

Abstract
High-fat diet has been implicated as a major cause of insulin resistance and dyslipidemia. The objective of this study was to evaluate the impact of dietary-supplementation of chromium (d-phenylalanine)3 [Cr(d-Phe)3] on -glucose and -insulin tolerance in high-fat diet fed mice. C57BL/6-mice were randomly assigned to orally receive vehicle or Cr(d-Phe)3 (45 μg of elemental chromium/kg/day) for 8-weeks. High-fat-fed mice exhibited impaired whole-body -glucose and- insulin tolerance and elevated serum triglyceride levels compared to normal chow-fed mice. Insulin-stimulated glucose up- take in the gastrocnemius muscles, assessed as 2-[3H-deoxyglucose] incorporation was markedly diminished in high-fat fed mice compared to control mice. Treatment with chromium reconciled the high-fat diet-induced alterations in carbohydrate and lipid metabolism. Treatment of cultured, differentiated myotubes with palmitic acid evoked insulin resistance as evidenced by lower levels of insulin-stimulated Akt-phosphorylation, elevated JNK-phosphorylation, (assessed by Western blotting), attenuation of phosphoinositol-3-kinase activity (determined in the insulin-receptor substrate-1-immunoprecipitates by measuring the extent of phosphorylation of phosphatidylinositol by γ-32P-ATP), and impairment in cellular glucose up-take, all of which were inhibited by Cr(d-Phe)3. These results suggest a beneficial effect of chromium-supplementation in insulin resistant conditions. It is likely that these effects of chromium may be mediated by augmenting downstream insulin signaling.
1. Introduction
Type 2 diabetes is increasing in epidemic proportions and it is predicted that > 366 million people will afflicted globally by the year 2030 [1]. In most cases, metabolic syndrome, characterized by insulin resistance, dyslipidemia and hypertension is often observed years before the development of frank observable diabetes. Although the etiology of type 2 diabetes remain poorly understood, lifestyle evolution and excessive intake of fat-enriched diet have been attributed as major determinants of this fast-growing epidemic [2]. Several human and animal studies have demonstrated an association between high-fat feeding and insulin resistance, suggesting a causal role of high-fat diet in the pathophysiology of type 2 diabetes [3-5].
Diet and exercise have beneficial effects in slowing the progression of the disease, although, alone they provided little reprieve from the current type-2 epidemic. Drugs such as sulfonylurea's and glitazones can be used to treat insulin resistance but are known to cause weight gain and adverse cardiovascular effects. Identification and development of novel molecules and molecular targets to treat or prevent insulin resistance and thus to pre-empt the development of type-2 diabetes is therefore a strategy that merits attention. In this context, attention has been drawn towards a variety of mineral and dietary supplements that may alleviate insulin resistance. Chromium is an ‘essential’ nutrient for the maintenance of normal glucose tolerance [6]. Consequently, chromium deficiency is thought to precipitate symptoms of type-2 diabetes and supplementation with chromium corrects some of these symptoms. Accordingly, several human and animal studies have demonstrated that chromium administration improves glucose tolerance and insulin sensitivity in type-2 diabetes and obesity [6, 7]. Despite these documented evidence of the benefits of chromium, there have been studies contradicting such findings and questioning the wisdom of using chromium in treating diabetic patients [8]. Recent studies indicating that the popular commercially available chromium supplement may cause developmental toxicities [9]. Although the exact mechanisms of toxicity of chromium complexes are unknown, studies by Shi and coworkers indicate that chromium undergoes redox activation to generate reactive oxygen species in the presence of hydrogen peroxide [10]. The fact that the picolinate ligand was attributed to these toxicities [11] prompted us to design and synthesize a novel chromium complex with the non-essential amino acid, d-phenylalanine, as ligand. Unlike chromium picolinate, Cr(d-Phe)3 did not cause DNA-damage [12] and was found to be more potent than picolinate in augmenting glucose-uptake in animal models of obesity [13]. However, the effect of Cr(d-Phe)3 on fatty acid-induced insulin resistance has not been evaluated thus far, although dietary fat is the major contributor of obesity and cardiovascular disease. Based on this background, the objective of this study was to examine the impact of oral supplementation with Cr(d-Phe)3 on glucose and insulin tolerance in a high-fat-fed mouse model of insulin resistance. In addition, in an effort to elucidate the potential molecular mechanisms, the effect of Cr(d-Phe)3 on free fatty acid-induced insulin resistance in cultured myotubes was also investigated.
2. Experimental Section
2.1. Materials and reagents
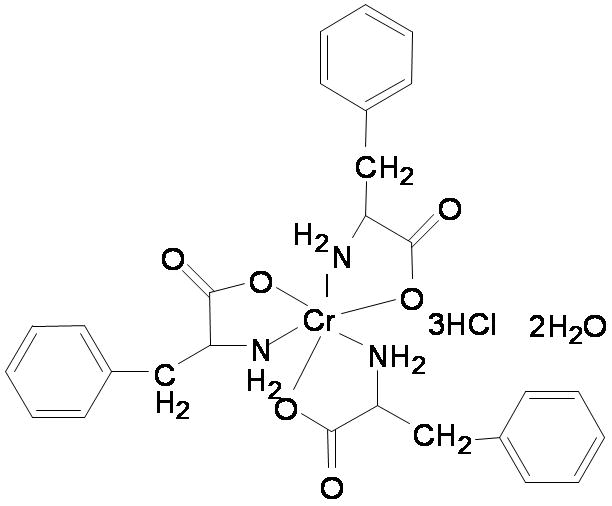
Cr(d-Phe)3 (Fig. 1) was synthesized and characterized as described previously [14]. Insulin receptor substrate-1 (IRS-1) antibody was obtained from Upstate NY, NY whereas all other antibodies were obtained from Cell Signaling Technology Inc. (Beverly, MA). Assay kits for IL-6 and C-reactive protein were purchased from Pierce Biotechnology (Rockford, IL) and GenWay Biotech, Inc. (San Diego, CA). Serum levels of triglycerides, total cholesterol and high-density-lipoprotein (HDL) were measured using commercial kits (from Equal Diagnostics, Exton, PA) per manufacturer's protocol.
Figure 1. Chemical structure of Cr(d-Phe)3.
2.2. Animal treatment protocol, intraperitoneal -glucose and -insulin tolerance tests, muscle glucose uptake, determination of serum lipids and inflammatory markers
The experimental procedure described here was approved by the University of Wyoming Institutional Animal Care and Use Committee. Four-month-old male C57BL/6 mice (n=24) were randomly assigned receive normal control chow (10% w/w fat, Research Diets, D12451), or high-fat diet (45.0% w/w lard fat, Research Diets 12450B) for a period of 8-weeks. One group of mice that received high-fat diet were supplemented with Cr(d-Phe)3, provided in the drinking water. On the basis of water intake, Cr(d-Phe)3 was administered to provide an intake of about 150 μg/kg/day corresponding to about 45μg of elemental chromium [14]. Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were performed as described previously [12, 14]. At the end of the 8-weeks feeding, the mice were fasted overnight and killed by cervical dislocation. The gastrocnemius muscle were dissected, frozen immediately in liquid nitrogen and stored at -80 °C until use. 2-deoxy-d-[3H] glucose deoxyglucose uptake into gastrocnemius muscles were determined as reported previously [15]. Body mass and organ mass were measured with a standard laboratory scale. Blood was collected in microfuge tubes and the serum separated by centrifuging the blood at 1000 g at 4 °C and stored at -80 °C. Serum levels of triglycerides, total cholesterol, HDL, TNF-α, IL-6 and C-reactive proteins were measured using commercial kits.
2.3. Cell culture, Western blotting, glucose uptake and PI3-kinase assay
Mouse C2C12 myoblasts (ATCC, Manassas, VA) were cultured and differentiated to myotubes as reported previously [16]. Fully, differentiated myotubes were rendered quiescent in the presence of serum-free media and were treated with or without Cr(d-Phe)3 (0-100 μM) and/or palmitate (0-2 mM). Palmitate-containing media were prepared by conjugation of palmitic acid with fatty acid-free bovine serum albumin (BSA), by a method described previously [17]. Following treatment, cells were homogenized and subjected to Western blot analysis using antibodies raised against phospho-cJun N-terminal kinases (pJNK), total JNK, phospho-Akt, total AKT and phospho-insulin receptor beta [18]. Wherever indicated, blots were stripped and reprobed with antibodies directed at the total protein. The intensities of bands were measured with a scanning densitometer (Model GS-800; Bio-Rad) coupled with Bio-Rad PC analysis software. Cellular 2-deoxy-d-[3H] glucose-uptake was assessed as described previously [14]. IRS-associated PI-3-kinase activity was determined in the immunoprecipitates of IRS-1 by assessing the [32P]-mediated phosphorylation of PI(3)P by thin layer chromatography (TLC) as reported previously [19].
2.4. Data analysis
Data are expressed as mean ± SEM and statistically evaluated using one-way analysis of variance (ANOVA) using Graph Pad Prism (Graph Pad Software, Inc. San Diego, CA, USA). A ‘p’ value of less than 0.05 was considered to be statistically significant.
3. Results
3.1. General features of high-fat-diet fed mice with or without Cr(d-Phe)3 supplementation
Treatment of mice for 8-weeks with a high-fat-diet failed to induce any overt gain in total body-mass or organ (heart, liver, and kidney) masses compared to the mice that received normal-chow-diet (Table 1). Furthermore, treatment with chromium failed to alter any of the aforementioned parameters. In contrast however, mice fed with high-fat-diet had elevated serum glucose and serum insulin levels which were normalized in the chromium-treated group. Food-intake did not vary among the groups tested (data not shown).
Table 1.
General features of standard chow and high-fat-diet- fed mice with or without 8-week treatment of Cr(d-Phe)3 at 45 μg/kg/d.
| Standard Chow | HF Diet | HF Diet + Cr(D-Phe)3 | |
|---|---|---|---|
| Fasting glucose (mg/dL) | 97.2 + 7.38 | 130.8 + 9.18* | 99.72 + 7.74 |
| Fasting insulin (ng/ml) | 0.24 ± 0.05 | 1.00 ± 0.27** | 0.13 ± 0.06 |
| Body mass (g) | 27.88 ± 0.67 | 28.11 ± 1.01 | 27.86 ± 1.68 |
| Heart mass (g) | 0.19 ± 0.01 | 0.18 ± 0.01 | 0.18 ± 0.01 |
| Liver mass (g) | 1.36 ± 0.05 | 1.34 ± 0.02 | 1.34 ± 0.06 |
| Kidney mass (g) | 0.31 ± 0.02 | 0.29 ± 0.02 | 0.27 ± 0.02 |
p < 0.05,
p < 0.001
3.2. Effect of Cr(d-Phe)3 on whole-body glucose and insulin tolerance
Following acute intraperitoneal glucose challenge, blood glucose levels in normal chow–fed mice started to decline after peaking at 30 min and nearly returned to baseline after 120 min (Fig. 2). In contrast, post-challenge blood glucose levels remained at much higher levels between 30 and 120 min in high-fat–fed mice indicating glucose intolerance. In high-fat-fed mice that received chromium supplementation, at all time points from 30-120 min, post-challenge blood glucose levels were significantly lower compared to high-fat-fed mice that received vehicle. The post blood glucose levels of the chromium treated high-fat-fed mice and were not distinguishable from the corresponding blood glucose levels of the mice that received normal chow. In mice receiving normal chow, acute challenge with intraperitoneal injections of insulin resulted in a significant drop in blood glucose levels during the first 60 min reverting back to basal levels in 120 min (Fig. 2). Mice receiving high-fat diet exhibited similar trend although the post-insulin-challenge blood glucose levels were higher at all points compared to the mice that received normal chow. Post-insulin glucose levels were unchanged in the chromium treated animals at 30, 60 and 90 min following insulin challenge. However at the 120 min time-point the glucose level was significantly lower in the chromium supplemented mice compared to the mice that received vehicle.
Figure 2.
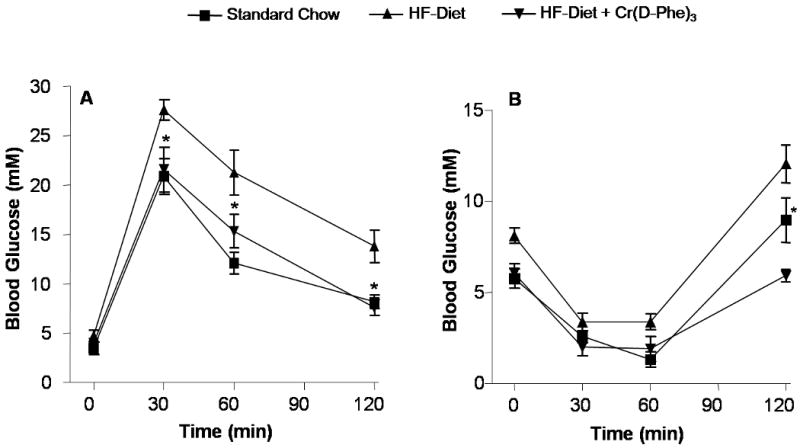
Effect oral supplementation with Cr(d-Phe)3 on glucose tolerance. (A) Blood glucose concentrations following glucose challenge (1 g/kg) at different time points. Values are means ± SEM (n = 8-12 per group), and *p < 0.05, compared to high-fat-fed, vehicle treated mice. (B) Blood glucose levels following insulin challenge (1.5 IU/Kg). Data are represented as means ± SEM (n = 8-12), *p < 0.01 compared to high-fat-fed, vehicle-treated mice.
3.3. Effect of Cr(d-Phe)3 on serum lipids and inflammatory markers
Mice that received the high-fat diet had elevated serum triglyceride, total cholesterol and low-density-lipoprotein (LDL) cholesterol without any changes in the levels of HDL-cholesterol (Fig. 3A-D). High-fat-fed mice that were supplemented with Cr(d-Phe)3 had significantly lower levels of triglycerides compared to those that received the vehicle. However, chromium treatment failed to negate the elevated LDL and total cholesterol-levels associated with high-fat feeding. Mice that received the high-fat diet had over four-fold higher levels of serum IL-6 compared to those that received normal chow which returned to basal levels following chromium supplementation (Fig 3 E.). In contrast however, no significant changes were observed in the serum levels of C-reactive with high-fat feeding or chromium supplementation (Fig 3 F). TNF-α levels were undetectable in the serum sample of these mice (data not shown).
Figure 3.
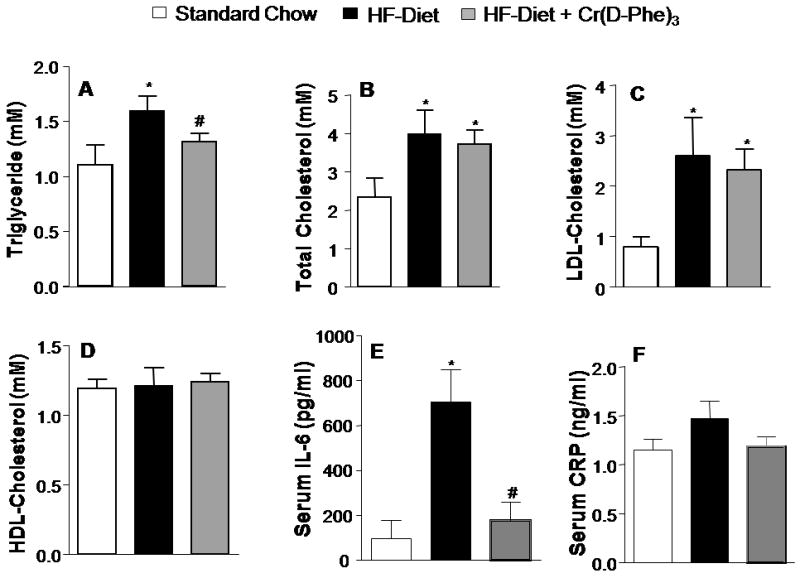
Effect of oral supplementation with Cr(d-Phe)3 on serum lipid profile and inflammatory markers (A) triglycerides (B) total cholesterol (C) LDL-cholesterol (D) Dr. HDL-cholesterol (E) IL-6 and (F) C-reactive protein. Data are presented as mean ± SEM (n = 8 per group), *p < 0.05 vs standard chow, #p < 0.05 vs high-fat diet, vehicle-treated animals).
3.4. Effect of Cr(d-Phe)3 glucose uptake in skeletal muscle and cultured myotubes
Glucose up-take in isolated gastrocnemius muscle was severely impaired in high-fat-fed mice compared to those that received normal chow. This high-fat-diet-induced peripheral insulin resistance was completely reversed in mice that received supplemental chromium (Fig. 4). In cultured, differentiated myotubes, insulin (100 nM, 10 min) induced a two-fold increase in cellular glucose up-take (Fig. 5). Pretreatment of the myotubes with the palmitic acid (0.4 mM, 6h), induced significant insulin resistance as evidenced by the blunting of the effects of insulin. Pre-treatment of the myotubes with Cr(d-Phe)3, negated the effects of palmitate on glucose up-take in a concentration-dependent manner, and a complete reversal of the effect of palmitate was observed in the presence of 100μM Cr(d-phe)3.
Figure 4.
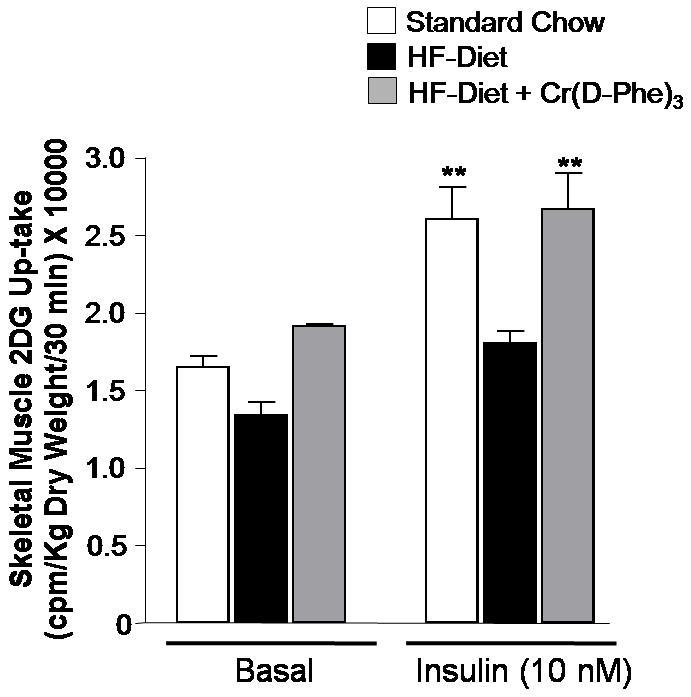
Effect of oral supplementation with Cr(d-Phe)3 on basal and insulin-stimulated skeletal muscle glucose up-take. Data are presented as mean ± SEM (n=8 per group), **p < 0.01 skeletal muscle glucose uptake in high-fat-fed mice compared to vehicle treatment.
Figure 5.
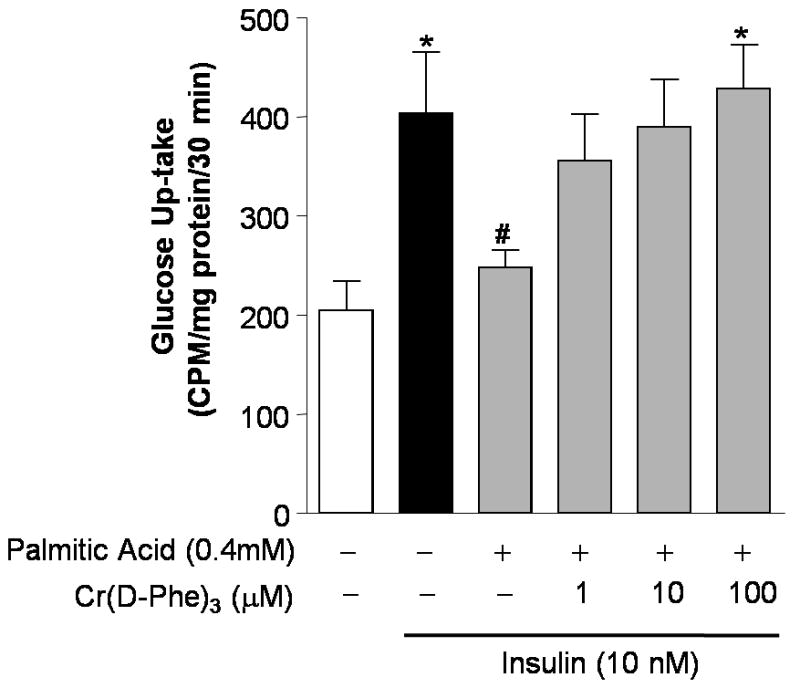
Effect of Cr(d-Phe)3 on glucose up-take in insulin resistant cultured myotubes. Cr(D-Phe)3 has caused concentration-dependent reversal of the blunting of glucose up-caused by palmitate. Data are represented as means ± SEM, *p<0.05 (n = 4) compared to palmitic acid 0.4mM, #p < 0.05 compared to insulin 100nM).
3.5. Effect of Cr(d-Phe)3-treatment on insulin-signaling proteins
Treatment of cultured myotubes with palmitic acid resulted in a concentration-dependent inhibition of insulin-stimulated phosphorylation of Akt and a concomitant increase in the phosphorylation of JNK (Fig. 6A). Both these effects were reversed by Cr(d-Phe)3 as shown in Fig. 6B. Figure 6C demonstrates a concentration-dependent reversal of the effect of palmitic acid on Akt phosphorylation by Cr(d-Phe)3. In contrast however, chromium treatment failed to reverse the palmitate-induced attenuation of insulin-stimulated phosphorylation of insulin receptor beta (Fig. 6C). Palmitic acid treatment also caused a reduction in PI3-kinase activity by about 40% which was inhibited in part by the presence of chromium (100 μM) (Fig 7). At 10 μM concentration chromium failed to show any effect on PI3-kinase activity.
Figure 6.
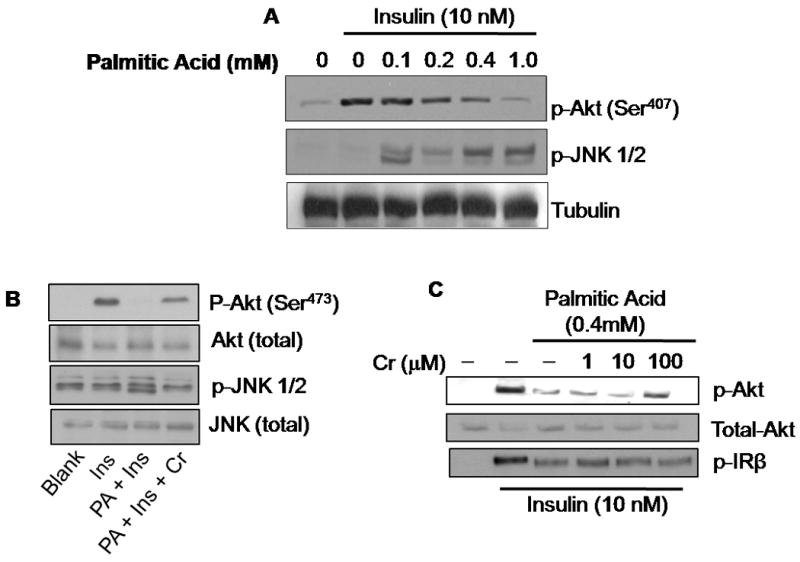
Effect of Cr(d-Phe)3 on insulin signaling in insulin resistant myotubes. Myotubes treated with palmitate (0-1 mM) for 16 h prior to stimulation with insulin (10 nM, 10 min), and immunoblotted with the indicated antibodies in the absence (A) or presence (B) of chromium (100 μM) prior to stimulation with insulin (10 nM, 10 min) and immunoblotted for p-Akt, and p-JNK. Blots were reprobed for total-Akt and total JNK respectively (C). Concentration dependent effect of chromium on the insulin stimulated- phosphorylation of Akt and insulin receptor β in the presence and absence of palmitate.
Figure 7.
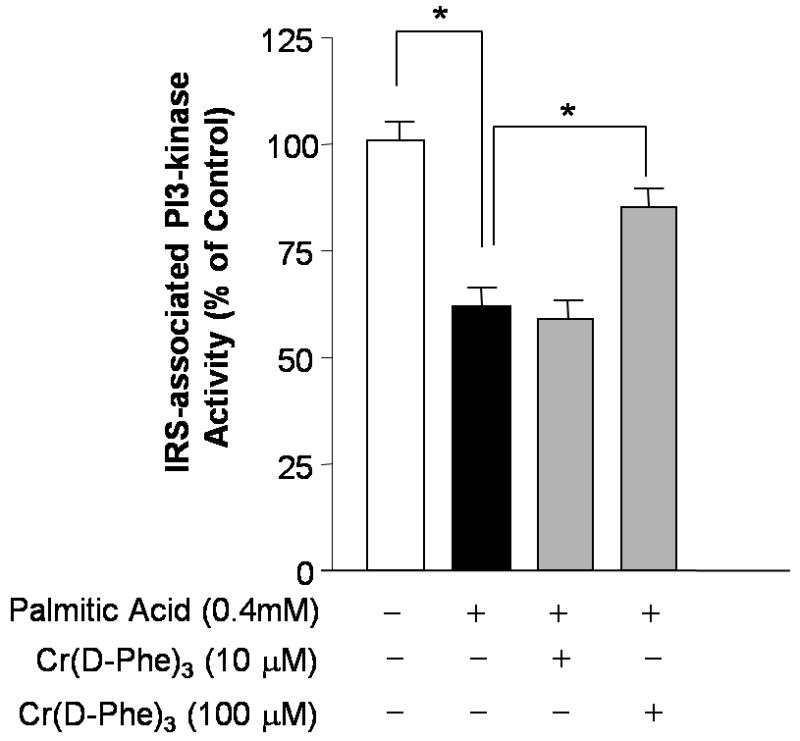
Effect of palmitate, in the absence and presence of Cr(d-Phe)3, on insulin-stimulated PI3-kinase activity in immunoprecipitates of IRS-1 complex from cultured myotubes. The data represent the mean ± SEM of triplicates from two independent experiments. *p < 0.05 (n = 3) between the indicated groups.
4. Discussion
The major findings of this study are that high-fat diet feeding elicits glucose intolerance, insulin insensitivity, and impaired skeletal muscle glucose uptake, all of which were attenuated by oral supplementation with Cr(d-Phe)3. In addition to correcting high-fat feeding-induced insulin and glucose resistance, supplementation with chromium resulted in a lowering of serum triglycerides which were elevated following high-fat feeding. In contrast, chromium feeding failed to lower the elevated levels of serum LDL and total cholesterol in high-fat fed mice.
Mice fed on a high-fat diet exhibited severe impairment in skeletal muscle glucose up-take compared to normal-chow-fed mice. Chromium supplementation caused a significant improvement in skeletal muscle glucose up-take in these mice. To further understand the mechanism of action of chromium in alleviating insulin resistance associated with high fat diet, insulin-resistance was evoked in cultured myotubes by treating them with palmitic acid. Insulin resistance in palmitate-treated cells was characterized by impaired 2-DG up-take, attenuation in Akt-phosphorylation, elevated JNK-phosphorylation and inhibition of PI3-kinase activity – all key regulators of insulin signal transduction. Insulin binds to the insulin receptor resulting in phosphorylation of insulin receptor beta. Phosphorylated insulin receptor beta functions as a kinase and phosphorylates downstream substrates such as IRS-1 resulting in the activation of PI3-kinase and phosphorylation of Akt. Phosphorylated Akt helps in the recruitment of Glut-4 vesicles to cell surface which mediates cellular glucose uptake. JNK is believed to cause insulin resistance by causing the phosphorylation of IRS-1 at the inhibitory site Ser-307 [20]. Phosphorylation of the serine residue prevents the insulin-stimulated tyrosine phosphorylation of IRS1, thereby preventing downstream signaling pathways, such as the phosphatidylinositol 3′-kinase (PI3K)-Akt cascade [21]. The results presented here demonstrate that chromium supplementation reconciled the effects of palmitate on insulin signaling proteins. These results are in accordance to that reported by Wang and coworkers who demonstrated that oral supplementation with chromium picolinate augments insulin-induced tyrosine phosphorylation of IRS-1 and augmentation of PI-3 kinase activity [22]. Palmitate has been shown to inhibit insulin-stimulated tyrosine phosphorylation of insulin receptor substrate 2 and serine phosphorylation of Akt, through c-Jun NH2-terminal kinase (JNK) activation [23]. Given that chromium did not alter the levels of insulin-stimulated phosphorylation of insulin receptor it is likely that reversal of insulin resistance by chromium may be mediated via the inhibition of JNK.
Human studies have shown that elevated levels of proinflammatory markers such as C-reactive proteins (CRP and interleukin-6 (IL-6) are predictors of type-2 diabetes [24]. Upregulation of inflammatory mediators also represent one of the several mechanisms by which saturated-fatty acids induce insulin resistance [25, 26]. Recent studies by Jain and co-workers have demonstrated that supplementation with chromium lowers levels of TNF-α, IL-6 and CRP in streptozotocin induced diabetes [27]. The robust upregulation of IL-6 in high-fat fed mice and the ability of chromium to reconcile the elevated levels of IL-6 observed in this study substantiate the view that chromium down-regulates inflammation associated with diabetes [28].
Together, these findings in high-fat-fed mice and in cultured myocytes rendered insulin resistant by treating with high-fatty acid indicate that Cr(d-Phe)3 alleviates insulin resistance by augmenting insulin signaling. This may, at least in part, explain some of the beneficial effects attributed to chromium complexes in the treatment of insulin resistance in type 2-diabetes.
Acknowledgments
This work was supported by grants from ADA (AMDIAB47595), NCRR and the Wyoming INBRE (P20RR016474).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smyth S, Heron A. Nat Med. 2006;12:75–80. doi: 10.1038/nm0106-75. [DOI] [PubMed] [Google Scholar]
- 2.Grundy SM. Arterioscler. Thromb Vasc Biol. 2008;28:629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 3.Ahren B, Scheurink AJ. Eur J Endocrinol. 1998;139:461–467. doi: 10.1530/eje.0.1390461. [DOI] [PubMed] [Google Scholar]
- 4.Chopra M, Galbraith S, Darnton-Hill I. Bull World Health Organ. 2002;80:952–958. [PMC free article] [PubMed] [Google Scholar]
- 5.Shoelson SE, Lee J, Goldfine AB. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau FC, Bagchi M, Sen CK, Bagchi D. Mol Cell Biochem. 2008;317:1–10. doi: 10.1007/s11010-008-9744-2. [DOI] [PubMed] [Google Scholar]
- 7.Wang ZQ, Cefalu WT. Curr Diab Rep. 2010;10:145–151. doi: 10.1007/s11892-010-0097-3. [DOI] [PubMed] [Google Scholar]
- 8.Kleefstra N, Houweling ST, Jansman FG, Groenier KH, Gans RO, Meyboom-de Jong B, Bakker SJ, Bilo HJ. Diabetes Care. 2006;29:521–525. doi: 10.2337/diacare.29.03.06.dc05-1453. [DOI] [PubMed] [Google Scholar]
- 9.Hepburn DD, Xiao J, Bindom S, Vincent JB, O'Donnell J. Proc Natl Acad Sci U S A. 2003;100:3766–3771. doi: 10.1073/pnas.0636646100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi X, Dalal NS, Kasprzak KS. Arch Biochem Biophys. 1993;302:294–299. doi: 10.1006/abbi.1993.1213. [DOI] [PubMed] [Google Scholar]
- 11.Bagchi D, Stohs SJ, Downs BW, Bagchi M, Preuss HG. Toxicology. 2002;180:5–22. doi: 10.1016/s0300-483x(02)00378-5. [DOI] [PubMed] [Google Scholar]
- 12.Yang X, Li SY, Dong F, Ren J, Sreejayan N. J Inorg Biochem. 2006;100:1187–1193. doi: 10.1016/j.jinorgbio.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 13.Dong F, Yang X, Sreejayan N, Ren J. Obesity (Silver Spring) 2007;15:2699–2711. doi: 10.1038/oby.2007.322. [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Palanichamy K, Ontko AC, Rao MN, Fang CX, Ren J, Sreejayan N. FEBS Lett. 2005;579:1458–1464. doi: 10.1016/j.febslet.2005.01.049. [DOI] [PubMed] [Google Scholar]
- 15.Kandadi MR, Rajanna PK, Unnikrishnan MK, Boddu SP, Hua Y, Li J, Du M, Ren J, Sreejayan N. Biochem Pharmacol. 2010;79:623–631. doi: 10.1016/j.bcp.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong F, Hua Y, Zhao P, Ren J, Du M, Sreejayan N. J Nutr Biochem. 2009;20:992–999. doi: 10.1016/j.jnutbio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA. J Biol Chem. 2003;278:10297–10303. doi: 10.1074/jbc.M212307200. [DOI] [PubMed] [Google Scholar]
- 18.Sreejayan N, Dong F, Kandadi MR, Yang X, Ren J. Obesity (Silver Spring) 2008;16:1331–1337. doi: 10.1038/oby.2008.217. [DOI] [PubMed] [Google Scholar]
- 19.Ridderstrale M, Degerman E, Tornqvist H. J Biol Chem. 1995;270:3471–3474. doi: 10.1074/jbc.270.8.3471. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre V, Uchida T, Yenush L, Davis R, White MF. J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi CM, Emanuelli B, Kahn CR. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 22.Wang ZQ, Zhang XH, Russell JC, Hulver M, Cefalu WT. J Nutr. 2006;136:415–420. doi: 10.1093/jn/136.2.415. [DOI] [PubMed] [Google Scholar]
- 23.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Proc Natl Acad Sci U S A. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. J Am Med Assn. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy A, Martinez K, Chuang CC, LaPoint K, McIntosh M. J Nutr. 2009;139:1–4. doi: 10.3945/jn.108.098269. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg GR. Cell Cycle. 2007;6:888–894. doi: 10.4161/cc.6.8.4135. [DOI] [PubMed] [Google Scholar]
- 27.Jain SK, Rains JL, Croad JL. Free Radic Biol Med. 2007;43:1124–1131. doi: 10.1016/j.freeradbiomed.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vinson JA. Free Radic Biol Med. 2007;43:1121–1123. doi: 10.1016/j.freeradbiomed.2007.06.029. [DOI] [PubMed] [Google Scholar]