bcl-2, cyclooxyngenase 2 (COX-2), and vascular endothelial growth factor; yet it also causes accumulation of p53 and induces G2/M phase arrest in NPC cells.67,68 Furthermore, activation of NF-κB by LMP1 leads to telomerase activation and cell immortalization.69 LMP1 induces binding of the NF-κB p65 subunit to human telomerase reverse transcriptase (hTERT) in NPC cells.69 This leads to nuclear translocation of both proteins and subsequent activation of telomerase. The ultimate effect of LMP1-induced NF-κB activation appears to be LMP1-mediated immortalization.
In contrast to LMP1, LMP2A decreases NF-κB levels.47 As NF-κB is also an important regulatory of the inflammatory response via expression of cytokines and chemokines,70 downregulation of NF-κB suppresses the immune response against NPC. So while LMP1 and 2A have opposite effects on NF-κB, these contradictory effects strike the balance between the antiapoptotic and anti-inflammatory levels of NF-κB that ultimately promotes tumor growth.47 However, the exact relationship of the contradictory roles of LMP1 and 2A on NF-κB remains to be fully elucidated.
APOPTOSIS DYSREGULATION
Aberrant apoptosis, as in all malignancies, is also required for NPC development. Inhibition of apoptosis seems to be critical to NPC tumorigenesis, second in importance only to cell proliferation. The upregulated antiapoptotic factors best identified in NPC are bcl-2, survivin, and telomerase.
Bcl-2 Overexpression Is Important for Cell Proliferation
Bcl-2 is an oncoprotein whose overactivation interferes with apoptosis.71 The best-known mechanism of bcl-2 upregulation is in follicular lymphomas, where t(14;18) translocation of the bcl-2 gene to a site adjacent to a heavy chain immunoglobulin gene results in bcl-2 overexpression.72 Bcl-2 is upregulated in NPC in the absence of the t(14;18) translocation.73,74 Overexpression is most likely EBV associated because EBV-positive NPC cells have greater bcl-2 expression than EBV-negative NPC cells.23 However, in contrast to other upregulated pathways in NPC, bcl-2 upregulation does not appear to be LMP1 dependent. Although LMP1 transfection of lymphocytes increases bcl-2 levels, this does not occur with transfection of epithelial cells such as those in NPC.24 Furthermore, inhibition of LMP1 does not affect bcl-2 expression in NPC.45 Although the mechanism of bcl-2 upregulation is unclear at this time, its overall role in promoting cell proliferation is well documented. In fact when compared with other head and neck cancers, bcl-2 is overexpressed in a higher percentage of NPC tumors and therefore appears to be more important in NPC development.23 In NPC, Bcl-2 acts synergistically with LMP1 to promote a more rapid cell growth than bcl-2 alone, LMP1 alone, or LMP1 with mutant p53.24 Previously it was thought that this allows tumor cells to overcome the apoptotic effects of increased wild-type p53.24 However, a more recent study shows that bcl-2 and wild-type p53 act synergistically to increase tumor cell growth via upregulation of proliferating cell nuclear antigen, a protein required for DNA synthesis and cell proliferation.75 These data suggest that bcl-2 is very important to NPC tumorigenesis and that further studies to elucidate the mechanism of bcl-2 upregulation as well as understanding its role in NPC development are warranted.
Survivin Overexpression Is Critical to NPC for Avoiding Apoptosis
Survivin is a key inhibitor of apoptosis and promoter of cell proliferation, notable for its absence in normal adult tissues but presence in embryonic cells and numerous tumors.76 Survivin inhibits apoptosis by associating with microtubules in mitotic spindles and inhibiting cell apoptosis mechanisms, though these have not been demonstrated in NPC cells specifically.77,78 In NPC, intranuclear survivin binds cyclin-dependent kinase 4 (cdk4) and displaces inhibitory proteins p21 and p16, thus allowing cdk4 to initiate transcription of S phase proteins.79 LMP1 induces survivin expression and nuclear translocation, though the mechanism is unknown at this time.80 The net effect is increased S-phase transitions and cell proliferation.80 Survivin over-expression is probably the most important antiapoptotic factor in NPC development given that a gene expression study found survivin overexpression in all study samples.18,81 Inhibition of survivin expression decreases NPC cell viability as well as increases NPC tumor radiosensitivity.18,82 Additionally, survivin levels have prognostic significance in NPC—patients who had tumors with low levels of survivin were less likely to have metastatic disease and had increased survival.83
High Telomerase Activity Contributes to NPC Cell Immortalization
Telomerases are enzymes responsible for the maintenance of eukaryotic chromosome telomeres and the continuous proliferation of cells, including neoplasms. Critical to telomerase activity is the hTERT. In most head and neck cancers, cell immortalization requires initial genomic instability, such as Robertsonian translocations or chromosomal losses or gains, and subsequent reactivation of telomerase via this genomic instability.84 Although the majority of primary NPC tumors display high telomerase activity as well, activation of telomerase is EBV dependent.22 LMP1 increases telomerase activity via NF-κB pathway and increased c-myc expression.69,85 NF-κB mediates transactivation of the hTERT gene and nuclear localization of the hTERT protein, whereas c-myc increases hTERT promoter activity.22 Telomerase is critical to the transformation of normal nasopharyngeal epithelia into malignancy. In NPC cells, 91% demonstrate hTERT expression and 85% have telomerase overactivity; conversely, cells from chronically inflamed nasopharyngeal epithelium show neither hTERT expression nor telomerase activity.21 Furthermore, normal nasopharyngeal epithelial cells can be transformed into immortalized cells by just activating telomerase.86 These findings show that telomerase is a necessary component in NPC development.
Curiously, LMP2A inhibits hTERT expression and subsequently decreases telomerase activity and cell proliferation in epithelial cells.87 These effects are paradoxical to the importance of EBV and telomerase to malignant transformation in NPC. Although it has been speculated that hTERT downregulation may interfere with normal B-cell function and therefore aid in the immune evasion of NPC, the role of telomerase inhibition in NPC development remains to be understood.87
OTHER ABNORMAL PROLIFERATION SIGNALING PATHWAYS
A variety of proliferative signals are aberrantly activated in NPC. Although Wnt pathway is perhaps the most important of these pathways that is dysregulated in NPC, aberrations in the PI3-K, MAP kinases, and EGFR signaling also contribute to tumorigenesis.
PI3K Is Activated via LMP1 and Inactivation of Phosphatase and Tensin Homolog
The PI3 kinases are a family of kinases that are involved in a wide variety of cellular pathways—PI3K activation may have an important role in the development of normal human keratinocytes.88 Akt, an important downstream target of PI3K, helps regulate cell proliferation and prevent apoptosis.89 Overactivation of PI3K has been implicated in various squamous cell carcinomas, in which the mechanism of PI3K activation is thought to be via the overexpression of a catalytic subunit of PI3K.90
While the overactivation of the PI3K pathway is critical to the development of NPC as well, its upregulation occurs by different mechanisms.19 LMP1 can directly activate PI3K, leading to Akt phosphorylation.66 As with NF-kB activation, the TRAF-binding domain of LMP1 is the active site. LMP2A can also directly activate Akt.57 Another potential mechanism of PI3K activation is through decreased levels of phosphatase and tensin homolog (PTEN),91 an inhibitor of PI3K.
PTEN is a protein tyrosine phosphatase92 that dephosphorylates phosphatidylinositol 3,4,5-tri-phosphate (PIP3),93 the lipid second messenger that activates Akt. It is a tumor suppressor gene with a rapidly growing importance in cancer development, especially in certain thyroid cancers that consistently demonstrated low PTEN levels.94,95 Downregulation of PTEN is found in about half of NPC tumors, making it partially responsible for the upregulation of the PI3-K/Akt pathway in NPC.91 Well-differentiated NPC shows a greater loss of PTEN than poorly differentiated cells.32 Mutation of the PTEN gene is extremely rare in HNSCC; therefore, other mechanisms are most likely responsible for the low PTEN levels observed in NPC.96 PTEN downregulation could be caused by epigenetic alterations to the PTEN genome (ie, promoter hypermethylation). PTEN hypermethylation has been demonstrated in laryngeal and thyroid cancer but not specifically in NPC97,98; however, given that a number of other genes are inactivated by hypermethylation in NPC, it is likely that the PTEN gene is epigenetically altered and warrants further investigation.99 Alternatively, lung cancer studies have found that nicotine stimulates PTEN degradation by phosphorylating the PTEN C-terminus.100 Although nicotine use does not have a strong association with NPC development, it is likely that an environmental factor with similar effects on PTEN exists for NPC.
Ultimately PTEN downregulation and Akt upregulation results in decreased activity of the cell division cycle 2/cyclin B (cdc2/cyclin B) complex, inducing G2/M phase arrest in NPC cells.67 It also results in the upregulation of c-fos expression101; c-fos is a proto-oncogene that dimerizes with Jun proteins to form the activator protein-1 (AP-1) transcription factor, which regulates expression of cell proliferation, migration, and survival.102 Interestingly, a study comparing malignancies with adjacent tissue found that though the adjacent tissue was histologically normal, those cells also expressed high Akt with low PTEN levels.91 Additionally, loss of PTEN is associated with metastatic disease—low PTEN is demonstrated in almost 80% of clinical stage III–IV tumors versus about 20% of clinical stage I–II tumors.32 This suggests that abnormalities in Akt and PTEN occur early in NPC tumorigenesis and contribute to disease advancement and metastasis.
MAP Kinases JNK and ERK Are Upregulated by LMP1
Located primarily in the nucleus, MAP kinases regulate gene expression by phosphorylating various transcription factors that are already bound to DNA.103 Their activity is regulated by a cascade involving MAPK kinase (MAPKK or MEK) and a MAPKK kinase (MAPKKK or MEKK).104 The MAP kinases most well studied in NPC are the c-Jun N-terminal kinase (JNK) and extracellular signal-related kinase (ERK).
Normal JNKs are activated by environmental stress and have important roles in determining cell survival or death.105 Prolonged JNK activation exerts a proapoptotic effect in normal cells via tumor necrosis factor (TNF)-α induced apoptosis, and transient JNK activation can also cause cell proliferation.105,106 Usually in tumors, JNK activity is depressed and thus has antiapoptotic effects on the cells.107 However, in NPC, JNK is consistently upregulated, and the activating effect of LMP1 on JNK is well documented.108,109 Though the reason for this remains to be fully explained, there are several possibilities. One is that the proapoptotic effect of prolonged JNK activation is overwhelmed by the numerous proliferative signals present in NPC. Another is that other influences provide the brief decreases in JNK activation to provide the transient JNK stimuli needed for cell proliferation. Wild-type p53 has been shown to decrease JNK activity in thyroid cells110; high levels of wild-type p53 are uniquely present in NPC and this may contribute to transient JNK activation.111 However, the most likely explanation for this pattern of JNK activation in NPC is that JNK may simply function differently in NPC and other head and neck cancers. Constitutive activation of JNK has been well demonstrated in NPC as well as oral squamous cell carcinoma (OSC) and thyroid cancers.112 In thyroid carcinoma, high basal JNK activation is associated with cell growth, not apoptosis.113 Furthermore, prolonged JNK activation in NPC has important roles in tumorigenesis, including increased p53 phosphorylation leading to its deactivation and activation of DNA methyltransferase, which is responsible for the hypermethylation and silencing of the E-cadherin gene.108,112 These data show that constitutive JNK is unique to head and neck cancers, including NPC, and it contributes to NPC tumorigenesis. However, the significance of JNK activation as well as its unique effects in NPC development remains unknown. Studies aimed at observing the effects of inhibiting JNK signaling on NPC apoptosis or cell growth would help elucidate this question and help determine if targeting JNK is a potential therapeutic strategy.
ERK is regulated by the Ras/Mek/ERK cascade. Normal ERK activation plays a role in cell growth and differentiation.114 ERK phosphorylates Ets transcription factors to induce fos gene expression,103 which ultimately leads to transactivation of NF-κB and AP-1.115 This pathway regulates cellular levels of cyclin D1 and c-myc, which are important cell cycle components.116 Therefore, it is no surprise that upregulation of this pathway has been demonstrated in a variety of malignancies, including hepatocellular carcinoma, gastric adenocarcinoma, and renal cell carcinoma.117–119 OSCs originating in southeast Asia have higher rates of Ras overexpression and mutations and stronger association with chewing tobacco.120 While NPC may have similar environmental influences, Ras mutations are rare in NPC and overactivation of the ERK is primarily LMP1 dependent.121 LMP1 can directly activate Ras, thus initiating the signaling cascade resulting in ERK activation.53 Additionally, LMP1 induces epigenetic alterations in Ras association domain family (RASSF) proteins, which are negative effectors of Ras, and can result in Ras overactivation.28,60 Because 80% of NPC cell lines demonstrated RASSF downregulation, it is likely that the latter of the 2 mechanisms is more critical to increased ERK activity.28 Ultimately the cell cycle is dysregulated and cell proliferation uncontrolled. Only about half of NPC exhibits upregulated ERK, suggesting that ERK is not critical to NPC.27 Nonetheless, ERK contributes to NPC development as tumors exhibiting high ERK levels have poorer prognoses, with shorter overall survival rates and faster disease progression.27
Epidermal Growth Factor Receptor Acts As An Intracellular Transcription Factor
The epidermal growth factor receptor (EGFR) is a 7-transmem-brane receptor whose overexpression has been demonstrated in a wide variety of tumors including lung, prostate, and breast.122–124 In these cancers, EGFR overexpression or mutation causes upregulation of its signaling cascade to ultimately cause uncontrolled cell proliferation.125 However, EGFR overexpression has been demonstrated in only half of NPC tumors.23,126 Additionally, suppression of EGFR signaling fails to fully inhibit NPC growth, suggesting that other proliferative signals such as Wnt may play a more important role for cell growth in NPC.127 Interestingly, EBV exerts a unique effect on EGFR: LMP1 causes the endocytosis and nuclear accumulation of EGFR.66 Intranuclear EGFR acts as a transcription factor to increase cell proliferation,66 and cytoplasmic EGFR binds to cyclin D1 and cyclin E proteins to accelerate G1/S transition.128 Therefore, it appears that EGFR has a more important role as a transcription factor than as a purely proliferative signal in NPC.
TUMOR SUPPRESSORS
As with all cancers, the development of NPC involves the loss of tumor suppressors. However, the mechanisms of inhibition of tumor suppressors may be somewhat unique in NPC.
High Levels of p53 Are Found in NPC
p53 is a hall-mark tumor suppressor that induces cell cycle arrest in response to DNA damage; its levels are traditionally decreased in tumors. In most head and neck cancers, low levels of p53 are due to mutations.129 However, p53 in NPC does not follow this classic pattern. NPC cells have increased levels of p53,111 with high LMP1 levels correlating with higher p53 expression.130,131 p53 mutations are relatively rare in NPC, so the vast majority of expressed p53 is wild type.24 The wild-type p53 fails to induce apoptosis in NPC because it is inactivated through 2 mechanisms—loss of p14 and excess ΔN-p63. p14 maintains p53 stability by inhibiting its proteolysis132; in NPC, p14 levels are low via promoter hypermethylation,99 thus allowing for more efficient p53 degradation. p63 is actually a homologue of p53 with similar DNA binding sequences to p53.133 There is a mutated version of p63 in NPC, called ΔN-p63, which lacks the N-terminal transactivation domain needed to activate apoptosis.134 The ΔN-p63 isoform binds the normal p63 (and p53) DNA sequences, thus preventing normal p63 or p53 from binding, but fails to induce apoptosis because of loss of the N-terminal sequence.134
The reason for high p53 levels in NPC is unclear. High levels of normal p53 may be advantageous to NPC development because tumor cells with normal p53 are immune to JNK-induced apoptosis.135 Alternatively the increased wild-type p53 may simply be the natural response to EBV infection.24 In light of this, it appears that the loss of p53 is not critical for NPC development, so deactivation of other tumor suppressors is likely required for NPC tumorigenesis.
p16 Activity Is Decreased
p16 is a cyclin-dependent kinase inhibitory protein (CKI) that suppresses activity and is frequently inactivated in cancer.136 The normal function of p16 is to suppress cdk4, an enzyme that controls the G1/S checkpoint by negatively regulating cyclin D1 activity.137 Therefore, loss of p16 results in cyclin D1 overactivation and subsequent increases in G1/S phase transitions.138 Traditionally, p16 transcription is mediated by the retinoblastoma protein (pRb); inactivation of pRb, as in retinoblastoma, leads to low levels of p16.139 However, in head and neck cancers, including NPC, this is not the case, as the majority of head and neck cancers exhibit low p16 levels with high pRb levels.140,141 In HNSCC, the most common mechanism of p16 inactivation is homozygous deletion of the gene followed by hypermethylation of the gene.142 Similarly, NPC cell lines have low levels of p16 secondary to hypermethylation of the p16,99 but this epigenetic alteration may be mediated by LMP1-induced formation of a c-Jun/JunB heterodimer causing the activation of DNA methyl-transferase.143 Additionally, LMP1 deactivates p16 by inducing cytoplasmic accumulation of E2F4/5 and Ets2, which are nuclear proteins required for normal p16 activity. Ets2 is a key transcription factor for p16 expression and LMP1 directly causes its translocation from the nucleus to the cytoplasm.144 E2F4/5 are proteins required for the activation of p16-induced cell cycle arrest in G1,144 and their nuclear localization is dependent on binding to nuclear pRb.145 LMP1 induces the translocation of E2F4/5 from the nucleus into the cytoplasm by causing E2F4/5 to dissociate from pRb proteins.144 LMP1 further promotes cytoplasmic accumulation of Ets2 and E2F4/5 by mediating their binding to a nuclear export protein called chromosome maintenance region 1 (CRM-1).144
About two thirds of NPCs exhibit low p16 levels, indicating that p16 downregulation is not critical to NPC development. However, the absence of p16 is still important to NPC. Patients with NPC tumors with low p16 levels have a worse prognosis because this is associated with decreased radiosensitivity and higher rates of tumor recurrence.26,30 The reason for this may be that the greatest radiosensitivity in cells is just prior to DNA synthesis and loss of p16 increases the number of cells in S phase.146 There is some data that pretreatment of NPC patients with p16 gene therapy before radiation can improve outcomes; the presumed mechanism behind this is that by normalizing p16 levels, the cell cycle is slowed at the G1/S checkpoint and the number of cells in G1 increased.147 However, further clinical trials are needed to evaluate the effectiveness of this treatment.
Activity of p27 Is Decreased
p27 is a CKI that binds to S-phase kinases (ie, cdk2) to inhibit cell cycle progression.148 Phosphorylated p27 cannot regulate cdk2 activity, thus allowing the cdk2/ cyclin E complex to remain activated and allows for progression of the cell cycle.149 Low p27 is well demonstrated in NPC as well as a number of tumors, including OSC, gastric cancer, and small cell lung cancer.150,151 In these latter cancers, low p27 levels are due to upregulation of ubiquitin–proteasome-mediated degradation mechanisms induced by high c-myc levels.150,152 However, in NPC cells, c-myc and p27 levels are not correlated, suggesting that other mechanisms are responsible for the low p27 activity in NPC.29 LMP1 causes this via upregulation of AKT-mediated phosphorylation of p27, thus targeting the protein for either degradation or cytoplasmic localization.45 Also, constitutively active ERK phosphorylates and deactivates p27.149 Because p27 negatively regulates cyclin E activity, reduced p27 activity results in more chromosomal instability and S-phase transitions. In addition, as elevated p27 activity is present in epithelial cells with cell–cell contact, lower levels of p27 may predispose to tumor cell metastasis.153 Low p27 is present in 68% of NPC, so it is unlikely that its downregulation is required for metastasis. However, its absence is still important to NPC progression, as reduced p27 activity has been linked to more aggressive NPC tumors as well as earlier disease reoccurrence after treatment.29 Therefore, p27 dysregulation plays a role in cell cycle dysregulation and chromosomal instability leading to higher-grade malignancy in NPC.
CELL CYCLE REGULATION
Like all cancers, development of NPC requires the derangement of the normal cell cycle. However, in NPC the abnormal expression of cell cycle proteins stems from LMP1-mediated upregulation of MAP kinases and downregulation of tumor suppressors.
High Cyclin D1 Levels May Give NPC Its Unique Radiosensitivity
Cyclin D1 is responsible for cell progression through G1 phase.154 Normal cyclin D1 activity is regulated by p16: cyclin D1 is active when bound to the cdk4/cdk6 complex, and displacement of cyclin D1 from this complex by p16 leads to cyclin D1 degradation and termination of G1.139 Overexpression of cyclin D1 enables cells with unrepaired structural or genomic damage to traverse the G1/S checkpoint, thus increasing the risk of tumor formation.155,156 Cyclin D1 is overexpressed in NPC.157 Underlying mechanisms include constitutively active Ras and Raf protein and, more importantly, low p16 levels.149,158 Additionally, LMP1-induced intranuclear accumulation of EGFR that can directly activate cyclin D1 transcription.128 The degree of cyclin D1 overexpression in NPC is comparable to that of other head and neck cancers—for example, 66%in NPC versus 64% in HNSCC.26,159 In HNSCC, high cyclin D1 levels are associated with increased local disease recurrence159; conversely, high cyclin D1 levels in NPC correlate with increased responsiveness to radiotherapy and fewer local tumor recurrences in NPC.26 This may be due to the fact that cyclin D1 affects the number of cells in G1/S phase transition and the radiosensitivity is highest just prior to DNA synthesis.146 Therefore, high cyclin D1 levels may be the molecular reason behind the unique sensitivity of NPC to radiation and chemotherapy. However, the exact effects of increased cyclin D1 levels in NPC is not known, and further investigation is warranted to determine whether upregulating cyclin D1 can enhance current therapeutic success.
Cyclin E Levels Are Increased
The cyclin E/cdk2 complex regulates cell entry into S phase and initiation of DNA synthesis.160 It also negatively regulates tumor suppressor p27 by phosphorylating and causing its degradation.161 Dysregulation of cyclin E activity results in rapid progression through S phase and increased chromosomal instability.162 Increased cyclin E activity has been demonstrated in a variety of head and neck tumors, including NPC and laryngeal and oral cancers.128,163,164 In most tumors, high cyclin E activity occurs due to impaired cyclin E degradation.154 But the increased cyclin E levels in NPC are secondary to LMP1-induced nuclear translocation of EGFR, which binds to the promoter of cyclin E and increases its expression.128 Cyclin E activity is further enhanced by the binding of cyto-solic EGFR.128 Additional increases in activity may be due to increased levels of activated E2F, which results from increased cdk4 levels.165 E2F binds to pRb to increase transcription of S phase proteins, including cyclin E. The net result of this is increased number of cells in S phase. While the mechanism of cyclin E upregulation is becoming clearer, the exact role cyclin E plays in NPC development remains largely unknown. The above data suggest that cyclin E plays an important role in the G1/S phase transition. Cyclin E, like cyclin D1, may have prognostic significance or may be a treatment target to enhance therapeutic success. Therefore, the mechanism of cyclin E in NPC tumorigenesis warrants further investigation. Gene studies could be done to elucidate what percentage of NPC tumors actually exhibit increased cyclin E levels. Additionally, it would be interesting to observe the effect inactivation of cyclin E has on NPC cell cycle progression.
C-myc Levels May Correlate with Disease Progression
C-myc is critical to the regulation of several important G1/S phase proteins; most notably, it sequesters inhibitory p27 from the cdk2/ cyclin E complex, thus allowing for cell proliferation and progression through the G1 phase.167 C-myc upregulation is a common occurrence in cancers and the mechanism of its dysregulation is diverse, secondary to chromosomal translocations and point mutations in lymphoid tumors and to mutations of its regulatory proteins in other cancers such as colon, breast, and melanoma.167 However, in NPC, there have been reports of both increased and decreased c-myc levels. Older studies show that when compared with normal nasopharyngeal epithelia, 90% of NPC showed increased c-myc expression, and this was correlated with decreased survival.25,168 It has been shown that LMP1 increases c-myc transcription via activation of the signal transducer and activator of transcription protein 3 (STAT3) and NF-κB transcription factors.17,169 Additionally, the Ras/ MEK/ERK pathway has also been shown to upregulate c-myc transcription in human fibroblasts149; although this association has not been specifically demonstrated in NPC cells, ERK over-activation is well established in NPC, making this a likely mechanism of c-myc overactivation. However, a more recent study showed that decreased c-myc levels are found in about 60% of NPC tumors and correlate with more aggressive NPC tumors with higher rates of lymph node metastasis.29 This study also showed that poorly differentiated (types 2 and 3) NPC is more associated with low c-myc levels, whereas well-differentiated (type 1) NPC has higher c-myc levels, which may explain this conflicting pattern of c-myc expression. This suggests that c-myc plays a role in disease progression, but further investigation is needed to better understand the role of c-myc in NPC development and to evaluate if targeting c-myc can slow NPC progression or metastases.
Checkpoint with Forkhead-Associated and RING Finger Domains is Downregulated via Promoter Hypermethylation
Checkpoint with forkhead-associated and ring finger domains (CHFR) is a mitotic checkpoint regulator that delays chromosome condensation in case of abnormal spindle formation.170 Gene expression of CHFR is reduced because of promoter hypermethylation in most tumors; however, the prevalence of CHFR hypermethylation is much higher in NPC—61%in NPC versus 30% in other primary head and neck cancers and 40% in colorectal cancer.31,171 This suggests that loss of CHFR expression is more common, and possibly more important, to NPC than other cancers. As a result, many NPC cells prematurely enter mitosis with unstable genomes, thus increasing the chance of chromosomal abnormalities and aneuploidy in daughter cells.31 Several chromosomal aberrations have been identified in NPC tumors—some sites correspond to proteins key to NPC development, including p16, RASSF1A, and CKIs, while a number of sites do not correspond to any known tumor suppressors or oncogenes.172,173 The prognostic and therapeutic value of CHFR remains unclear. CHFR activity can be restored in methylated cells treated with a methyltransferase inhibitor; fewer of the treated cells entered mitosis when compared with methylated cells.171 This indicates that targeting CHFR may slow the progression of tumors by reducing aneuploidy and chromosome damage. However, other studies show that detection of methylated CHFR genes is better used as a prognostic indicator because cancer cells with methylated CHFR genes actually have better prognoses and increased chemosensitivity to microtubule inhibitors such as paclitaxel.174,175 Further studies about the role of CHFR in NPC are warranted to better understand this relationship and determine if CHFR methylation can be used as a prognostic factor or treatment modality.
CELL ADHESION
Abnormal cell adhesion is critical to the invasive and metastatic potential of tumor cells. In NPC, E-cadherin and matrix metalloproteinases (MMPs) are 2 important components of cell adhesion that are dysregulated.
Decreased E-Cadherin Levels Correlate with Metastatic Disease
E-cadherin is a transmembrane glycoprotein that mediates cell communication in normal cells and as a metastasis suppressor in tumor cells.176 The downregulation of E-cadherin in NPC is due to aberrant promoter methylation of the E-cadherin gene.58 E-cadherin requires cytoplasmic β-catenin to maintain cell adhesion51; because cytoplasmic levels of β-catenin is decreased in NPC,20 cell adhesion via E-cadherin is compromised in NPC. Furthermore, E-cadherin levels are inversely proportional to disease progression: metastatic NPC tumors display lower E-cadherin mRNA and protein levels than primary NPC tumors (19% in primary tumors vs 42% in metastatic tumors).33,58 However, the proportion of NPCs that exhibit decreased E-cadherin levels is lower than for other primary HNSCC, where 88% of tumors show decreased E-cadherin expression.177 This suggests that while E-cadherin is important to NPC development, it is not as critical to metastases in NPC as in other head and neck cancers.
MMPs Are Upregulated and Correlate with Disease Progression
MMPs are type IV collagenases whose overexpression has been implicated in a number of cancers. There are 16 subtypes of MMPs that differ in structural domains that confer substrate specificity, cell-surface localization, and inhibitor binding.178 The complexity of MMP function in cancer metastasis is just now becoming clear: not only do they degrade basement membranes and extracellular matrices to allow for tumor invasion, they are also involved in activation of growth factors to promote cell growth and angiogenesis and also protect tumor cells from apoptotic signals.179–182 Interestingly, although tumor cells produce MMPs, the vast majority of MMPs come from inflammatory cells in the surrounding stroma of tumors. In HNSCC, the high level of MMP activity is secondary to overexpression of membrane type-1 MMP, a protease responsible for activating MMPs.183 In NPC, LMP1 is the driving force behind high MMP activity.
While a number of MMPs are upregulated by LMP1, current data suggest that MMP1 is the most important to NPC development. The upregulation of MMP1 in NPC far surpasses that in other cancers: when compared with controls, MMP1 expression is increased 124-fold in NPC versus 9-fold in other head and neck cancers.184 LMP1 upregulates MMP1 by increasing transcription with a new Ets binding site in the MMP1 promoter and by increasing expression of MMP3, an activator of latent MMP1.185 A recent study found that some NPC tumors contain a MMP1 promotor polymorphism that prevents binding of the promoter repressor protein; patients with tumors that are homozygous for this polymorphism tended to have more aggressive tumors and late-stage disease at presentation.186 Additionally, MMP1 has the ability to activate MMP2, further enhancing overall MMP activity.187
Two other MMPs consistently upregulated in NPC are MMP2 and MMP9, but considerably less is known about these 2 enzymes than MMP1. High MMP2 expression correlates with poor survival in NPC.188 An activating MMP2 promoter polymorphism, similar to that in MMP1, has been discovered and correlates with an increased susceptibility to developing NPC; additionally, heavy smokers with the polymorphism had the highest risk for NPC, suggesting that environmental factors such as smoking may interact with MMP2 in NPC development.189 MMP9 expression is associated with lymph node metastases.190 While polymorphisms for MMP9 have also been discovered, these do not correlate with an increased NPC risk like those for MMP1 and MMP2, suggesting that MMP9 polymorphisms are not activated.186 MMP9 transcription is more EBV dependent than MMP1 and 2 as their expression is upregulated by LMP1 via NF-κB and AP-1 activation.191,192
These findings show that MMPs have prognostic significance in NPC and may also have therapeutic implications. Antibodies or blocking peptides directed against MMPs may prevent metastases and increase treatment efficacy. Given the diversity of effects MMPs have in other tumors, the role of MMPs in NPC pathogenesis should first be better understood and the findings applied to develop novel therapeutics for NPC.
NEW TREATMENTS FOR NPC
While the traditional treatment for NPC has been radiation and chemotherapy, there are several new potential treatments that specifically target the molecular aberrations of NPC.
BRD7 is a protein expressed in normal nasopharyngeal cells but is downregulated in NPC.193 Interestingly, its expression is unchanged in gastric and colorectal cancers, suggesting that its upregulation is unique to NPC and it is exclusively involved in NPC tumorigenesis.194 The uniqueness of BRD7 lies in its ability to affect multiple pathways critical to NPC development. By regulating the transcription of signaling proteins in the ERK and Rb/E2F pathways, BRD7 normalizes cyclin D1 and E activity and, therefore, the G1/S progression.193 BRD7 also stabilizes cell proliferation by downregulating c-jun and inhibiting the accumulation of intracellular β-catenin.195 Finally, BRD7 downregulates AP-1 activity, which subsequently lowers MMP activity and helps regain normal cell adhesion.195 A recent study confirmed these properties when ectopic administration of BRD7 to NPC cells resulted in cell growth inhibition and delayed G1/S phase progression.193 These data show that methods targeting the upregulation of BRD7 are promising, targeted treatment modalities for NPC. Current research has elucidated various molecular properties of BRD7, including nuclear localization sequences and promoter regulators.196,197 Further studies are needed to discover clinically applicable methods of increasing BRD7 in NPC, whether by simply increasing protein levels or upregulating expression. Also all studies on BRD7 thus far have used NPC cell lines; animal studies looking at the effect of BRD7 on tumor growth would be useful in determining the efficacy of this treatment modality.
Cyclooxygenase (COX)-2 overexpression has been demonstrated in many cancers, including NPC.126 The subsequent increase in prostacyclins are key mediators of cell proliferation, angiogenesis, and apoptosis.198–200 COX-2 inhibition has shown promise as a new treatment modality for other head and neck cancers: nonsteroidal anti-inflammatory drugs were protective against chemically induced tongue cancers in animals, and selective COX-2 inhibitors prevented HNSCC xenograft growth in nude mice.201,202 More recently, COX-2 inhibitors have been shown to decrease growth of NPC cell lines in a dose-dependent manner and reduce cyclin D1 levels.203 Additionally, the use of COX-2 inhibitors in NPC is promising because of their tumor suppressor effects and ability to potentiate the effects of radiotherapy, the current primary treatment for NPC.161
As overactivation of the Wnt pathway is highly important to NPC development, inhibition of this pathway is a potential treatment modality for NPC. Because nearly all NPC exhibits Wnt over-expression or WIF underexpression and this pathway is critical to the abnormal β-catenin that is also observed in all NPC, blockade of the Wnt pathway appears to be the most promising molecular treatment modality. Blocking the Wnt pathway can be achieved either by anti-Wnt antibodies or Wnt antagonists. Wnt antibodies have been studied in HNSCC cells and resulted in inhibited cell growth with induction of apoptosis.204 Various Wnt antagonists have been developed, the most notable of which is WIF-1. As downregulation of WIF-1 has been specifically demonstrated in NPC, attempts to normalize WIF-1 in NPC would be the most promising of Wnt-targeted treatment. The WIF-1 protein has been successfully transfected into colorectal, melanoma, and non-small cell lung cancer (NSCLC) cells and resulted in significant tumor suppression.205,206 WIF-1 gene therapy in xenograft mouse models of melanoma and NSCLC resulted in lower tumor volume and weight.205,207 The adenovirus vector used for Wnt gene therapies has been successful at blocking Wnt expression; although it is associated with significant intestinal toxicity and high mortality rates with other Wnt antagonists, these adverse effects are not seen with WIF-1 delivery.208,209 This evidence points to WIF-1 as an effective and safe potential therapy for NPC.
CONCLUSION
The mechanism of NPC tumorigenesis is complex, involving the aberrations of a large variety of pathways and the alteration in expression of numerous proteins. Normal regulation of apoptosis, cell proliferation, and cell adhesion are dysregulated. The understanding of these mechanisms has greatly increased in the past decade, generating enough data to begin the development of prognostic factors and targeted treatments for NPC. Refining prognostic factors into clinically applicable assays may aid in the detection of NPC in asymptomatic patients, in addition to staging and monitoring disease. The search for molecularly based treatments for NPC has only just begun, and so much is yet to be discovered. Improved understanding of the unique molecular mechanisms behind NPC will hopefully lead to the development of targeted treatments against NPC that slow disease progression and improve survival.
Acknowledgments
This work was partially supported by the Larry Hall and Zygielbaum Memorial Trusts, and the Kazan, McClain, Edises, Abrams, Fernandez, Lyons and Farrise Foundation.
Contract grant sponsor: National Institutes of Health; contract grant number: RO1 CA 093708-01A3.
References
- 1.Sham JS, Wei WI, Zong YS, et al. Detection of subclinical nasopharyngeal carcinoma by fibreoptic endoscopy and multiple biopsy. Lancet. 1990;335:371–374. doi: 10.1016/0140-6736(90)90206-k. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Muir CS. Cancer incidence in five continents. Comparability and quality of data. IARC Sci Publ. 1992;120:45–173. [PubMed] [Google Scholar]
- 3.Nielsen NH, Mikkelsen F, Hansen JP. Nasopharyngeal cancer in Greenland. The incidence in an Arctic Eskimo population. Acta Pathol Microbiol Scand [A] 1977;85:850–858. [PubMed] [Google Scholar]
- 4.Buell P. The effect of migration on the risk of nasopharyngeal cancer among Chinese. Cancer Res. 1974;34:1189–1191. [PubMed] [Google Scholar]
- 5.Yu MC, Ho JH, Ross RK, Henderson BE. Nasopharyngeal carcinoma in Chinese—salted fish or inhaled smoke? Prev Med. 1981;10:15–24. doi: 10.1016/0091-7435(81)90002-5. [DOI] [PubMed] [Google Scholar]
- 6.Goldsmith DB, West TM, Morton R. HLA associations with nasopharyngeal carcinoma in Southern Chinese: a meta-analysis. Clin Otolaryngol Allied Sci. 2002;27:61–67. doi: 10.1046/j.0307-7772.2001.00529.x. [DOI] [PubMed] [Google Scholar]
- 7.Shanmugaratnam K, Sobin L. Histologic typing of tumours of the upper respiratory tract and ear. 2. Geneva: WHO; 1991. [DOI] [PubMed] [Google Scholar]
- 8.Marks JE, Phillips JL, Menck HR. The National Cancer Data Base report on the relationship of race and national origin to the histology of nasopharyngeal carcinoma. Cancer. 1998;83:582–588. doi: 10.1002/(sici)1097-0142(19980801)83:3<582::aid-cncr29>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 9.Vasef MA, Ferlito A, Weiss LM. Nasopharyngeal carcinoma, with emphasis on its relationship to Epstein-Barr virus. Ann Otol Rhinol Laryngol. 1997;106:348–356. doi: 10.1177/000348949710600416. [DOI] [PubMed] [Google Scholar]
- 10.Al-Sarraf M, LeBlanc M, Giri PG, et al. Chemoradio-therapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16:1310–1317. doi: 10.1200/JCO.1998.16.4.1310. [DOI] [PubMed] [Google Scholar]
- 11.Sham JS, Choy D. Prognostic factors of nasopharyngeal carcinoma: a review of 759 patients. Br J Radiol. 1990;63:51–58. doi: 10.1259/0007-1285-63-745-51. [DOI] [PubMed] [Google Scholar]
- 12.Altun M, Fandi A, Dupuis O, Cvitkovic E, Krajina Z, Eschwege F. Undifferentiated nasopharyngeal cancer (UCNT): current diagnostic and therapeutic aspects. Int J Radiat Oncol Biol Phys. 1995;32:859–877. doi: 10.1016/0360-3016(95)00516-2. [DOI] [PubMed] [Google Scholar]
- 13.Chow E, Payne D, Keane T, Panzarella T, Izard MA. Enhanced control by radiotherapy of cervical lymph node metastases arising from nasopharyngeal carcinoma compared with nodal metastases from other head and neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys. 1997;39:149–154. doi: 10.1016/s0360-3016(97)00313-1. [DOI] [PubMed] [Google Scholar]
- 14.Cheng SH, Jian JJ, Tsai SY, et al. Long-term survival of nasopharyngeal carcinoma following concomitant radiotherapy and chemotherapy. Int J Radiat Oncol Biol Phys. 2000;48:1323–1330. doi: 10.1016/s0360-3016(00)00779-3. [DOI] [PubMed] [Google Scholar]
- 15.Yamashita S, Kondo M, Hashimoto S. Squamous cell carcinoma of the nasopharynx. An analysis of failure patterns after radiation therapy. Acta Radiol Oncol. 1985;24:315–320. doi: 10.3109/02841868509136058. [DOI] [PubMed] [Google Scholar]
- 16.Lee AW, Foo W, Law SC, et al. Nasopharyngeal carcinoma: presenting symptoms and duration before diagnosis. Hong Kong Med J. 1997;3:355–361. [PubMed] [Google Scholar]
- 17.Lo AK, Lo KW, Tsao SW, et al. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia. 2006;8:173–180. doi: 10.1593/neo.05625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi W, Bastianutto C, Li A, et al. Multiple dysregulated pathways in nasopharyngeal carcinoma revealed by gene expression profiling. Int J Cancer. 2006;119:2467–2475. doi: 10.1002/ijc.22107. [DOI] [PubMed] [Google Scholar]
- 19.Morrison J, Gulley M, Pathmanathan R, Raab-Traub N. Differential signaling pathways are activated in the Epstein-Barr virus-associated malignancies nasopharyngeal carcinoma and Hodgkin lymphoma. Cancer Res. 2004;64:5251–5260. doi: 10.1158/0008-5472.CAN-04-0538. [DOI] [PubMed] [Google Scholar]
- 20.Zeng ZY, Zhou YH, Zhang WL, et al. Gene expression profiling of nasopharyngeal carcinoma reveals the abnormally regulated Wnt signaling pathway. Hum Pathol. 2007;38:120–133. doi: 10.1016/j.humpath.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Xiao J, Zhao S, Tian Y, Wang G. Expression of telomerase subunits and its relationship with telomerase activity in nasopharyngeal carcinoma. Zhonghua Yi Xue Za Zhi. 2001;81:553–556. [PubMed] [Google Scholar]
- 22.Liu JP, Cassar L, Pinto A, Li H. Mechanisms of cell immortalization mediated by EB viral activation of telomerase in nasopharyngeal carcinoma. Cell Res. 2006;16:809–817. doi: 10.1038/sj.cr.7310098. [DOI] [PubMed] [Google Scholar]
- 23.Yang HJ, Cho YJ, Kim HS, Chang MS, Sung MW, Kim WH. Association of p53 and BCL-2 expression with Epstein-Barr virus infection in the cancers of head and neck. Head Neck. 2001;23:629–636. doi: 10.1002/hed.1089. [DOI] [PubMed] [Google Scholar]
- 24.Sheu L, Chen A, Lee H, Hsu H, Yu D. Cooperative interactions among p53, bcl-2 and Epstein-Barr virus latent membrane protein 1 in nasopharyngeal carcinoma cells. Pathol Int. 2004;54:475–485. doi: 10.1111/j.1440-1827.2004.01654.x. [DOI] [PubMed] [Google Scholar]
- 25.Luo J, Xiao J, Tao Z, Li X. Detection of c-myc gene expression in nasopharyngeal carcinoma by nonradioactive in situ hybridization and immunohistochemistry. Chin Med J (Engl) 1997;110:229–232. [PubMed] [Google Scholar]
- 26.Hwang CF, Cho CL, Huang CC, et al. Loss of cyclin D1 and p16 expression correlates with local recurrence in nasopharyngeal carcinoma following radiotherapy. Ann Oncol. 2002;13:1246–1251. doi: 10.1093/annonc/mdf215. [DOI] [PubMed] [Google Scholar]
- 27.Wang SS, Guan ZZ, Xiang YQ, et al. Significance of EGFR and p-ERK expression in nasopharyngeal carcinoma. Zhonghua Zhong Liu Za Zhi. 2006;28:28–31. [PubMed] [Google Scholar]
- 28.Zhang Z, Sun D, Van do N, Tang A, Hu L, Huang G. Inactivation of RASSF2A by promoter methylation correlates with lymph node metastasis in nasopharyngeal carcinoma. Int J Cancer. 2007;120:32–38. doi: 10.1002/ijc.22185. [DOI] [PubMed] [Google Scholar]
- 29.Hwang CF, Su CY, Huang SC, et al. Low expression levels of p27 correlate with loco-regional recurrence in nasopharyngeal carcinoma. Cancer Lett. 2003;189:231–236. doi: 10.1016/s0304-3835(02)00508-6. [DOI] [PubMed] [Google Scholar]
- 30.Makitie AA, MacMillan C, Ho J, et al. Loss of p16 expression has prognostic significance in human nasopharyngeal carcinoma. Clin Cancer Res. 2003;9:2177–2184. [PubMed] [Google Scholar]
- 31.Cheung H, Ching Y, Nicholls J, et al. Epigenetic inactivation of CHFR in nasopharyngeal carcinoma through promoter methylation. Mol Carcinog. 2005;43:237–245. doi: 10.1002/mc.20106. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Yang H, Huo X. Expression and significance of PTEN in nasopharyngeal carcinoma. Lin Chuang Er Bi Yan Hou Ke Za Zhi. 2004;18:658–659. [PubMed] [Google Scholar]
- 33.Huang GW, Mo WN, Kuang GQ, et al. Expression of p16, nm23-H1, E-cadherin, and CD44 gene products and their significance in nasopharyngeal carcinoma. Laryngoscope. 2001;111:1465–1471. doi: 10.1097/00005537-200108000-00025. [DOI] [PubMed] [Google Scholar]
- 34.Wan J, Sun L, Mendoza J, et al. Elucidation of the c-Jun N-terminal kinase pathway mediated by Epstein-Barr virus-encoded latent membrane protein 1. Mol Cell Biol. 2004;24:192–199. doi: 10.1128/MCB.24.1.192-199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henle G, Henle W, Clifford P, et al. Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control groups. J Natl Cancer Inst. 1969;43:1147–1157. [PubMed] [Google Scholar]
- 36.Macsween K, Crawford D. Epstein-Barr virus—recent advances. Lancet Infect Dis. 2003;3:131–140. doi: 10.1016/s1473-3099(03)00543-7. [DOI] [PubMed] [Google Scholar]
- 37.Wei W, Sham J. Nasopharyngeal carcinoma. Lancet. 2005;365:2041–2054. doi: 10.1016/S0140-6736(05)66698-6. [DOI] [PubMed] [Google Scholar]
- 38.Brooks L, Yao QY, Rickinson AB, Young LS. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol. 1992;66:2689–2697. doi: 10.1128/jvi.66.5.2689-2697.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 40.Mainou BA, Raab-Traub N. LMP1 strain variants: biological and molecular properties. J Virol. 2006;80:6458–6468. doi: 10.1128/JVI.00135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozyar E, Ayhan A, Korcum A, Atahan I. Prognostic role of Epstein-Barr virus latent membrane protein-1 and interleukin-10 expression in patients with nasopharyngeal carcinoma. Cancer Invest. 2004;22:483–491. doi: 10.1081/cnv-200026386. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Peng J, Chang H, Hung W. RECK is a target of Epstein-Barr virus latent membrane protein 1. Oncogene. 2003;22:8263–8270. doi: 10.1038/sj.onc.1207157. [DOI] [PubMed] [Google Scholar]
- 43.Keryer-Bibens C, Pioche-Durieu C, Villemant C, et al. Exosomes released by Epstein-Barr virus infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer. 2006;6:283. doi: 10.1186/1471-2407-6-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HS, Kim JS, Kim JS, et al. The association between CD99 and LMP-1 expression in nasopharyngeal carcinoma. Exp Oncol. 2006;28:40–43. [PubMed] [Google Scholar]
- 45.Mei YP, Zhou JM, Wang Y, et al. Silencing of LMP1 induces cell cycle arrest and enhances chemosensitivity through inhibition of AKT signaling pathway in EBV-positive nasopharyngeal carcinoma cells. Cell Cycle. 2007;6:1379–1385. doi: 10.4161/cc.6.11.4274. [DOI] [PubMed] [Google Scholar]
- 46.Miller CL, Lee JH, Kieff E, Longnecker R. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc Natl Acad Sci U S A. 1994;91:772–776. doi: 10.1073/pnas.91.2.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart S, Dawson CW, Takada K, et al. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-κB transcription factor pathway. Proc Natl Acad Sci U S A. 2004;101:15730–15735. doi: 10.1073/pnas.0402135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pegtel DM, Subramanian A, Sheen TS, Tsai CH, Golub TR, Thorley-Lawson DA. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: possible role in nasopharyngeal carcinoma metastasis. J Virol. 2005;79:15430–15442. doi: 10.1128/JVI.79.24.15430-15442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yates JL, Warren N, Sugden B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985;313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- 50.Wang F, Tsang SF, Kurilla MG, Cohen JI, Kieff E. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J Virol. 1990;64:3407–3416. doi: 10.1128/jvi.64.7.3407-3416.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jou T, Stewart D, Stappert J, Nelson W, Marrs J. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc Natl Acad Sci U S A. 1995;92:5067–5071. doi: 10.1073/pnas.92.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mazieres J, He B, You L, Xu Z, Jablons D. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1–10. doi: 10.1016/j.canlet.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 53.Lo A, Liu Y, Wang X, et al. Alterations of biologic properties and gene expression in nasopharyngeal epithelial cells by the Epstein-Barr virus-encoded latent membrane protein 1. Lab Invest. 2003;83:697–709. doi: 10.1097/01.lab.0000067480.44925.10. [DOI] [PubMed] [Google Scholar]
- 54.Sriuranpong V, Mutirangura A, Gillespie J, et al. Global gene expression profile of nasopharyngeal carcinoma by laser capture microdissection and complementary DNA microarrays. Clin Cancer Res. 2004;10:4944–4958. doi: 10.1158/1078-0432.CCR-03-0757. [DOI] [PubMed] [Google Scholar]
- 55.Lin YC, You L, Xu Z, et al. Wnt signaling activation and WIF-1 silencing in nasopharyngeal cancer cell lines. Biochem Biophys Res Commun. 2006;341:635–640. doi: 10.1016/j.bbrc.2005.12.220. [DOI] [PubMed] [Google Scholar]
- 56.Everly D, Kusano S, Raab-Trau N. Accumulation of cytoplasmic B-catenin and nuclear glycogen synthase kinase 3B in Epstein-Barr virus-infected cells. J Virol. 2004;78:11648–11655. doi: 10.1128/JVI.78.21.11648-11655.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morrison J, Raab-Traub N. Roles of the ITAM and PY motifs of Epstein-Barr virus latent membrane protein 2A in the inhibition of epithelial cell differentiation and activation of {β}-catenin signaling. J Virol. 2005;79:2375–2382. doi: 10.1128/JVI.79.4.2375-2382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Ren Y, Lin S, Liang Y, Liang H. Association of E-cadherin and β-catenin with metastasis in nasopharyngeal carcinoma. Chin Med J (Engl) 2004;117:1232–1239. [PubMed] [Google Scholar]
- 59.Ren Q, Sato H, Murono S, Furukawa M, Yoshizaki T. Epstein-Barr virus (EBV) latent membrane protein 1 induces interleukin-8 through the nuclear factor-κB signaling pathway in EBV-infected nasopharyngeal carcinoma cell line. Laryngoscope. 2004;114:855–859. doi: 10.1097/00005537-200405000-00012. [DOI] [PubMed] [Google Scholar]
- 60.Man C, Rosa J, Lee LT, et al. Latent membrane protein 1 suppresses RASSF1A expression, disrupts microtubule structures and induces chromosomal aberrations in human epithelial cells. Oncogene. 2007;26:3069–3080. doi: 10.1038/sj.onc.1210106. [DOI] [PubMed] [Google Scholar]
- 61.Bargou R, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-κB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest. 1997;100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang W, Abbruzzese J, Evans D, Larry L, Cleary K, Chiao P. The nuclear factor-κB RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 63.Sovak M, Bellas R, Kim D, et al. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolf JS, Chen Z, Dong G, et al. IL (interleukin)-1α promotes nuclear factor-κB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res. 2001;7:1812–1820. [PubMed] [Google Scholar]
- 65.Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr Selective activation of NF-κB subunits in human breast cancer: potential roles for NF-κB2/p52 and for Bcl-3. Oncogene. 2000;19:1123–1131. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- 66.Mainou B, Everly D, Jr, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene. 2005;24:6917–6924. doi: 10.1038/sj.onc.1208846. [DOI] [PubMed] [Google Scholar]
- 67.Deng l, Yang J, Zhao X, et al. Cells in G2/M phase increased in human nasopharyngeal carcinoma cell line by EBV-LMP1 through activation of NF-κB and AP-1. Cell Res. 2003;13:187–194. doi: 10.1038/sj.cr.7290163. [DOI] [PubMed] [Google Scholar]
- 68.Murono S, Inoue H, Tanabe T, et al. Induction of cyclo-oxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci U S A. 2001;98:6905–6910. doi: 10.1073/pnas.121016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ding L, Li L, Yang J, et al. Epstein-Barr virus encoded latent membrane protein 1 modulates nuclear translocation of telomerase reverse transcriptase protein by activating nuclear factor-κB p65 in human nasopharyngeal carcinoma cells. Int J Biochem Cell Biol. 2005;37:1881–1889. doi: 10.1016/j.biocel.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 70.Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 71.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 72.Yunis JJ, Oken MM, Kaplan ME, Ensrud KM, Howe RR, Theologides A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N Engl J Med. 1982;307:1231–1236. doi: 10.1056/NEJM198211113072002. [DOI] [PubMed] [Google Scholar]
- 73.Fan SQ, Ma J, Zhou J, et al. Differential expression of Epstein-Barr virus-encoded RNA and several tumor-related genes in various types of nasopharyngeal epithelial lesions and nasopharyngeal carcinoma using tissue microarray analysis. Hum Pathol. 2006;37:593–605. doi: 10.1016/j.humpath.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 74.Harn HJ, Ho LI, Liu CA, et al. Down regulation of bcl-2 by p53 in nasopharyngeal carcinoma and lack of detection of its specific t(14;18) chromosomal translocation in fixed tissues. Histopathology. 1996;28:317–323. doi: 10.1046/j.1365-2559.1996.d01-431.x. [DOI] [PubMed] [Google Scholar]
- 75.Niemhom S, Kitazawa S, Murao S, Kunachak S, Maeda S. Co-expression of p53 and bcl-2 may correlate to the presence of Epstein-Barr virus genome and the expression of proliferating cell nuclear antigen in nasopharyngeal carcinoma. Cancer Lett. 2000;160:199–208. doi: 10.1016/s0304-3835(00)00582-6. [DOI] [PubMed] [Google Scholar]
- 76.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 77.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 78.Tamm I, Wang Y, Sausville E, et al. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 79.Ai MD, Li LL, Zhao XR, Wu Y, Gong JP, Cao Y. Regulation of survivin and CDK4 by Epstein-Barr virus encoded latent membrane protein 1 in nasopharyngeal carcinoma cell lines. Cell Res. 2005;15:777–784. doi: 10.1038/sj.cr.7290347. [DOI] [PubMed] [Google Scholar]
- 80.Faqing T, Zhi H, Liqun Y, et al. Epstein-Barr virus LMP1 initiates cell proliferation and apoptosis inhibition via regulating expression of survivin in nasopharyngeal carcinoma. Exp Oncol. 2005;27:96–101. [PubMed] [Google Scholar]
- 81.Yip KW, Shi W, Pintilie M, et al. Prognostic significance of the Epstein-Barr virus, p53, Bcl-2, and survivin in nasopharyngeal cancer. Clin Cancer Res. 2006;12:5726–5732. doi: 10.1158/1078-0432.CCR-06-0571. [DOI] [PubMed] [Google Scholar]
- 82.Jiang W, Liao Y, Zhao S, et al. Role of enhanced radio-sensitivity and the tumor-specific suicide gene vector in gene therapy of nasopharyngeal carcinoma. J Radiat Res (Tokyo) 2007;48:211–218. doi: 10.1269/jrr.06056. [DOI] [PubMed] [Google Scholar]
- 83.Xiang Y, Yao H, Wang S, et al. Prognostic value of survivin and livin in nasopharyngeal carcinoma. Laryngoscope. 2006;116:126–130. doi: 10.1097/01.mlg.0000187392.87904.35. [DOI] [PubMed] [Google Scholar]
- 84.McCaul JA, Gordon KE, Clark LJ, Parkinson EK. Telomerase inhibition and the future management of head-and-neck cancer. Lancet Oncol. 2002;3:280–288. doi: 10.1016/s1470-2045(02)00729-5. [DOI] [PubMed] [Google Scholar]
- 85.Yang J, Deng X, Deng L, Gu H, Fan W, Cao Y. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J Exp Clin Cancer Res. 2004;23:495–506. [PubMed] [Google Scholar]
- 86.Li HM, Man C, Jin Y, et al. Molecular and cytogenetic changes involved in the immortalization of nasopharyngeal epithelial cells by telomerase. Int J Cancer. 2006;119:1567–1576. doi: 10.1002/ijc.22032. [DOI] [PubMed] [Google Scholar]
- 87.Chen F, Liu C, Lindvall C, Xu D, Ernberg I. Epstein-Barr virus latent membrane 2A (LMP2A) down-regulates telomerase reverse transcriptase (hTERT) in epithelial cell lines. Int J Cancer. 2005;113:284–289. doi: 10.1002/ijc.20594. [DOI] [PubMed] [Google Scholar]
- 88.Dackour R, Carter T, Steinberg BM. Phosphatidylinositol 3-kinase regulates early differentiation in human laryngeal keratinocytes. In Vitro Cell Dev Biol Anim. 2005;41:111–117. doi: 10.1290/0501003.1. [DOI] [PubMed] [Google Scholar]
- 89.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Worsham MJ, Pals G, Schouten JP, et al. Delineating genetic pathways of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2003;129:702–708. doi: 10.1001/archotol.129.7.702. [DOI] [PubMed] [Google Scholar]
- 91.Pedrero J, Carracedo D, Pinto C, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114:242–248. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 93.Maehama T, Dixon J. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 94.Ohigashi T, Muzuno R, Nakashima J, Marumo K, Murai M. Inhibition of wnt signaling downregulates Akt activity and induces chemosensitivity in PTEN-mutated prostate cancer cells. Prostate. 2005;52:61–68. doi: 10.1002/pros.20117. [DOI] [PubMed] [Google Scholar]
- 95.Gimm O. Thyroid cancer. Cancer Lett. 2001;163:143–156. doi: 10.1016/s0304-3835(00)00697-2. [DOI] [PubMed] [Google Scholar]
- 96.Chen Q, Samaranayake L, Zhou H, Xiao L. Homozygous deletion of the PTEN tumor-suppressor gene is not a feature in oral squamous cell carcinoma. Oral Oncol. 2000;36:95–99. doi: 10.1016/s1368-8375(99)00068-8. [DOI] [PubMed] [Google Scholar]
- 97.Schagdarsurengin U, Gimm O, Dralle H, Hoang-Vu C, Dammann R. CpG island methylation of tumor-related promoters occurs preferentially in undifferentiated carcinoma. Thyroid. 2006;16:633–642. doi: 10.1089/thy.2006.16.633. [DOI] [PubMed] [Google Scholar]
- 98.Bai W, Li W, Chen X, Wang T. The relationship between hypermethylation of the PTEN promoter and laryngeal squamous cell carcinoma. Lin Chuang Er Bi Yan Hou Ke Za Zhi. 2006;20:254–256. [PubMed] [Google Scholar]
- 99.Kwong J, Lo K, To K, Teo P, Johnson P, Huang D. Promoter hypermethylation of multiple genes in nasopharyngeal carcinoma. Clin Cancer Res. 2002;8:131–137. [PubMed] [Google Scholar]
- 100.West K, Brognard J, Clark A, et al. Rapid AKT activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111:81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, Sun L, Liu N, Yu H, Zhang Y, Shan Y. Effects of basic fibroblast growth factor on protein kinase B activity and c-fos expression in CNE- I nasopharyngeal carcinoma cell line. Zhonghua Er Bi Yan Hou Ke Za Zhi. 2004;39:679–682. [PubMed] [Google Scholar]
- 102.Milde-Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer. 2005;41:2449. doi: 10.1016/j.ejca.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 103.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 104.English J, Pearson G, Wilsbacher J, et al. New insights into the control of MAP kinase pathways. Exp Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- 105.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 106.Tang F, Tang G, Xiang J, Dai Q, Rosner MR, Lin A. The absence of NF-κB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor α-induced apoptosis. Mol Cell Biol. 2002;22:8571–8579. doi: 10.1128/MCB.22.24.8571-8579.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Potapova O, Anisimov SV, Gorospe M, et al. Targets of c-Jun NH(2)-terminal kinase 2-mediated tumor growth regulation revealed by serial analysis of gene expression. Cancer Res. 2002;62:3257–3263. [PubMed] [Google Scholar]
- 108.Tsai CL, Li HP, Lu YJ, et al. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006;66:11668–11676. doi: 10.1158/0008-5472.CAN-06-2194. [DOI] [PubMed] [Google Scholar]
- 109.Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) Oncogene. 1998;16:1731–1742. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- 110.Shklyaev SS, Namba H, Sautin Y, et al. Involvement of wild-type p53 in radiation-induced c-Jun N-terminal kinase activation in human thyroid cells. Anticancer Res. 2001;21:2569–2575. [PubMed] [Google Scholar]
- 111.Chen M, Lee H, Chang J, Chang C. Expression of p53 protein and primary tumour volume in patients with nasopharyngeal carcinoma. J Otolaryngol. 2004;33:304–317. doi: 10.2310/7070.2004.00304. [DOI] [PubMed] [Google Scholar]
- 112.Li L, Guo L, Tao Y, et al. Latent membrane protein 1 of Epstein-Barr virus regulates p53 phosphorylation through MAP kinases. Cancer Lett. 2007;255:219–231. doi: 10.1016/j.canlet.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 113.Shklyaev SS, Namba H, Mitsutake N, et al. Transient activation of c-Jun NH2-terminal kinase by growth factors influences survival but not apoptosis of human thyrocytes. Thyroid. 2001;11:629–636. doi: 10.1089/105072501750362691. [DOI] [PubMed] [Google Scholar]
- 114.Schmidt M, Goebeler M, Posern G, et al. Ras-independent activation of the Raf/MEK/ERK pathway upon calcium-induced differentiation of keratinocytes. J Biol Chem. 2000;275:41011–41017. doi: 10.1074/jbc.M003716200. [DOI] [PubMed] [Google Scholar]
- 115.Bancroft C, Chen Z, Dong G, et al. Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-κB signal pathways. Clin Cancer Res. 2001;7:435–442. [PubMed] [Google Scholar]
- 116.Treinies I, Paterson H, Hooper S, Wilson R, Marshall C. Activated MEK stimulates expression of AP-1 components independently of phosphatidylinositol 3-kinase (PI3-kinase) but requires a PI3-kinase signal to stimulate DNA synthesis. Mol Cell Biol. 1999;19:321–329. doi: 10.1128/mcb.19.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. Increased MAPK expression and activity in primary human hepatocellular carcinoma. Biochem Biophys Res Commun. 1997;236:54–58. doi: 10.1006/bbrc.1997.6840. [DOI] [PubMed] [Google Scholar]
- 118.Bang YJ, Kwon JH, Kang SH, Kim JW, Yang YC. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem Biophys Res Commun. 1998;250:43–47. doi: 10.1006/bbrc.1998.9256. [DOI] [PubMed] [Google Scholar]
- 119.Oka H, Chatani Y, Hoshino R, et al. Constitutive activation of mitogen-activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res. 1995;55:4182–4187. [PubMed] [Google Scholar]
- 120.Xu J, Gimenez-Conti IB, Cunningham JE, et al. Alterations of p53, cyclin D1, rb, and H-ras in human oral carcinomas related to tobacco use. Cancer. 1998;83:204–212. doi: 10.1002/(sici)1097-0142(19980715)83:2<204::aid-cncr2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 121.Yung WC, Sham JS, Choy DT, Ng MH. ras mutations are uncommon in nasopharyngeal carcinoma. Eur J Cancer B Oral Oncol B. 1995;31:399–400. doi: 10.1016/0964-1955(95)00046-1. [DOI] [PubMed] [Google Scholar]
- 122.Haeder M, Rotsch M, Bepler G, et al. Epidermal growth factor receptor expression in human lung cancer cell lines. Cancer Res. 1988;48:1132–1136. [PubMed] [Google Scholar]
- 123.Di Lorenzo G, Tortora G, D’Armiento F, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8:3438–3444. [PubMed] [Google Scholar]
- 124.Slamon D, Clark G, Wong S, Levin W, Ullrich A, McGuire W. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 125.Goustin A, Leof E, Shipley G, Moses H. Growth factors and cancer. Cancer Res. 1986;46:1015–1029. [PubMed] [Google Scholar]
- 126.Soo R, Putti T, Tao Q, Goh B. Overexpression of cyclo-oxygenase-2 in nasopharyngeal carcinoma and association with epidermal growth factor receptor expression. Arch Otol Head Neck Surg. 2005;131:147–152. doi: 10.1001/archotol.131.2.147. [DOI] [PubMed] [Google Scholar]
- 127.Hsu C, Gao M, Chen C, Yeh P, Cheng A. Inhibitors of epidermoid growth factor receptor suppress cell growth and enhance chemosensitivity of nasopharyngeal cancer cells in vitro. Oncology. 2005;68:538–547. doi: 10.1159/000086998. [DOI] [PubMed] [Google Scholar]
- 128.Tao Y, Song X, Deng X, et al. Nuclear accumulation of epidermal growth factor receptor and acceleration of G1/S stage by Epstein-Barr-encoded oncoprotein latent membrane protein 1. Exp Cell Res. 2005;303:240–251. doi: 10.1016/j.yexcr.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 129.Gasco M, Crook T. The p53 network in head and neck cancer. Oral Oncol. 2003;39:222–231. doi: 10.1016/s1368-8375(02)00163-x. [DOI] [PubMed] [Google Scholar]
- 130.Wu H, Lu T, Lee J, et al. MDM2 expression in EBV-infected nasopharyngeal carcinoma cells. Lab Invest. 2004;84:1547–1556. doi: 10.1038/labinvest.3700183. [DOI] [PubMed] [Google Scholar]
- 131.Burgos J. Involvement of the Epstein-Barr virus in the nasopharyngeal carcinoma pathogenesis. Med Oncol. 2005;22:113–122. doi: 10.1385/MO:22:2:113. [DOI] [PubMed] [Google Scholar]
- 132.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 133.Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 134.Crook T, Nicholls JM, Brooks L, O’Nions J, Allday MJ. High level expression of deltaN-p63: a mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene. 2000;19:3439–3444. doi: 10.1038/sj.onc.1203656. [DOI] [PubMed] [Google Scholar]
- 135.Potapova O, Gorospe M, Dougherty R, Dean N, Gaarde W, Holbrook N. Inhibition of c-Jun N-terminal kinase 2 expression suppresses growth and induces apoptosis of human tumor cells in a p53-dependent manner. Mol Cell Biol. 2000;20:1713–1722. doi: 10.1128/mcb.20.5.1713-1722.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Merlo A, Herman J, Mao L, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 137.Cooper GM, Hausman RE. The cell: a molecular approach. 4. Washington DC: ASM Press and Sinauer Associates, Inc; 2006. [Google Scholar]
- 138.Shibosawa E, Tsutsumi K, Koizuka I. Absence of nuclear p16 from Epstein-Barr virus-associated undifferentiated nasopharyngeal carcinomas. Laryngoscope. 2000;110:93–97. doi: 10.1097/00005537-200001000-00017. [DOI] [PubMed] [Google Scholar]
- 139.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 140.El-Naggar AK, Lai S, Clayman GL, et al. Expression of p16, Rb, and cyclin D1 gene products in oral and laryngeal squamous carcinoma: biological and clinical implications. Hum Pathol. 1999;30:1013–1018. doi: 10.1016/s0046-8177(99)90217-4. [DOI] [PubMed] [Google Scholar]
- 141.Gulley ML, Nicholls JM, Schneider BG, Amin MB, Ro JY, Geradts J. Nasopharyngeal carcinomas frequently lack the p16/MTS1 tumor suppressor protein but consistently express the retinoblastoma gene product. Am J Pathol. 1998;152:865–869. [PMC free article] [PubMed] [Google Scholar]
- 142.Reed AL, Califano J, Cairns P, et al. High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res. 1996;56:3630–3633. [PubMed] [Google Scholar]
- 143.Song X, Tao Y, Deng X, et al. Heterodimer formation between c-Jun and Jun B proteins mediated by Epstein-Barr virus encoded latent membrane protein 1. Cell Signal. 2004;16:1153–1162. doi: 10.1016/j.cellsig.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 144.Ohtani N, Brennan P, Gaubatz S, et al. Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J Cell Biol. 2003;162:173–183. doi: 10.1083/jcb.200302085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rayman J, Takahashi Y, Indjeian V, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hill AA, Wan F, Acheson DK, Skarsgard LD. Lack of correlation between G1 arrest and radiation age-response in three synchronized human tumour cell lines. Int J Radiat Biol. 1999;75:1395–1408. doi: 10.1080/095530099139269. [DOI] [PubMed] [Google Scholar]
- 147.Wang GL, Lo KW, Tsang KS, et al. Inhibiting tumorigenic potential by restoration of p16 in nasopharyngeal carcinoma. Br J Cancer. 1999;81:1122–1126. doi: 10.1038/sj.bjc.6690818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Macri E, Loda M. Role of p27 in prostate carcinogenesis. Cancer Metastasis Rev. 1998/1999;17:337–344. doi: 10.1023/a:1006133620914. [DOI] [PubMed] [Google Scholar]
- 149.Kerkhoff E, Rapp U. Cell cycle targets of Ras/Raf signalling. Oncogene. 1998;17:1457–1462. doi: 10.1038/sj.onc.1202185. [DOI] [PubMed] [Google Scholar]
- 150.Kudo Y, Kitajima S, Ogawa I, Miyauchi M, Takata T. Down-regulation of Cdk inhibitor p27 in oral squamous cell carcinoma. Oral Oncol. 2005;41:105–116. doi: 10.1016/j.oraloncology.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 151.Baba Y, Tsukuda M, Mochimatsu I, et al. Reduced expression of p16 and p27 proteins in nasopharyngeal carcinoma. Cancer Detect Prev. 2001;25:414–419. [PubMed] [Google Scholar]
- 152.Muller D, Bouchard C, Rudolph B, et al. Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene. 1997;15:2561–2576. doi: 10.1038/sj.onc.1201440. [DOI] [PubMed] [Google Scholar]
- 153.St Croix B, Florenes VA, Rak JW, et al. Impact of the cyclin-dependent kinase inhibitor p27Kip1 on resistance of tumor cells to anticancer agents. Nat Med. 1996;2:1204–1210. doi: 10.1038/nm1196-1204. [DOI] [PubMed] [Google Scholar]
- 154.Lee MH, Yang HY. Regulators of G1 cyclin-dependent kinases and cancers. Cancer Metastasis Rev. 2003;22:435–449. doi: 10.1023/a:1023785332315. [DOI] [PubMed] [Google Scholar]
- 155.Robles A, Larcher F, Whalin R, et al. Expression of cyclin D1 in epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad Sci U S A. 1996;93:7634–7638. doi: 10.1073/pnas.93.15.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhou B, Elledge S. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 157.Xie L, Xu L, He Z, et al. Identification of differentially expressed genes in nasopharyngeal carcinoma by means of the Atlas human cancer cDNA expression array. J Cancer Res Clin Oncol. 2000;126:400–406. doi: 10.1007/PL00008488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Song X, Tao Y, Zeng L, et al. Latent membrane protein 1 encoded by Epstein-Barr virus modulates directly and synchronously cyclin D1 and p16 by newly forming a c-Jun/Jun B heterodimer in nasopharyngeal carcinoma cell line. Virus Res. 2005;113:89–99. doi: 10.1016/j.virusres.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 159.Michalides R, van Veelen N, Hart A, Loftus B, Wient-jens E, Balm A. Overexpression of cyclin D1 correlates with recurrence in a group of forty-seven operable squamous cell carcinomas of the head and neck. Cancer Res. 1995;55:975–978. [PubMed] [Google Scholar]
- 160.Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73–92. [PubMed] [Google Scholar]
- 161.Tan K, Putti T. Cyclooxygenase 2 expression in nasopharyngeal carcinoma: immunohistochemical findings and potential implications. J Clin Pathol. 2005;58:535–538. doi: 10.1136/jcp.2004.021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 163.Shintani S, Mihara M, Nakahara Y, et al. Expression of cell cycle control proteins in normal epithelium, premalignant and malignant lesions of oral cavity. Oral Oncol. 2002;38:235–243. doi: 10.1016/s1368-8375(01)00048-3. [DOI] [PubMed] [Google Scholar]
- 164.Ioachim E, Peschos D, Goussia A, et al. Expression patterns of cyclins D1, E in laryngeal epithelial lesions: correlation with other cell cycle regulators (p53, pRb, Ki-67 and PCNA) and clinicopathological features. J Exp Clin Cancer Res. 2004;23:277–283. [PubMed] [Google Scholar]
- 165.Hermeking H, Rago C, Schuhmacher M, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Boxer L, Dang C. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 167.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 168.Porter MJ, Field JK, Leung SF, et al. The detection of the c-myc and ras oncogenes in nasopharyngeal carcinoma by immunohistochemistry. Acta Otolaryngol. 1994;114:105–109. doi: 10.3109/00016489409126025. [DOI] [PubMed] [Google Scholar]
- 169.Chen H, Hutt-Fletcher L, Cao L, Hayward SD. A positive autoregulatory loop of LMP1 expression and STAT activation in epithelial cells latently infected with Epstein-Barr virus. J Virol. 2003;77:4139–4148. doi: 10.1128/JVI.77.7.4139-4148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Scolnick D, Halazonetis T. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406:430–435. doi: 10.1038/35019108. [DOI] [PubMed] [Google Scholar]
- 171.Toyota M, Sasaki Y, Satoh A, et al. Epigenetic inactivation of CHFR in human tumors. Proc Natl Acad Sci U S A. 2003;100:7818–7823. doi: 10.1073/pnas.1337066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hui A, Lo K, Leung S, et al. Detection of recurrent chromosomal gains and losses in primary nasopharyngeal carcinoma by comparative genomic hybridisation. Int J Cancer. 1999;82:498–503. doi: 10.1002/(sici)1097-0215(19990812)82:4<498::aid-ijc5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 173.Lo KW, Huang DP. Genetic and epigenetic changes in nasopharyngeal carcinoma. Semin Cancer Biol. 2002;12:451–462. doi: 10.1016/s1044579x02000883. [DOI] [PubMed] [Google Scholar]
- 174.Koga Y, Kitajima Y, Miyoshi A, Sato K, Sato S, Miyazaki K. The significance of aberrant CHFR methylation for clinical response to microtubule inhibitors in gastric cancer. J Gastroenterol. 2006;41:133–139. doi: 10.1007/s00535-005-1732-7. [DOI] [PubMed] [Google Scholar]
- 175.Chaturvedi P, Sudakin V, Bobiak ML, et al. Chfr regulates a mitotic stress pathway through its RING-finger domain with ubiquitin ligase activity. Cancer Res. 2002;62:1797–1801. [PubMed] [Google Scholar]
- 176.Vleminckx K, Vakaet LJ, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 177.Franchi A, Gallo O, Boddi V, Santucci M. Prediction of occult neck metastases in laryngeal carcinoma: role of proliferating cell nuclear antigen, MIB-1, and E-cad-herin immunohistochemical determination. Clin Cancer Res. 1996;2:1801–1808. [PubMed] [Google Scholar]
- 178.Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. 1997;89:1260–1270. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 179.Rosenthal EL, Johnson TM, Allen ED, Apel IJ, Punturieri A, Weiss SJ. Role of the plasminogen activator and matrix metalloproteinase systems in epidermal growth factor- and scatter factor-stimulated invasion of carcinoma cells. Cancer Res. 1998;58:5221–5230. [PubMed] [Google Scholar]
- 180.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 181.Fingleton B, Vargo-Gogola T, Crawford HC, Matrisian LM. Matrilysin (MMP-7) expression selects for cells with reduced sensitivity to apoptosis. Neoplasia. 2001;3:459–468. doi: 10.1038/sj.neo.7900190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10:415–433. doi: 10.1006/scbi.2000.0379. [DOI] [PubMed] [Google Scholar]
- 183.Rosenthal EL, Matrisian LM. Matrix metalloproteases in head and neck cancer. Head Neck. 2006;28:639–648. doi: 10.1002/hed.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lu J, Chua HH, Chen SY, Chen JY, Tsai CH. Regulation of matrix metalloproteinase-1 by Epstein-Barr virus proteins. Cancer Res. 2003;63:256–262. [PubMed] [Google Scholar]
- 185.Kondo S, Wakisaka N, Schell M, et al. Epstein-Barr virus latent membrane protein 1 induces the matrix met-alloproteinase-1 promoter via an Ets binding site formed by a single nucleotide polymorphism: enhanced susceptibility to nasopharyngeal carcinoma. Int J Cancer. 2005;115:368–376. doi: 10.1002/ijc.20849. [DOI] [PubMed] [Google Scholar]
- 186.Nasr HB, Mestiri S, Chahed K, et al. Matrix metallo-proteinase-1 (−1607) 1G/2G and -9 (−1562) C/T promoter polymorphisms: susceptibility and prognostic implications in nasopharyngeal carcinomas. Clin Chim Acta. 2007;384:57–63. doi: 10.1016/j.cca.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 187.Crabbe T, O’Connell JP, Smith BJ, Docherty AJ. Reciprocated matrix metalloproteinase activation: a process performed by interstitial collagenase and progelatinase A. Biochemistry. 1994;33:14419–14425. doi: 10.1021/bi00252a007. [DOI] [PubMed] [Google Scholar]
- 188.Wong T, Kwong D, Sham J, Wei W, Kwong Y, Yuen A. Clinicopathologic significance of plasma matrix metallo-proteinase-2 and -9 levels in patients with undifferentiated nasopharyngeal carcinoma. Eur J Surg Oncol. 2004;30:560–564. doi: 10.1016/j.ejso.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 189.Zhou G, Zhai Y, Cui Y, et al. Functional polymorphisms and haplotypes in the promoter of the MMP2 gene are associated with risk of nasopharyngeal carcinoma. Hum Mutat. 2007;28:1091–1097. doi: 10.1002/humu.20570. [DOI] [PubMed] [Google Scholar]
- 190.Horikawa T, Yoshizaki T, Sheen TS, Lee SY, Furukawa M. Association of latent membrane protein 1 and matrix metalloproteinase 9 with metastasis in nasopharyngeal carcinoma. Cancer. 2000;89:715–723. doi: 10.1002/1097-0142(20000815)89:4<715::aid-cncr1>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 191.Takeshita H, Yoshizaki T, Miller W, et al. Matrix metal-loproteinase 9 expression is induced by Epstein-Barr virus latent membrane protein 1 C-terminal activation regions 1 and 2. J Virol. 1999;73:5548–5555. doi: 10.1128/jvi.73.7.5548-5555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zeng L, Liu Y, Tao Y, Ai M, Zhao X, Cao Y. Cross-talk between c-Jun/Ets1 involved in EB virus-encoded latent membrane protein 1 regulates expression of matrix metalloproteinase-9 in nasopharyngeal carcinoma. Zhonghua Zhong Liu Za Zhi. 2005;27:204–208. [PubMed] [Google Scholar]
- 193.Zhou J, Ma J, Zhang B, et al. BRD7, a novel bromodo-main gene, inhibits G1-S progression by transcriptionally regulating some important molecules involved in ras/MEK/ERK and Rb/E2F pathways. J Cell Physiol. 2004;200:89–98. doi: 10.1002/jcp.20013. [DOI] [PubMed] [Google Scholar]
- 194.Zhang XM, Wang XY, Sheng SR, Wang JR, Li J. Expression of tumor related genes NGX6, NAG-7, BRD7 in gastric and colorectal cancer. World J Gastroenterol. 2003;9:1729–1733. doi: 10.3748/wjg.v9.i8.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Peng C, Liu HY, Zhou M, et al. BRD7 suppresses the growth of nasopharyngeal carcinoma cells (HNE1) through negatively regulating beta-catenin and ERK pathways. Mol Cell Biochem. 2007;303:141–149. doi: 10.1007/s11010-007-9466-x. [DOI] [PubMed] [Google Scholar]
- 196.Zhou M, Liu H, Xu X, et al. Identification of nuclear localization signal that governs nuclear import of BRD7 and its essential roles in inhibiting cell cycle progression. J Cell Biochem. 2006;98:920–930. doi: 10.1002/jcb.20788. [DOI] [PubMed] [Google Scholar]
- 197.Liu H, Peng C, Zhou M, et al. Cloning and characterization of the BRD7 gene promoter. DNA Cell Biol. 2006;25:346–358. doi: 10.1089/dna.2006.25.346. [DOI] [PubMed] [Google Scholar]
- 198.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 199.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prosta-glandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 200.Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J Clin Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cornwall H, Odukoya O, Shklar G. Oral mucosal tumor inhibition by ibuprofen. J Oral Maxillofac Surg. 1983;41:795–800. doi: 10.1016/s0278-2391(83)80046-9. [DOI] [PubMed] [Google Scholar]
- 202.Nishimura G, Yanoma S, Mizuno H, Kawakami K, Tsukuda M. A selective cyclooxygenase-2 inhibitor suppresses tumor growth in nude mouse xenografted with human head and neck squamous carcinoma cells. Jpn J Cancer Res. 1999;90:1152–1162. doi: 10.1111/j.1349-7006.1999.tb00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chan CM, Ma BB, Wong SC, Chan AT. Celecoxib induces dose dependent growth inhibition in nasopharyngeal carcinoma cell lines independent of cyclooxygen-ase-2 expression. Biomed Pharmacother. 2005;59(Suppl 2):S268–S271. doi: 10.1016/s0753-3322(05)80043-5. [DOI] [PubMed] [Google Scholar]
- 204.Rhee C, Sen M, Lu D, et al. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002;21:6598–6605. doi: 10.1038/sj.onc.1205920. [DOI] [PubMed] [Google Scholar]
- 205.Lin YC, You L, Xu Z, et al. Wnt inhibitory factor-1 gene transfer inhibits melanoma cell growth. Hum Gene Ther. 2007;18:379–386. doi: 10.1089/hum.2006.005. [DOI] [PubMed] [Google Scholar]
- 206.He B, Reguart N, You L, et al. Blockade of Wnt-1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene. 2005;24:3054–3058. doi: 10.1038/sj.onc.1208511. [DOI] [PubMed] [Google Scholar]
- 207.Kim J, You L, Xu Z, et al. Wnt inhibitory factor inhibits lung cancer cell growth. J Thorac Cardiovasc Surg. 2007;133:733–737. doi: 10.1016/j.jtcvs.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 208.Kuhnert F, Davis CR, Wang HT, et al. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci U S A. 2004;101:266–271. doi: 10.1073/pnas.2536800100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lee M, Hoffman J, Kuhnert F, et al. Systemically expressed Wnt inhibitors as probes for Wnt pathway functions in cancer and normal adult physiology. Presented at the Keystone Symposium on Wnt and Beta-Catenin Signaling in Developmental and Disease; Snowbird, UT. April 7–12, 2006; Abstract 214. [Google Scholar]